





Abstract
Follistatin is a plasma protein recently reported to increase under conditions with negative energy balance, such as exercise and fasting in humans. Currently, the perception is that circulating follistatin is a result of para/autocrine actions from various tissues. The large and acute increase in circulating follistatin in response to exercise suggests that it may function as an endocrine signal.
We assessed origin and regulation of circulating follistatin in humans.
First, we assessed arterial-to-venous difference of follistatin over the splanchnic bed at rest and during exercise in healthy humans. To evaluate the regulation of plasma follistatin we manipulated glucagon-to-insulin ratio in humans at rest as well as in cultured hepatocytes. Finally, the impact of follistatin on human islets of Langerhans was assessed.
We demonstrate that in humans the liver is a major contributor to circulating follistatin both at rest and during exercise. Glucagon increases and insulin inhibits follistatin secretion both in vivo and in vitro, mediated via the secondary messenger cAMP in the hepatocyte. Short-term follistatin treatment reduced glucagon secretion from islets of Langerhans, whereas long-term follistatin treatment prevented apoptosis and induced proliferation of rat β cells.
In conclusion, in humans, the liver secretes follistatin at rest and during exercise, and the glucagon-to-insulin ratio is a key determinant of circulating follistatin levels. Circulating follistatin may be a marker of the glucagon-to-insulin tone on the liver.
Follistatin is a secreted protein that inactivates members of the TGF-β family including activins, growth and differentiation factors, and bone morphogenetic proteins (BMP) (1–5). Because follistatin was discovered in high amounts in follicular fluids (5) and binds activin in the pituitary, thereby inhibiting follicle stimulating hormone secretion (6), it has mostly been linked to reproductive physiology. Other functions of circulating follistatin have, however, emerged because it is also associated with the regulation of muscle growth (7) and inflammation (8). In addition, follistatin has been linked to metabolic diseases because it is elevated in plasma in patients with type 2 diabetes (9), nonalcoholic fatty liver disease (NAFLD) (10), and nonalcoholic steatohepatitis (NASH) (10). Follistatin is expressed in a broad range of cells, but no specific tissue or organ is identified as the source of circulating follistatin. Consequently, plasma follistatin is often regarded as spillover from autocrine or paracrine actions (6).
The auto- and paracrine views of circulating follistatin are challenged by the observation that plasma follistatin acutely increases about 7-fold after an exercise bout (11), which suggests an endocrine function. In mice, exercise-induced follistatin is most likely liver-derived (11), which further supports that follistatin functions as a signal between the liver and peripheral tissues.
The regulatory mechanism for exercise-induced follistatin secretion has not been elucidated, but expression of follistatin is induced by glucagon in cultured hepatocytes (12). Glucagon acts on the liver via glucagon receptors abundantly expressed on the hepatocytes (13, 14), and during exercise plasma glucagon increases to stimulate hepatic glucose production (15). Although the main function of glucagon is believed to be glucoregulation, recent data demonstrate that glucagon acts as a secretagogue for hepatic FGF-21 secretion (16–18). The ability of glucagon to promote secretion of hepatic signaling molecules reveals perspectives regarding endocrine signaling between the liver and other organs.
Recent data suggest that follistatin may influence β-cell survival as β-cell–specific delivery of follistatin results in increased proliferation (19). In addition, when β cells are exposed to follistatin for 72 hours, the gene expression changes to a pattern associated with more mature β-cell phenotype (20). Thus, follistatin may interact with the endocrine pancreas.
In the present study the following hypotheses are tested: 1) in man, follistatin is secreted from the liver both at rest and during exercise; 2) follistatin secretion depends on the glucagon-to-insulin ratio; and 3) in vitro, follistatin interacts with cells of the endocrine pancreas to modulate hormone secretion and/or cell β-cell survival.
