





Abstract
Familial short stature (FSS) is a term describing a growth disorder that is vertically transmitted. Milder forms may result from the combined effect of multiple genes; more severe short stature is suggestive of a monogenic condition. The etiology of most FSS cases has not been thoroughly elucidated to date.
To identify the genetic etiology of severe FSS in children treated with GH because of the diagnosis of small for gestational age or GH deficiency (SGA/GHD).
Of 736 children treated with GH because of GHD/SGA, 33 with severe FSS (life-minimum height −2.5 SD or less in both the patient and shorter parent) were included in the study. The genetic etiology was known in 5 of 33 children prior to the study [ACAN (in 2], NF1, PTPN11, and SOS1). In the remaining 28 of 33, whole-exome sequencing was performed. The results were evaluated using American College of Medical Genetics and Genomics standards and guidelines.
In 30 of 33 children (90%), we found at least one variant with potential clinical significance in genes known to affect growth. A genetic cause was elucidated in 17 of 33 (52%). Of these children, variants in growth plate-related genes were found in 9 of 17 [COL2A1, COL11A1, and ACAN (all in 2), FLNB, FGFR3, and IGF1R], and IGF-associated proteins were affected in 2 of 17 (IGFALS and HMGA2). In the remaining 6 of 17, the discovered genetic mechanisms were miscellaneous (TRHR, MBTPS2, GHSR, NF1, PTPN11, and SOS1).
Single-gene variants are frequent among families with severe FSS, with variants affecting the growth plate being the most prevalent.
Children with familial short stature (FSS) form a substantial proportion of patients in pediatric endocrinology clinics. The term is commonly used to describe a growth disorder that is vertically transmitted; however, it has not been strictly defined yet. Traditionally, a short child is classified as having FSS if at least one of his or her parents is also short (height −2 SD or less in both the child and his or her shorter parent). The etiology of FSS is heterogeneous. Milder forms especially may result from the combined effect of multiple genes (polygenic inheritance) (1). On the other hand, children with more severe growth disorders often carry a single gene variant that has a major effect on their height: monogenic autosomal dominant (AD) inheritance (1).
In the evaluation of monogenic causes of FSS, multiple genes must be considered. SHOX deficiency is a prevalent growth disorder found in 2% to 15% of individuals who had been originally classified as having idiopathic short stature (2). SHOX protein is a transcriptional factor essential for the proper functioning of the growth plate. Because the disorder typically exhibits AD inheritance (except for the extremely rare autosomal recessive Langer mesomelic dysplasia), most cases present as FSS. The wide phenotypic spectrum in affected children ranges from asymmetric short stature with mesomelic shortening of the limbs and typical Madelung deformity (Léri-Weil syndrome) to apparently symmetric short stature (2). Multiple other genes responsible for AD growth plate disorders have been discovered. Examples include the COL2A1, COL11A1, COL9A2, COMP, MATN3, and ACAN genes that encode important components of the growth plate’s extracellular matrix (3–6) or the FGFR3 and NPR2 gene products representing proteins essential for paracrine signaling within the cartilage (7, 8).
Apart from growth plate disorders, there are multiple other causes of monogenic FSS. AD inherited GH deficiency (GHD) type II caused by mutations in the GH1 gene resulting in exon 3 skipping is one an example. A shortened GH isoform has a dominant-negative effect on the secretion of a full-length GH molecule (9). Among the genetic causes of multiple pituitary hormone deficiency (genes affecting pituitary morphogenesis or differentiation), several conditions can have AD inheritance (e.g., HESX1, POU1F1, LHX4, GLI2, and PITX2 gene variants) and were described as rare causes of FSS (10). GH insensitivity (IGF deficiency and IGF resistance) may also be inherited in an AD manner. Virtually all reported cases of IGF1R defects are heterozygotes; dominant-negative mutations of GHR have also been reported (1). To highlight the heterogeneity of the etiology of monogenic FSS, it is important to mention Noonan syndrome and additional AD RASopathies, e.g., PTPN11, SOS1, RAF1, and other genes that encode components of the RAS-MAPK signaling pathway, that affect cell proliferation, differentiation, survival, and metabolism (11).
Despite the substantial advance in understanding the genetic background of short stature, current practice still relies mostly on physical examination, laboratory parameters, and imaging methods. The standard medical evaluation identifies a pathological cause of short stature in 1% to 40% of cases (1). Thus, most short children with short parents do not know the real cause of their growth disorder and are classified using only the descriptive diagnosis of FSS.
The aim of our study was to reveal the genetic etiology of the growth disorder in children with GHD or small for gestational age (SGA), treated with GH, who were from families with FSS using next-generation sequencing (NGS) methods in a country with low consanguinity level.
Materials and Methods
Patients
Inclusion criteria
At our center, 736 children were being treated with GH. Of 555 children who were treated because of SGA or primary GHD, 33 children with severe FSS (life-minimum height −2.5 SD or less in both the patient and his or her shorter parent) were enrolled into the study. The detailed work flow of patient selection is summarized in Fig. 1. All study participants or their legal guardians signed written informed consent forms prior to genetic testing. The study was approved by the institutional ethics committees of the 2nd Faculty of Medicine, Charles University in Prague and University Hospital Motol, Czech Republic.
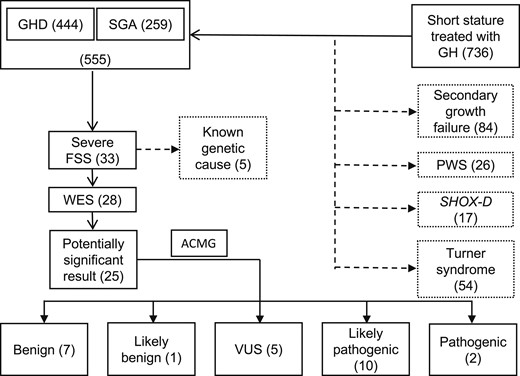
Work flow summarizing the selection of patients for the study and the genetic results. ACMG, American College of Medical Genetics; PWS; Prader-Willi syndrome; SHOX-D, short stature homeobox deficiency.
Evaluation prior to the study
All children’s heights were obtained during anthropometric measurement focused also on body proportionality (sitting height, arm span, subischiadical leg length), and information about their birth parameters were obtained from medical records. All the parents’ heights were measured to the nearest 1 mm, and the heights of more distant relatives were obtained from the parents. All the data were standardized according to recent normative values (12, 13). GHD and SGA were evaluated according to current guidelines. In all short children with auxological data suggestive of GHD and/or IGF-1 levels below −2 SD using reference ranges standardized for age and sex, GH provocation tests were performed. Children with peak GH concentration below 10 μg/L in two different provocation tests were classified as GHD (14). Sex-steroid priming was used in all children of 7 years of age or older. Children with birth weight and/or birth length below −2 SD using the reference ranges standardized for sex and gestational week who showed no evidence of catch-up growth at the age of 4 years (height −2.5 SD or less and growth velocity before treatment <0 SD) were treated with GH in SGA indication (15).
The median age of the study group was 10 years (range: 5 to 16 years), and GH treatment was initiated at 7 years (median; range: 3 to 13 years). The life-minimum height was −3.27 SD (−2.53 to −4.95 SD), and the shorter parent’s height was −2.89 SD (−2.59 to −4.03 SD). Twenty-one children (64%) were born with SGA, and their birth weight ranged from −0.60 to −3.01 SD (median −2.16) and birth length from −1.23 to −4.14 SD (median −3.00). Twenty-three children (70%) were classified as GHD. Their maximum GH level after stimulation was 6.7 µg/L (2.5-9.4 µg/L); 1 girl had ectopic posterior pituitary, and no child had multiple pituitary hormone deficiency. Eleven children (33%) fit both criteria—GHD and SGA. Of all 33 subjects, 18 (55%) had apparent symmetric FSS with no clinical signs of additional diseases. The clinical features of individual children are described in Table 1 in detail.
Clinical Features and Genetic Results of Children With Severe FSS Examined by WES
| Patient No. | Sex | Age, y | Patient`s Height (SDS) | Shorter Parent`s Height (SDS) | IGF-1 (SDS) | Stimulated GH, μg/L | Birth Weight (SDS) | Birth Length (SDS) | Additional Phenotypic Features | Gene | Mutation Status | Transcript Variant | Protein Variant | Parents Available for Genetic Testing | Conclusion |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SGA only – 5/8 (62.5%) with P/LP variant | |||||||||||||||
| 1 | F | 10 | −4.32 | −3.54 | −1.68 | 11.0 | −1.64 | −3.05 | — | COL11A1 | M/n | c.2921C>A | p.Pro974Gln | Yes | Likely pathogenic |
| 2 | M | 14 | −4.10 | −2.89 | −3.68 | 10.0 | −1.64 | −2.39 | Central hypothyroidism | TRHR | M/M | c.392T>C | p.Ile131Thr | Yes | Likely pathogenic (16) |
| 3 | M | 12 | −3.49 | −3.00 | −0.26 | 11.5 | −3.01 | −3.87 | — | THRA | M/n | c.455G>A | p.Arg152Gln | Yes | Uncertain significance |
| 4 | M | 10 | −3.42 | −3.03 | −0.98 | 10.2 | −1.62 | −3.26 | Advanced bone age (+1.9 years) | IHH | M/n | c.857C>T | p.Pro286Leu | No | Uncertain significance |
| 5 | M | 9 | −3.34 | −2.67 | −0.92 | NA | −2.13 | −3.52 | — | COL9A2 | M/n | c.1834G>A | p.Gly612Arg | Yes | Benign |
| 6 | M | 16 | −3.06 | −2.75 | 0.16 | 19.3 | −2.39 | −2.57 | Scoliosis | COL11A1 | M/n | c.1543C>G | p.Gln515Glu | Yes | Likely pathogenic |
| 7 | M | 10 | −2.92 | −3.30 | −0.63 | 29.7 | −1.35 | −2.13 | — | IGF1R | M/n | c.158C>T | p.Thr53Met | Yes | Likely pathogenic |
| 8 | F | 11 | −2.76 | −2.75 | −1.04 | NA | −2.56 | −4.14 | Frequent OMA, genua valga, advanced bone age (+3.4 y) | COL2A1 | M1/M2 | c.410G>A / c.3106C>G | p.Arg137His/ p.Arg1036Gly | Yes | Likely pathogenic (6) / Likely pathogenic |
| GHD only – 4/9 (44.4%) with P/LP variant | |||||||||||||||
| 9 | M | 14 | −4.95 | −2.89 | −2.48 | 7.22 | −0.94 | −1.78 | Minor stigmata, bilateral cryptorchidism | IGFALS | M/n | c.860C>T | p.Pro287Leu | No | Uncertain significance (17) |
| 10 | M | 8 | −4.48 | −2.73 | −1.11 | 6.36 | −1.06 | −1.75 | B-cell immunity disorder, skin problems (hyperkeratosis, alopecia), corneal ulceration | MBTPS2 | M/- | c.1538T>C | p.Leu513Pro | Yes | Likely pathogenic (19) |
| PIK3CD | M/n | c.1775T>C | p.Val952Ala | Yes | Uncertain significance | ||||||||||
| 11 | M | 7 | −4.37 | −2.89 | −1.86 | 9.40 | −0.56 | −1.04 | Limb shortening | FGFR3 | M/n | c.1612A>G | p.Ile538Val | Yes | Likely pathogenic (18) |
| 12 | F | 8 | −3.50 | −2.60 | −1.57 | 2.50 | 0.04 | −0.95 | Limb shortening | HSPG2 | M/n | c.8044C>T | p.Arg2682Trp | Yes | Benign |
| 13 | M | 13 | −3.10 | −2.59 | −1.91 | 5.92 | −0.85 | −0.65 | — | PAX6 | M/n | c.13C>G c.392C>G | p.His5Asp p.Ala131Gly | Yes | Benign |
| 14 | M | 5 | −3.06 | −3.86 | −2.18 | 8.34 | −1.94 | −1.42 | — | HMGA2 | M/n | c.223C>T | p.Arg75Trp | Yes | Likely pathogenic |
| 15 | M | 9 | −3.04 | −2.75 | −1.82 | 6.06 | −1.57 | −1.68 | Minor stigmata | IGFALS | M/n | c.589C>T | p.Arg197Cys | Yes | Pathogenic |
| IHH | M/n | c.1169G>A | p.Arg390His | Yes | Uncertain significance | ||||||||||
| 16 | M | 8 | −2.75 | −2.75 | −1.23 | 7.79 | −1.02 | −1.35 | — | IGFALS | M/n | c.1195G>A | p.Gly399Arg | No | Uncertain significance |
| 17 | M | 12 | −2.53 | −2.75 | −1.28 | 8.30 | −1.60 | −0.65 | — | GH1 | M/n | c.478C>T | p.Arg160Trp | Yes | Benign |
| FBN1 | M/n | c.902G>T | p.Gly301Val | Yes | Benign | ||||||||||
| SGA + GHD – 3/11 (27.3%) with P/LP variant | |||||||||||||||