Materials and Methods
Liver vein catheterization trial
This study has been described elsewhere (18). In brief, 10 healthy males (age, 22.9 ± 0.8; body mass index [BMI], 22.6 ± 0.5; maximum oxygen consumption, 51.0 ± 1.2 ml/kg/min) performed 2 hours of bicycle exercise at 60% maximum oxygen consumption in the semisupine position with catheters placed in an antecubital vein, a hepatic vein, and brachial artery, allowing for arterial-venous measurements over the liver. After the exercise bout, the subjects rested in the supine position for 4 hours. The subjects arrived after an overnight fast and remained fasted throughout the trial, but had free access to water. Splanchnic blood flow was assessed by the indocyanine green method (21). Splanchnic follistatin production was calculated as ([follistatin (ng/l)]artery − [follistatin (ng/l)]hepatic vein) × splanchnic plasma flow (liter/min).
Glucagon infusion trial
This study has been described elsewhere (18). Ten healthy males went through four experimental protocols (test days 1–4). For test day 3 (somatostatin infusion), four additional subjects were recruited because four of the original participants were not available. Characteristics of subjects in trials 1, 2, and 4: age, 22.9 ± 0.4; BMI, 22.3 ± 0.5; subjects in trial 3: age, 23.0 ± 0.5; BMI, 22.3 ± 0.5. Test day 1: 1-hour glucagon infusion (GlucaGen, Novo Nordisk) at 6 ng/kg/min. Test day 2: 2-hour somatostatin infusion (Octreotide, Hospira Nordic) at 100 ng/kg/min (started 10 minutes before the glucagon infusion, in total 130 minutes) and 1-hour glucagon infusion at 6 ng/kg/min. Test day 3: 2 hours (130 minutes) of somatostatin infusion at 100 ng/kg/min (same as test day 2). Test day 4: saline infusion at the same rate as the glucagon infusion rate. In all four trials, the duration of the experiments was 8 hours and the subjects remained fasted throughout the trial but had free access to water.
Intralipid infusion trial
This study has been described (22). Fourteen subjects were recruited to the trials; however, blood samples were only available for seven subjects for this report. In brief, the subjects went through two protocols in fasted conditions: 1) saline infusion with heparin and 2) intralipid infusion in combination with heparin. Only time points before the bolus of endotoxin (at 6 hours) were included in this report. In both trials, the subjects remained fasted throughout the trial, but had free access to water.
Islet and β-cell experiments
Cells and culture conditions.
Islets of Langerhans were isolated by collagenase digestion of pancreas from adult male Wistar rats followed by Ficoll (Histopaque-1077, SigmaeAldrich) purification (23). Rat β cells were sorted as previously described (24). Human intact islets were cultured on 804G matrix-coated dishes. Acute insulin and glucagon secretion: human islets were washed, preincubated, and incubated for insulin or glucagon secretion assays as described in detail elsewhere (25). Data are mean of six for insulin secretion experiments and mean of four for glucagon secretion experiments.
Detection of apoptosis and proliferation.
Cell death was measured by terminal deoxynucleotidyl transferase assay and proliferation was assessed by bromodeoxyuridine incorporation (26). Data are the mean of six experiments.
Hepatocyte and HepG2 cells experiments
Human hepatocytes were isolated from surgical liver biopsies of patients undergoing segmental hepatectomies (n = 2). Human liver tissue (between 15 and 20 g) was removed during liver resection for benign or malignant diseases. Human hepatocyte isolation was performed using a two-step collagenase perfusion method (27). Briefly, resected liver was perfused with Ca2+-free buffer washing solution (10 ml/min for 20 minutes) followed by collagenase solution (type I, 1 mg/ml at 5 ml/min for 15 minutes; Sigma). Hepatocytes were released by mincing and shaking the liver in a Petri dish containing cold RPMI (Invitrogen) and 10% FBS (Invitrogen). The cell suspension was filtered through a sterile 100-μm nylon mesh into a beaker placed on ice and then transported to the Surgical Research Unit, University of Geneva Medical School, where human hepatocytes were purified by sedimentation on 30% Percoll isodensity solution (Pharmacia Biotech AB) (28). Viability was assessed by trypan blue exclusion. Cells were cultured in Dulbecco's modified Eagle's medium/F12 medium (Invitrogen) containing 2% FBS (Invitrogen), 1 × 10−6 mol/liter dexamethasone (Sigma-Aldrich GmbH), 1 × 10−8 mol/l 3,3′ triiodo-L-thyronine, 1 × 10−8 mol/liter human insulin (Huminsulin, Lilly France S.A.S.), 5 μg/ml apotransferrin (Sigma-Aldrich GmbH), and 15 × 10−3 mol/liter Hepes.