| 18 | M | 9 | −4.17 | −3.81 | −1.98 | 5.18 | −1.95 | −3.09 | Hypospadia, minor stigmata | HSPG2 | M/n | c.12874G>A | p.Glu4292Lys | Yes | Benign |
| 19 | F | 8 | −3.82 | −4.03 | −0.44 | 7.77 | −0.84 | −2.14 | Limb shortening | GLI2 GLI2 | M/n | c.4332G>A c.4333C>T | p.Met1444Ile p.Leu1445Phe | Yes | Benign |
| 20 | M | 6 | −3.61 | −3.25 | −1.14 | 6.4 | −2.16 | −3.12 | — | — | — | — | — | — | No variant found |
| 21 | M | 14 | −3.47 | −2.89 | −1.63 | 8.39 | −2.39 | −3.00 | Microcephaly | COL2A1 | M/n | c.3106C>G | p.Arg1036Gly | Yes | Likely pathogenic |
| 22 | M | 8 | −3.28 | −2.89 | −1.83 | 6.37 | −0.60 | −2.39 | — | — | — | — | — | — | No variant found |
| 23 | M | 13 | −3.26 | −2.81 | −1.22 | 7.44 | −2.18 | −2.22 | — | SHH | M/n | c.424G>A | p.Glu142Lys | Yes | Benign |
| 24 | F | 14 | −3.18 | −2.75 | −2.1 | 7.15 | −2.56 | −3.13 | Lower-limb shortening, minor stigmata | GHSR | M/n | c.526G>A | p.Gly176Arg | Yes | Pathogenic |
| 25 | M | 8 | −2.84 | −2.87 | −0.98 | 5.91 | −2.59 | −2.83 | Minor stigmata | COL10A1 | M/n | c.1581G>C | p.Lys527Asn | Yes | Likely benign |
| 26 | F | 12 | −2.76 | −2.89 | −3.45 | 3.62 | −2.78 | −1.23 | Shorter trunk, longer limbs, blue sclera, teeth decay, ectopic posterior pituitary | — | — | — | — | — | No variant found |
| 27 | M | 10 | −2.70 | −2.75 | −0.66 | 3.44 | −2.39 | −3.43 | — | EXT2 | M/n | c.2034G>C | p.Lys678Asn | No | Uncertain significance |
| 28 | M | 5 | −2.62 | −3.60 | −2.25 | 6.99 | −1.40 | −2.22 | — | FLNB | M/n | c.1601T>G | p.Ile534Ser | Yes | Likely pathogenic |
| Patient No. | Sex | Age, y | Patient`s Height (SDS) | Shorter Parent`s Height (SDS) | IGF-1 (SDS) | Stimulated GH, μg/L | Birth Weight (SDS) | Birth Length (SDS) | Additional Phenotypic Features | Gene | Mutation Status | Transcript Variant | Protein Variant | Parents Available for Genetic Testing | Conclusion |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SGA only – 5/8 (62.5%) with P/LP variant | |||||||||||||||
| 1 | F | 10 | −4.32 | −3.54 | −1.68 | 11.0 | −1.64 | −3.05 | — | COL11A1 | M/n | c.2921C>A | p.Pro974Gln | Yes | Likely pathogenic |
| 2 | M | 14 | −4.10 | −2.89 | −3.68 | 10.0 | −1.64 | −2.39 | Central hypothyroidism | TRHR | M/M | c.392T>C | p.Ile131Thr | Yes | Likely pathogenic (16) |
| 3 | M | 12 | −3.49 | −3.00 | −0.26 | 11.5 | −3.01 | −3.87 | — | THRA | M/n | c.455G>A | p.Arg152Gln | Yes | Uncertain significance |
| 4 | M | 10 | −3.42 | −3.03 | −0.98 | 10.2 | −1.62 | −3.26 | Advanced bone age (+1.9 years) | IHH | M/n | c.857C>T | p.Pro286Leu | No | Uncertain significance |
| 5 | M | 9 | −3.34 | −2.67 | −0.92 | NA | −2.13 | −3.52 | — | COL9A2 | M/n | c.1834G>A | p.Gly612Arg | Yes | Benign |
| 6 | M | 16 | −3.06 | −2.75 | 0.16 | 19.3 | −2.39 | −2.57 | Scoliosis | COL11A1 | M/n | c.1543C>G | p.Gln515Glu | Yes | Likely pathogenic |
| 7 | M | 10 | −2.92 | −3.30 | −0.63 | 29.7 | −1.35 | −2.13 | — | IGF1R | M/n | c.158C>T | p.Thr53Met | Yes | Likely pathogenic |
| 8 | F | 11 | −2.76 | −2.75 | −1.04 | NA | −2.56 | −4.14 | Frequent OMA, genua valga, advanced bone age (+3.4 y) | COL2A1 | M1/M2 | c.410G>A / c.3106C>G | p.Arg137His/ p.Arg1036Gly | Yes | Likely pathogenic (6) / Likely pathogenic |
| GHD only – 4/9 (44.4%) with P/LP variant | |||||||||||||||
| 9 | M | 14 | −4.95 | −2.89 | −2.48 | 7.22 | −0.94 | −1.78 | Minor stigmata, bilateral cryptorchidism | IGFALS | M/n | c.860C>T | p.Pro287Leu | No | Uncertain significance (17) |
| 10 | M | 8 | −4.48 | −2.73 | −1.11 | 6.36 | −1.06 | −1.75 | B-cell immunity disorder, skin problems (hyperkeratosis, alopecia), corneal ulceration | MBTPS2 | M/- | c.1538T>C | p.Leu513Pro | Yes | Likely pathogenic (19) |
| PIK3CD | M/n | c.1775T>C | p.Val952Ala | Yes | Uncertain significance | ||||||||||
| 11 | M | 7 | −4.37 | −2.89 | −1.86 | 9.40 | −0.56 | −1.04 | Limb shortening | FGFR3 | M/n | c.1612A>G | p.Ile538Val | Yes | Likely pathogenic (18) |
| 12 | F | 8 | −3.50 | −2.60 | −1.57 | 2.50 | 0.04 | −0.95 | Limb shortening | HSPG2 | M/n | c.8044C>T | p.Arg2682Trp | Yes | Benign |
| 13 | M | 13 | −3.10 | −2.59 | −1.91 | 5.92 | −0.85 | −0.65 | — | PAX6 | M/n | c.13C>G c.392C>G | p.His5Asp p.Ala131Gly | Yes | Benign |
| 14 | M | 5 | −3.06 | −3.86 | −2.18 | 8.34 | −1.94 | −1.42 | — | HMGA2 | M/n | c.223C>T | p.Arg75Trp | Yes | Likely pathogenic |
| 15 | M | 9 | −3.04 | −2.75 | −1.82 | 6.06 | −1.57 | −1.68 | Minor stigmata | IGFALS | M/n | c.589C>T | p.Arg197Cys | Yes | Pathogenic |
| IHH | M/n | c.1169G>A | p.Arg390His | Yes | Uncertain significance | ||||||||||
| 16 | M | 8 | −2.75 | −2.75 | −1.23 | 7.79 | −1.02 | −1.35 | — | IGFALS | M/n | c.1195G>A | p.Gly399Arg | No | Uncertain significance |
| 17 | M | 12 | −2.53 | −2.75 | −1.28 | 8.30 | −1.60 | −0.65 | — | GH1 | M/n | c.478C>T | p.Arg160Trp | Yes | Benign |
| FBN1 | M/n | c.902G>T | p.Gly301Val | Yes | Benign | ||||||||||
| SGA + GHD – 3/11 (27.3%) with P/LP variant | |||||||||||||||
| 18 | M | 9 | −4.17 | −3.81 | −1.98 | 5.18 | −1.95 | −3.09 | Hypospadia, minor stigmata | HSPG2 | M/n | c.12874G>A | p.Glu4292Lys | Yes | Benign |
| 19 | F | 8 | −3.82 | −4.03 | −0.44 | 7.77 | −0.84 | −2.14 | Limb shortening | GLI2 GLI2 | M/n | c.4332G>A c.4333C>T | p.Met1444Ile p.Leu1445Phe | Yes | Benign |
| 20 | M | 6 | −3.61 | −3.25 | −1.14 | 6.4 | −2.16 | −3.12 | — | — | — | — | — | — | No variant found |
| 21 | M | 14 | −3.47 | −2.89 | −1.63 | 8.39 | −2.39 | −3.00 | Microcephaly | COL2A1 | M/n | c.3106C>G | p.Arg1036Gly | Yes | Likely pathogenic |
| 22 | M | 8 | −3.28 | −2.89 | −1.83 | 6.37 | −0.60 | −2.39 | — | — | — | — | — | — | No variant found |
| 23 | M | 13 | −3.26 | −2.81 | −1.22 | 7.44 | −2.18 | −2.22 | — | SHH | M/n | c.424G>A | p.Glu142Lys | Yes | Benign |
| 24 | F | 14 | −3.18 | −2.75 | −2.1 | 7.15 | −2.56 | −3.13 | Lower-limb shortening, minor stigmata | GHSR | M/n | c.526G>A | p.Gly176Arg | Yes | Pathogenic |
| 25 | M | 8 | −2.84 | −2.87 | −0.98 | 5.91 | −2.59 | −2.83 | Minor stigmata | COL10A1 | M/n | c.1581G>C | p.Lys527Asn | Yes | Likely benign |
| 26 | F | 12 | −2.76 | −2.89 | −3.45 | 3.62 | −2.78 | −1.23 | Shorter trunk, longer limbs, blue sclera, teeth decay, ectopic posterior pituitary | — | — | — | — | — | No variant found |
| 27 | M | 10 | −2.70 | −2.75 | −0.66 | 3.44 | −2.39 | −3.43 | — | EXT2 | M/n | c.2034G>C | p.Lys678Asn | No | Uncertain significance |
| 28 | M | 5 | −2.62 | −3.60 | −2.25 | 6.99 | −1.40 | −2.22 | — | FLNB | M/n | c.1601T>G | p.Ile534Ser | Yes | Likely pathogenic |
Abbreviations: F, female; GHD, GH deficiency; LP, likely pathogenic; M, male; M/n, heterozygote; M/M, homozygote; M1/M2, compound heterozygote; M/-, hemizygote; OMA, otitis media acuta; P, pathogenic; SDS, SD score; SGA, small for gestational age.
Clinical Features and Genetic Results of Children With Severe FSS Examined by WES
| Patient No. | Sex | Age, y | Patient`s Height (SDS) | Shorter Parent`s Height (SDS) | IGF-1 (SDS) | Stimulated GH, μg/L | Birth Weight (SDS) | Birth Length (SDS) | Additional Phenotypic Features | Gene | Mutation Status | Transcript Variant | Protein Variant | Parents Available for Genetic Testing | Conclusion |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SGA only – 5/8 (62.5%) with P/LP variant | |||||||||||||||
| 1 | F | 10 | −4.32 | −3.54 | −1.68 | 11.0 | −1.64 | −3.05 | — | COL11A1 | M/n | c.2921C>A | p.Pro974Gln | Yes | Likely pathogenic |
| 2 | M | 14 | −4.10 | −2.89 | −3.68 | 10.0 | −1.64 | −2.39 | Central hypothyroidism | TRHR | M/M | c.392T>C | p.Ile131Thr | Yes | Likely pathogenic (16) |
| 3 | M | 12 | −3.49 | −3.00 | −0.26 | 11.5 | −3.01 | −3.87 | — | THRA | M/n | c.455G>A | p.Arg152Gln | Yes | Uncertain significance |
| 4 | M | 10 | −3.42 | −3.03 | −0.98 | 10.2 | −1.62 | −3.26 | Advanced bone age (+1.9 years) | IHH | M/n | c.857C>T | p.Pro286Leu | No | Uncertain significance |
| 5 | M | 9 | −3.34 | −2.67 | −0.92 | NA | −2.13 | −3.52 | — | COL9A2 | M/n | c.1834G>A | p.Gly612Arg | Yes | Benign |
| 6 | M | 16 | −3.06 | −2.75 | 0.16 | 19.3 | −2.39 | −2.57 | Scoliosis | COL11A1 | M/n | c.1543C>G | p.Gln515Glu | Yes | Likely pathogenic |
| 7 | M | 10 | −2.92 | −3.30 | −0.63 | 29.7 | −1.35 | −2.13 | — | IGF1R | M/n | c.158C>T | p.Thr53Met | Yes | Likely pathogenic |
| 8 | F | 11 | −2.76 | −2.75 | −1.04 | NA | −2.56 | −4.14 | Frequent OMA, genua valga, advanced bone age (+3.4 y) | COL2A1 | M1/M2 | c.410G>A / c.3106C>G | p.Arg137His/ p.Arg1036Gly | Yes | Likely pathogenic (6) / Likely pathogenic |
| GHD only – 4/9 (44.4%) with P/LP variant | |||||||||||||||
| 9 | M | 14 | −4.95 | −2.89 | −2.48 | 7.22 | −0.94 | −1.78 | Minor stigmata, bilateral cryptorchidism | IGFALS | M/n | c.860C>T | p.Pro287Leu | No | Uncertain significance (17) |