HepG2 cells (human hepatocarcinoma, DMSZ) were grown in RPMI 1640 containing 11 mM glucose and 10% FBS, 1% glutamine, and 1% penicillin/streptomycin. Cells were starved for 3 hours in RPMI 1640 before insulin (100 nM) or forskolin (20 μM) was added. In a second set of experiments, cells were starved in RPMI 1640 without glucose. The Quantitect Primer assay Hs_FST_1_SG was used for real-time quantitative PCR analysis. Data are mean of six experiments.
Ethical committee approval
The human in vivo studies were approved by the Scientific Ethics Committee of the capital region of Denmark (Hormone infusion trial and exercise trial with liver vein catheterization: H-1-2012-129, Intralipid infusion trial: (22)) in accordance with the Helsinki Declaration. All subjects provided written informed consent to participate.
Plasma measurements
Blood samples were obtained in tubes containing EDTA for analysis of hormones and in tubes containing aprotinin for analysis of glucagon. All blood samples were immediately spun at 4 C at 3000 g for 15 minutes and the plasma fractions were stored at −80 C until analysis. Follistatin (Figures 1, 2, and 3F) was measured by ELISA (R&D Systems) with an intra-assay coefficient of variation of less than 5% and interassay coefficient of variation of less than 5%. According to R&D Systems, this assay measures total follistatin concentration (follistatin 288, follistatin 300, and follistatin 315). All samples were run in duplicate in accordance with the protocol from the manufacturer.
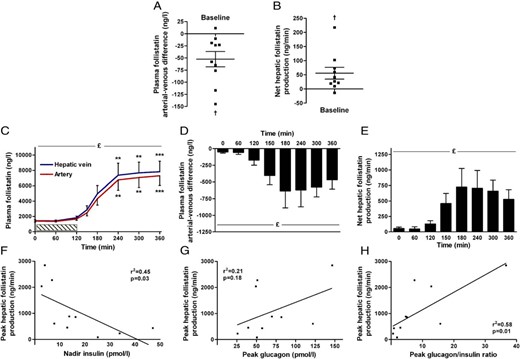
Splanchnic plasma follistatin regulation during exercise in humans (n = 10). At rest, the arterial-venous difference (A) and net production of follistatin (B) are different from zero. Exercise increases plasma follistatin (C), and the arterial-venous difference of the splanchnic circulation (D) and net hepatic production is increased (E). Peak hepatic follistatin production correlate with nadir insulin (F) and peak glucagon/insulin ratio (H), but not with peak glucagon (G). £Significant by one-way ANOVA and *significant by one-way ANOVA and Dunnett's post hoc test (***P < .0001, **P < .01, *P < .05). †Denotes significant by Student's t test. Data are mean ± SEM. (A) and (B) present individual data. Hatched bar (C) indicates duration of the exercise bout.
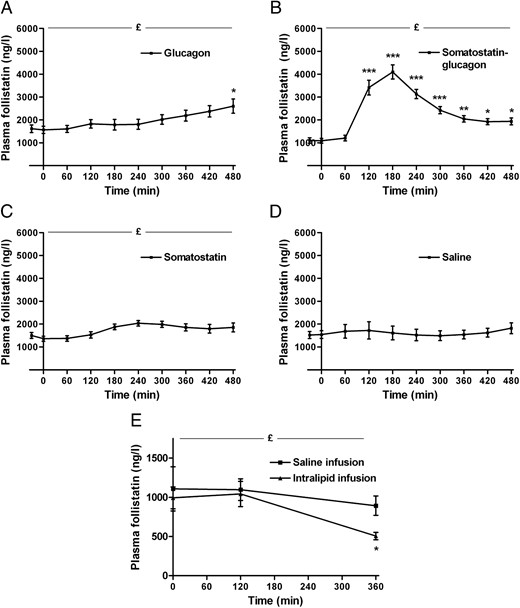
Regulation of plasma follistatin by glucagon and insulin in humans (n = 10). Infusion of glucagon increases follistatin slightly (A), whereas a combined somatostatin-glucagon infusion increases plasma follistatin markedly (B). Infusion of somatostatin (C) only increases plasma follistatin marginally. Infusion of saline does not change plasma follistatin (D). Infusion of intralipid decreases plasma follistatin (E), whereas there no change during the saline infusion (E). £Significant by one-way ANOVA, *significant by one-way ANOVA and Dunnett's post hoc test (***P < .0001, **P < .01, *P < .05). Data are mean ± SEM.