| 10 | M | 8 | −4.48 | −2.73 | −1.11 | 6.36 | −1.06 | −1.75 | B-cell immunity disorder, skin problems (hyperkeratosis, alopecia), corneal ulceration | MBTPS2 | M/- | c.1538T>C | p.Leu513Pro | Yes | Likely pathogenic (19) |
| PIK3CD | M/n | c.1775T>C | p.Val952Ala | Yes | Uncertain significance | ||||||||||
| 11 | M | 7 | −4.37 | −2.89 | −1.86 | 9.40 | −0.56 | −1.04 | Limb shortening | FGFR3 | M/n | c.1612A>G | p.Ile538Val | Yes | Likely pathogenic (18) |
| 12 | F | 8 | −3.50 | −2.60 | −1.57 | 2.50 | 0.04 | −0.95 | Limb shortening | HSPG2 | M/n | c.8044C>T | p.Arg2682Trp | Yes | Benign |
| 13 | M | 13 | −3.10 | −2.59 | −1.91 | 5.92 | −0.85 | −0.65 | — | PAX6 | M/n | c.13C>G c.392C>G | p.His5Asp p.Ala131Gly | Yes | Benign |
| 14 | M | 5 | −3.06 | −3.86 | −2.18 | 8.34 | −1.94 | −1.42 | — | HMGA2 | M/n | c.223C>T | p.Arg75Trp | Yes | Likely pathogenic |
| 15 | M | 9 | −3.04 | −2.75 | −1.82 | 6.06 | −1.57 | −1.68 | Minor stigmata | IGFALS | M/n | c.589C>T | p.Arg197Cys | Yes | Pathogenic |
| IHH | M/n | c.1169G>A | p.Arg390His | Yes | Uncertain significance | ||||||||||
| 16 | M | 8 | −2.75 | −2.75 | −1.23 | 7.79 | −1.02 | −1.35 | — | IGFALS | M/n | c.1195G>A | p.Gly399Arg | No | Uncertain significance |
| 17 | M | 12 | −2.53 | −2.75 | −1.28 | 8.30 | −1.60 | −0.65 | — | GH1 | M/n | c.478C>T | p.Arg160Trp | Yes | Benign |
| FBN1 | M/n | c.902G>T | p.Gly301Val | Yes | Benign | ||||||||||
| SGA + GHD – 3/11 (27.3%) with P/LP variant | |||||||||||||||
| 18 | M | 9 | −4.17 | −3.81 | −1.98 | 5.18 | −1.95 | −3.09 | Hypospadia, minor stigmata | HSPG2 | M/n | c.12874G>A | p.Glu4292Lys | Yes | Benign |
| 19 | F | 8 | −3.82 | −4.03 | −0.44 | 7.77 | −0.84 | −2.14 | Limb shortening | GLI2 GLI2 | M/n | c.4332G>A c.4333C>T | p.Met1444Ile p.Leu1445Phe | Yes | Benign |
| 20 | M | 6 | −3.61 | −3.25 | −1.14 | 6.4 | −2.16 | −3.12 | — | — | — | — | — | — | No variant found |
| 21 | M | 14 | −3.47 | −2.89 | −1.63 | 8.39 | −2.39 | −3.00 | Microcephaly | COL2A1 | M/n | c.3106C>G | p.Arg1036Gly | Yes | Likely pathogenic |
| 22 | M | 8 | −3.28 | −2.89 | −1.83 | 6.37 | −0.60 | −2.39 | — | — | — | — | — | — | No variant found |
| 23 | M | 13 | −3.26 | −2.81 | −1.22 | 7.44 | −2.18 | −2.22 | — | SHH | M/n | c.424G>A | p.Glu142Lys | Yes | Benign |
| 24 | F | 14 | −3.18 | −2.75 | −2.1 | 7.15 | −2.56 | −3.13 | Lower-limb shortening, minor stigmata | GHSR | M/n | c.526G>A | p.Gly176Arg | Yes | Pathogenic |
| 25 | M | 8 | −2.84 | −2.87 | −0.98 | 5.91 | −2.59 | −2.83 | Minor stigmata | COL10A1 | M/n | c.1581G>C | p.Lys527Asn | Yes | Likely benign |
| 26 | F | 12 | −2.76 | −2.89 | −3.45 | 3.62 | −2.78 | −1.23 | Shorter trunk, longer limbs, blue sclera, teeth decay, ectopic posterior pituitary | — | — | — | — | — | No variant found |
| 27 | M | 10 | −2.70 | −2.75 | −0.66 | 3.44 | −2.39 | −3.43 | — | EXT2 | M/n | c.2034G>C | p.Lys678Asn | No | Uncertain significance |
| 28 | M | 5 | −2.62 | −3.60 | −2.25 | 6.99 | −1.40 | −2.22 | — | FLNB | M/n | c.1601T>G | p.Ile534Ser | Yes | Likely pathogenic |
| Patient No. | Sex | Age, y | Patient`s Height (SDS) | Shorter Parent`s Height (SDS) | IGF-1 (SDS) | Stimulated GH, μg/L | Birth Weight (SDS) | Birth Length (SDS) | Additional Phenotypic Features | Gene | Mutation Status | Transcript Variant | Protein Variant | Parents Available for Genetic Testing | Conclusion |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SGA only – 5/8 (62.5%) with P/LP variant | |||||||||||||||
| 1 | F | 10 | −4.32 | −3.54 | −1.68 | 11.0 | −1.64 | −3.05 | — | COL11A1 | M/n | c.2921C>A | p.Pro974Gln | Yes | Likely pathogenic |
| 2 | M | 14 | −4.10 | −2.89 | −3.68 | 10.0 | −1.64 | −2.39 | Central hypothyroidism | TRHR | M/M | c.392T>C | p.Ile131Thr | Yes | Likely pathogenic (16) |
| 3 | M | 12 | −3.49 | −3.00 | −0.26 | 11.5 | −3.01 | −3.87 | — | THRA | M/n | c.455G>A | p.Arg152Gln | Yes | Uncertain significance |
| 4 | M | 10 | −3.42 | −3.03 | −0.98 | 10.2 | −1.62 | −3.26 | Advanced bone age (+1.9 years) | IHH | M/n | c.857C>T | p.Pro286Leu | No | Uncertain significance |
| 5 | M | 9 | −3.34 | −2.67 | −0.92 | NA | −2.13 | −3.52 | — | COL9A2 | M/n | c.1834G>A | p.Gly612Arg | Yes | Benign |
| 6 | M | 16 | −3.06 | −2.75 | 0.16 | 19.3 | −2.39 | −2.57 | Scoliosis | COL11A1 | M/n | c.1543C>G | p.Gln515Glu | Yes | Likely pathogenic |
| 7 | M | 10 | −2.92 | −3.30 | −0.63 | 29.7 | −1.35 | −2.13 | — | IGF1R | M/n | c.158C>T | p.Thr53Met | Yes | Likely pathogenic |
| 8 | F | 11 | −2.76 | −2.75 | −1.04 | NA | −2.56 | −4.14 | Frequent OMA, genua valga, advanced bone age (+3.4 y) | COL2A1 | M1/M2 | c.410G>A / c.3106C>G | p.Arg137His/ p.Arg1036Gly | Yes | Likely pathogenic (6) / Likely pathogenic |
| GHD only – 4/9 (44.4%) with P/LP variant | |||||||||||||||
| 9 | M | 14 | −4.95 | −2.89 | −2.48 | 7.22 | −0.94 | −1.78 | Minor stigmata, bilateral cryptorchidism | IGFALS | M/n | c.860C>T | p.Pro287Leu | No | Uncertain significance (17) |
| 10 | M | 8 | −4.48 | −2.73 | −1.11 | 6.36 | −1.06 | −1.75 | B-cell immunity disorder, skin problems (hyperkeratosis, alopecia), corneal ulceration | MBTPS2 | M/- | c.1538T>C | p.Leu513Pro | Yes | Likely pathogenic (19) |
| PIK3CD | M/n | c.1775T>C | p.Val952Ala | Yes | Uncertain significance | ||||||||||
| 11 | M | 7 | −4.37 | −2.89 | −1.86 | 9.40 | −0.56 | −1.04 | Limb shortening | FGFR3 | M/n | c.1612A>G | p.Ile538Val | Yes | Likely pathogenic (18) |
| 12 | F | 8 | −3.50 | −2.60 | −1.57 | 2.50 | 0.04 | −0.95 | Limb shortening | HSPG2 | M/n | c.8044C>T | p.Arg2682Trp | Yes | Benign |
| 13 | M | 13 | −3.10 | −2.59 | −1.91 | 5.92 | −0.85 | −0.65 | — | PAX6 | M/n | c.13C>G c.392C>G | p.His5Asp p.Ala131Gly | Yes | Benign |
| 14 | M | 5 | −3.06 | −3.86 | −2.18 | 8.34 | −1.94 | −1.42 | — | HMGA2 | M/n | c.223C>T | p.Arg75Trp | Yes | Likely pathogenic |
| 15 | M | 9 | −3.04 | −2.75 | −1.82 | 6.06 | −1.57 | −1.68 | Minor stigmata | IGFALS | M/n | c.589C>T | p.Arg197Cys | Yes | Pathogenic |
| IHH | M/n | c.1169G>A | p.Arg390His | Yes | Uncertain significance | ||||||||||
| 16 | M | 8 | −2.75 | −2.75 | −1.23 | 7.79 | −1.02 | −1.35 | — | IGFALS | M/n | c.1195G>A | p.Gly399Arg | No | Uncertain significance |
| 17 | M | 12 | −2.53 | −2.75 | −1.28 | 8.30 | −1.60 | −0.65 | — | GH1 | M/n | c.478C>T | p.Arg160Trp | Yes | Benign |
| FBN1 | M/n | c.902G>T | p.Gly301Val | Yes | Benign | ||||||||||
| SGA + GHD – 3/11 (27.3%) with P/LP variant | |||||||||||||||
| 18 | M | 9 | −4.17 | −3.81 | −1.98 | 5.18 | −1.95 | −3.09 | Hypospadia, minor stigmata | HSPG2 | M/n | c.12874G>A | p.Glu4292Lys | Yes | Benign |
| 19 | F | 8 | −3.82 | −4.03 | −0.44 | 7.77 | −0.84 | −2.14 | Limb shortening | GLI2 GLI2 | M/n | c.4332G>A c.4333C>T | p.Met1444Ile p.Leu1445Phe | Yes | Benign |
| 20 | M | 6 | −3.61 | −3.25 | −1.14 | 6.4 | −2.16 | −3.12 | — | — | — | — | — | — | No variant found |
| 21 | M | 14 | −3.47 | −2.89 | −1.63 | 8.39 | −2.39 | −3.00 | Microcephaly | COL2A1 | M/n | c.3106C>G | p.Arg1036Gly | Yes | Likely pathogenic |
| 22 | M | 8 | −3.28 | −2.89 | −1.83 | 6.37 | −0.60 | −2.39 | — | — | — | — | — | — | No variant found |
| 23 | M | 13 | −3.26 | −2.81 | −1.22 | 7.44 | −2.18 | −2.22 | — | SHH | M/n | c.424G>A | p.Glu142Lys | Yes | Benign |
| 24 | F | 14 | −3.18 | −2.75 | −2.1 | 7.15 | −2.56 | −3.13 | Lower-limb shortening, minor stigmata | GHSR | M/n | c.526G>A | p.Gly176Arg | Yes | Pathogenic |
| 25 | M | 8 | −2.84 | −2.87 | −0.98 | 5.91 | −2.59 | −2.83 | Minor stigmata | COL10A1 | M/n | c.1581G>C | p.Lys527Asn | Yes | Likely benign |
| 26 | F | 12 | −2.76 | −2.89 | −3.45 | 3.62 | −2.78 | −1.23 | Shorter trunk, longer limbs, blue sclera, teeth decay, ectopic posterior pituitary | — | — | — | — | — | No variant found |
| 27 | M | 10 | −2.70 | −2.75 | −0.66 | 3.44 | −2.39 | −3.43 | — | EXT2 | M/n | c.2034G>C | p.Lys678Asn | No | Uncertain significance |
| 28 | M | 5 | −2.62 | −3.60 | −2.25 | 6.99 | −1.40 | −2.22 | — | FLNB | M/n | c.1601T>G | p.Ile534Ser | Yes | Likely pathogenic |
Abbreviations: F, female; GHD, GH deficiency; LP, likely pathogenic; M, male; M/n, heterozygote; M/M, homozygote; M1/M2, compound heterozygote; M/-, hemizygote; OMA, otitis media acuta; P, pathogenic; SDS, SD score; SGA, small for gestational age.
All patients underwent basic genetic examination. In all of the girls, Turner syndrome and SHOX-haploinsufficiency was excluded by fluorescence in situ hybridization. In all boys with confirmed disproportionate short stature, SHOX deficiency including point mutations was excluded using Sanger sequencing and multiplex ligation probe amplification. Children born with SGA were scored by the Netchine-Harbison Clinical scoring system (20) for Silver-Russel syndrome (SRS) exclusion. In children with positive Netchine-Harbison Clinical scoring system, genetic examination for Silver-Russel syndrome was performed.