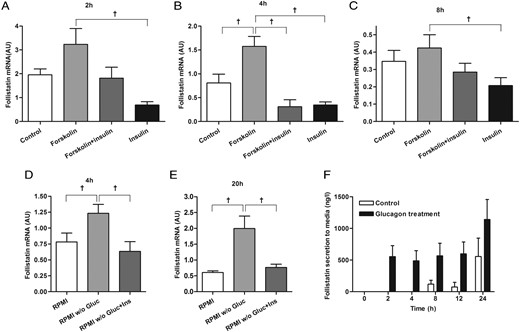
Regulation of follistatin in hepatocytes. In HepG2 cells, follistatin mRNA expression is induced by forskolin, whereas it is inhibited by insulin (ins) at 2 (A), 4 (B), and 8 hours (C). In HepG2 cells, starvation induces follistatin mRNA expression, which is inhibited by insulin at 4 (D) and 20 hours (E). In primary human hepatocytes, glucagon (gluc) stimulates follistatin secretion (F, black bars) compared with control conditions (F, white bars). †Significant by Student's t test. Data are mean ± SEM.
Statistical analysis
Data are presented as means ± (SEM). One-way ANOVAs with Dunnett's post hoc tests were applied for the analysis of hormones over time.
Results
Follistatin is secreted from the splanchnic circulation at rest and during exercise
Here, we evaluated the splanchnic release of follistatin in humans by measuring arterial-to-hepatic vein (a-hv) differences at rest and during exercise in 10 healthy human subjects. At rest, both the a-hv difference of −52.4 ± 15.7 ng/liter (P = .009) and the net hepatic follistatin production (a-hv × plasma flow) of 55.7 ± 20.9 ng/min (P = .03) (Figure 1, A and B, respectively) indicates a constant hepatic follistatin secretion.
Follistatin increases both in the hepatic vein and artery in response to exercise (Figure 1C). Plasma follistatin remains at baseline level during the exercise bout (0–120 minutes) and then increases 5-fold (P < .0001) the first 2 hours into the recovery. During the last 2 hours of recovery, follistatin reaches a plateau of approximately 7600 ng/liter (P < .0001). The a-hv difference is negative during the entire trial (significant by Student's t test, P < .05, except t = 60 minutes, P = .09) (Figure 1D), demonstrating a constant hepatic secretion of follistatin from the splanchnic bed. During exercise, the a-hv difference of follistatin increases markedly and peaks at 636 ng/liter at 180 minutes (P = .02, post hoc test at 180 minutes P = .05). During the last 3 hours of recovery, the a-hv of follistatin remains stable, although with a small decrease at time point 360 minutes. When accounting for the hepatic plasma flow, the splanchnic production of follistatin is constant at ∼650 ng/min (mean) during the recovery (Figure 1E).
Because glucagon stimulation induces follistatin expression in hepatocytes (12), we analyzed the correlation between the exercise-induced plasma glucagon and insulin responses and plasma follistatin. Peak hepatic follistatin production correlates with nadir insulin (r2 = 0.45, P = .03) and the peak glucagon-to-insulin ratio (r2 = 0.58, P = .01) (Figure 1, F and H), whereas there is no significant correlation with the glucagon peak (r2 = 0.21, P = .18) (Figure 1G).
Glucagon-to-insulin ratio regulates circulating follistatin
To investigate whether glucagon and insulin acutely regulate circulating follistatin, the glucagon-to-insulin ratio was manipulated in physiologically relevant doses and durations in four different trials (see reference (18) for details); 1) high glucagon/high insulin, 2) high glucagon/low insulin, 3) low glucagon/low insulin, and 4) control trial with saline infusion.