Genetic examination
Genomic DNA was extracted from peripheral blood using the QIAmp DNA Blood Mini system (Qiagen, Hilden, Germany). DNA was analyzed using whole-exome sequencing (WES). WES was performed using SureSelect Human All Exon Kit V6 + UTRs (Agilent Technologies, Santa Clara, CA), and the indexed products were sequenced by synthesis in an Illumina NextSeq 500 analyzer (San Diego, CA) with 100× average coverage. FastQC was used for quality control (21). The obtained sequences were aligned to the reference human genome (hg19 build) using BWA software (22). The detected variants were subsequently filtered using Ingenuity Variant Analysis (Qiagen, Hilden, Germany) as follows: the initial set up of the filters removed variants with a call quality below 20 and variants the frequency of which in the public databases (1000 Genome Project, ExAc or NHLBI ESP) exceeded 1%. The remaining variants were selected according to whether they were experimentally observed to be associated with a phenotype: pathogenic, likely pathogenic, or frameshift, in-frame indel, or start/stop codon change, or missense unless predicted tolerated by SIFT and PolyPhen2, or predicted deleterious by having a Combined Annotation-Dependent Depletion score greater than 20, or disrupt splice site loss up to 2.0 bases into an intron or as predicted by MaxEntScan.
Evaluation of the genetic results
All the variants with potential clinical importance obtained from WES were confirmed using Sanger sequencing as described previously (23) and further evaluated using the American College of Medical Genetics and Genomics standards and guidelines (24). For the evaluation of the segregation of the genetic variants with short stature within the families, Sanger sequencing was performed in relatives who had agreed to genetic testing. In only 4 of 25 families were the parents not available for the testing. Last, all the variants were classified as pathogenic, likely pathogenic, benign, likely benign, or as variants of uncertain significance (VUS). The evaluation process is summarized in Fig. 1.
Results
Of 33 probands with severe FSS, 5 (15%) had an already known genetic diagnosis prior to the start of the study. In 2 children, we previously reported casual variants in the ACAN gene (p.Val478Serfs* and p.Ser306Cys) (25), 2 children had Noonan syndrome (SOS1 and PTPN11 genes), and 1 had neurofibromatosis type 1 (NF1 gene). The remaining 28 patients with severe FSS with unknown genetic diagnosis underwent WES examination.
In 89% (25/28) of children examined by WES, we found at least one genetic variant with potential clinical importance in genes with a known impact on growth. Pathogenic or likely pathogenic variants were revealed in 43% of children (12/28); three variants were classified as pathogenic, nine as likely pathogenic. Genetic variants in eight children were classified as (likely) benign, and variants in the remaining five children were classified as VUS. The segregation of variants with short stature within the families was one of the major criteria allowing the classification of variants as pathogenic and excluding nonsegregating ones. Results are summarized in Table 1; for detailed information about variant evaluation in specific patients refer to information provided in an online repository (26).
The additional clinical features in patients generally correlated with the elucidated genetic diagnosis. Collagenopathies are known to be associated with scoliosis (patient 6) (27), joint deformities and frequent middle ear infections (patient 8) (28, 29). Limb shortening in patient 11 is typical for FGFR3 gene mutations (30). Partial GH deficiency in patient 24 is typical for patients with GHSR mutations (31). On the other hand, limb shortening and being born SGA is not generally associated with GHSR mutations. However, patient 24 has a younger brother with the same genetic variant who was born appropriate for gestational age (AGA). He was diagnosed with GHD at the age of 5 years. At diagnosis, he had body height −3 SD, low IGF-1, and GH levels after stimulation lower than 1 μg/L. Intrauterine growth restriction in patient 24 could have been caused by his mother’s preeclampsia during pregnancy.
Altogether, causative gene variants were identified in 17 of 33 (52%) children of the study cohort. Of these, 9 of 17 carried gene variants affecting the growth plate: ACAN (2 children), COL2A1 (2 children), and COL11A1 (2 children) gene products are the structural proteins of the cartilaginous extracellular matrix, and the FLNB gene encodes filamin B, a protein that helps to build cytoskeleton in the chondrocytes. The products of FGFR3 and IGF1R play an important role in endocrine or paracrine regulation of the growth plate. In 2 of 17 families, affected genes encode IGF-associated proteins (IGFALS and HMGA2 genes). In the remaining 6 of 17 families, the genetic mechanisms of growth disorders were miscellaneous. TRHR is a gene for the TRH receptor, GHSR coregulates GH secretion, MBTPS2 is associated with ichthyosis follicularis, alopecia, and photophobia syndrome, NF1 is associated with neurofibromatosis type 1, and PTPN11 and SOS1 are associated with Noonan syndrome (16, 19, 31–34). The tissue and cellular localization of several gene products are summarized in Fig. 2, and the variants discovered in our study are fully colored in the figure.
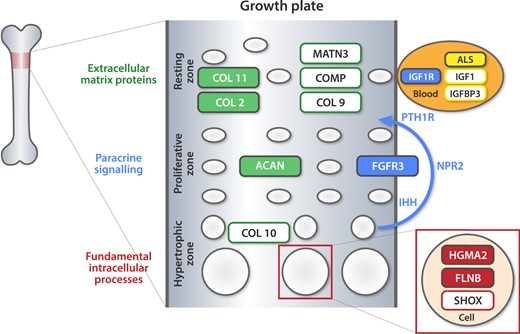
Tissue and cellular localization of proteins encoded by causative genes. The variants identified in our study are fully colored in the figure. ALS, acid-labile subunit; COL 2, collagen type II; COL 9, collagen type IX; COL 10, collagen type X; COL 11, collagen type XI; COMP, cartilage oligomeric matrix protein precursor; FGFR3, fibroblast growth factor receptor 3; IGF1 – insulinlike growth factor-1; IGF1R, insulinlike growth factor-1 receptor; IGFBP3, insulinlike growth factor-binding protein 3; IHH, Indian hedgehog; MATN3, matrilin 3; NPR2, natriuretic peptide receptor 2; PTH1R, parathyroid hormone 1 receptor.
In the study, we discovered an important difference in the spectrum of genetic disorders elucidated in SGA children compared with children born AGA. A majority (8/9; 89%) of growth plate disorders was found among SGA children. Among GHD children born with AGA, the etiology of their FSS was different. The most frequent findings were variants in genes encoding IGF-associated proteins (2/9 children; 22%).
Discussion
This study evaluates the complex genetic etiology of FSS using NGS methods in a population with a low rate of consanguinity. We have revealed that monogenic conditions are frequent among children with severe FSS (17/33 children; 52%). Interestingly, growth plate disorders play an important role, especially among children with SGA.
In clinical practice, growth plate disorders have traditionally been categorized as bone dysplasias. They form a heterogeneous group of more than 400 disorders. A few (e.g., SHOX deficiency, Turner syndrome, achondroplasia) have distinct clinical characteristics that make a correct diagnosis relatively easy (2, 35, 36). In other cases, the clinical diagnosis may not be feasible. However, the genetic etiology can be discovered using NGS methods. Recently, Zhang et al. (35) elucidated the monogenic causes of growth plate disorders in 44 of 82 (54%) Chinese patients with clinically suspicious bone dysplasia. Our study has shown that monogenic growth plate disorders are frequent not only among patients with clear signs of bone dysplasia but also in children with FSS who do not have substantial disproportionality.
GHD evaluation is, in clinical practice, performed using provocative tests (37) that are known to show low specificity (14.9% to 49%) (38). It is presumed that idiopathic GHD does not have to be a disease per se but rather a description of heterogeneous and poorly understood conditions. Our study has supported these assumptions by observation that short children in our cohort had been frequently misdiagnosed as having GHD. In fact, among 23 children with clinically diagnosed GHD, only 1 carried a causative genetic variant affecting GH secretion (GHSR). In the remaining 7 GHD children with a known genetic cause of their short stature, different molecular mechanisms were elucidated: 3 had growth plate disorders (ACAN, FGFR3 and FLNB genes), 2 carried causative variants in genes encoding IGF-associated proteins (IGFALS and HMGA2 genes), and 2 had Noonan syndrome (PTPN11 and SOS1 genes). Recognizing the etiology of short stature and clarifying the gene-specific reaction to GH treatment poses one of the important current challenges of pediatric endocrinology. Until this is achieved, provocative tests for GH secretion will often remain (despite their poor specificity) the only possibility how to allow short children to start GH treatment in countries in which idiopathic short stature is not an approved indication.
Except for genes causing growth plate disorders or GHD and genes affecting IGF-associated proteins, completely different monogenic mechanisms of short stature have been elucidated. Our findings support the new paradigm in the understanding of short stature presented recently by Baron et al. (32). Within this novel concept, the GH-IGF-1 axis represents only one part of the multiple regulatory systems controlling chondrogenesis in the growth plate. Thus, understanding the pathogenesis of short stature should be centered on the chondrocyte and its complex endocrine, paracrine, and endogenous regulation (32).
Interpretation of the genetic findings in our study was not always simple. Some genetic variants did not explain all the features of the complex phenotype. For example, TRHR homozygous, likely pathogenic variant in patient 2 certainly explains central hypothyroidism. However, as only four patients with pathogenic TRHR variant have been described so far, it is difficult to evaluate if this variant also causes short stature. Although growth is definitely affected by thyroid hormones, only two of four patients with pathogenic TRHR variants had short stature (the other two had normal growth parameters) (16). Moreover, as the variant is inherited in autosomal recessive pattern, it cannot explain the short stature in proband’s parents. We must concede that the growth disorder in patient 2 may be caused by different mechanisms than by TRHR gene variant. Patient 10 is another interesting case. Apart from his short stature, he presented with severe immunodeficiency, generalized hyperkeratosis, alopecia, and corneal ulceration. He carried a likely pathogenic hemizygous variant in MBTPS2 explaining his short stature and the combination of a dermatologic disorder, alopecia, and corneal ulceration (ichthyosis follicularis, alopecia, and photophobia syndrome). This variant was inherited from his mother (X-linked inheritance) with normal stature and mild hyperkeratosis. However, the variant does not explain his severe immune defect or his father’s short stature. It is possible that the immune defect results from his variant in PIK3CD classified as VUS. Moreover, patient 1 in our study cohort carried a p.Pro974Gln variant in COL11A1 gene classified as likely pathogenic that was rather frequent in population databases (ExAC European frequency 0.32%). A greater frequency than expected alone is not the reason to classify the variant as (likely) benign (24). Because other proof supporting the pathogenicity was convincing (26), we are confident to present this variant as likely pathogenic. We presume that short stature may be not noticeable, and these people can easily be included in population databases.
Although our study revealed information to better understand the pathogenesis of FSS, we must acknowledge that it had several limitations. First, no functional studies have been performed in our study. However, according to current guidelines other methods can be used to prove the pathogenicity of genetic variants (24). In our cohort of patients with severe FSS, the most important was the segregation of the variants with short stature within the families. In most cases, we have performed genetic testing in multiple family members from different generations. If the family members were not available for genetic testing, the variants usually had to be classified as VUS even if they were interesting. The supportive methods to evaluate the genetic variants were, e.g., their frequency in population databases or various in silico studies [for information about specific families see data provided in an online repository supplemental materials (26)]. Second, noncoding variants (except for disruptions in the exon-intron boundaries) and epigenetic and somatic changes were not captured by WES, therefore, they were not considered in our study.
Conclusions
Variants in a single gene are frequent causes of severe FSS. Although the etiology is heterogeneous, we have demonstrated that the genetic variants responsible for normal function of the growth plate play an important role, especially in those with SGA. The results of our study support the controversies related to the clinical diagnosis of GHD. Further research should be performed to test the causative gene-specific GH responsiveness as a necessary first step to better personalize the management of short stature.
Abbreviations:
- AD
autosomal dominant
- AGA
appropriate for gestational age
- FSS
familial short stature
- GHD
GH deficiency
- NGS
next-generation sequencing
- SGA
small for gestational age
- VUS
variants of uncertain significance
- WES
whole-exome sequencing
Acknowledgments
We thank Professor Ondrej Cinek (Charles University, Prague, Czech Republic), who created the database of patients treated with GH at our center, and Jana Kaprova (Charles University, Prague, Czech Republic) for her help in completing the database. We also thank Pavla Kankova and Klara Vesela, laboratory workers at our center, for their work and dedication to the study. Finally, we thank Shenali Amaratunga (Charles University, Prague, Czech Republic) for her advice with the formulation of the manuscript.
Financial Support: This work was supported by the Ministry of Health, Czech Republic, Grants 16-31211A (to J. L.) and NV18-07-00283 (to J. L.), by Project for Conceptual Development of Research Organization Grant 00064203/6001, and by the research project of the Grant Agency of Charles University, Prague, GAUK 976718 (to L. P.).
Disclosure Summary: The authors have nothing to disclose.
References
Plachy L, Strakova V, Elblova L, Obermannova B, Kolouskova S, Snajderova M, Zemkova D, Dusatkova P, Sumnik Z, Lebl J, Pruhova S. Data from: High prevalence of growth plate gene variants in children with familial short stature treated with GH. Open Science Framework 2019. Deposited 15 January 2019. https://osf.io/9npcf/?view_only=d42f62f99c464cf1967fd21be54225c0.

{kind=link}
{kind=link}