In the high glucagon/high insulin setting, both glucagon and insulin increases to approximately 100 pmol/liter during the 1-hour glucagon infusion. Initially, there is no regulation of plasma follistatin, whereas there is a slow approximate 1.7-fold increase (P = .01) in the last part of the trial (Figure 2A). However, this kinetic profile does not resemble exercise-induced follistatin secretion. During high glucagon/low insulin (as observed during exercise), glucagon increases to approximately 100 pmol/liter, whereas insulin decreases to below baseline level. Follistatin increases rapidly with a kinetic profile similar to the exercise response; plasma follistatin increases from ∼1200 ng/liter at 60 minutes to its peak at approximately 4100 ng/liter at 180 minutes (approximate 4-fold increase, P < .0001) (Figure 2B). In contrast to the exercise response, plasma follistatin then decreases from 180 minutes, which indicates a different termination of the follistatin secretion than observed with exercise. During low glucagon/low insulin, both glucagon and insulin are suppressed to below baseline level. Only a minor increase (1.4-fold) in plasma follistatin is observed (P = .003) (Figure 2C). During saline infusion, there is no regulation of plasma follistatin which remains at approximately 1600 ng/liter (P = 1.0) (Figure 2D).
Because both glucagon infusion (29) and exercise (30) stimulate lipolysis in the adipose tissue with a subsequent increase in plasma free fatty acids (FFAs) (29), we evaluated the effect of FFAs on follistatin secretion in healthy males. During intralipid infusion with a prime bolus of heparin, both triglycerides and plasma FFAs increase substantially to 3000 and 2500 μmol/liter (22), whereas circulating follistatin decreases by approximately 50% (P = .01) (Figure 2E). During the control trial (saline infusion), there is no change in circulating follistatin (P = .7) (Figure 2E).
Glucagon-stimulated follistatin expression and secretion in hepatocytes
To further investigate the regulation of follistatin in liver cells, we stimulated HepG2 cells with forskolin (an adenylate cyclase activator) and insulin. Forskolin increases follistatin mRNA expression at 4 hours, whereas this expression is reduced by insulin at 2, 4, and 8 hours (P < .05) (Figure 3, A-C). Starvation of HepG2 cells by glucose withdrawal for 4 and 20 hours increases hepatic follistatin mRNA expression (P < .05), which is inhibited by insulin (P < .05) (Figure 3, D and E). Furthermore, we stimulated human primary hepatocytes from two independent donors with glucagon. In the presence of 10 nM glucagon, follistatin was detected in the media 2 and 4 hours after stimulation, whereas the control cells did not produce detectable amounts of follistatin at these time points (Figure 3F). After 8 hours, control cells produced detectable amount of follistatin demonstrating that the liver secretes follistatin under normal conditions (Figure 3F). At 8, 12, and 24 hours, there is no difference between glucagon-stimulated and controls cells. Collectively, these data demonstrate that hepatocytes secrete follistatin, and that cAMP and insulin have opposing effects on its expression.
In vitro effects of follistatin on cells of the endocrine pancreas
Next, we evaluated the acute effects of follistatin exposure on pancreatic islets within a relevant time course (hours) that mimic the human exercise response. The effect of follistatin on pancreatic insulin and glucagon secretion was tested using whole human islets from healthy donors in static culture. The cells were treated with follistatin (50 ng/ml) for 1, 2, 6, and 24 hours in the presence of high and low glucose concentrations. Follistatin treatment does not alter insulin secretion at high or low glucose (Figure 4A). In contrast, follistatin treatment acutely suppresses basal glucagon secretion at high glucose, whereas it has no effect on glucagon secretion at low glucose (Figure 4B). In addition, we investigated the impact of long-term follistatin exposure on β-cell survival and apoptosis. Sorted rat β cells were treated with follistatin for 24 hours, and cell death and proliferation was assessed by terminal deoxynucleotidyl transferase and bromodeoxyuridine assays, respectively. At both doses of follistatin treatment increased the proliferative rate of rat β cells equally (Figure 4C). In addition, at both doses follistatin decreased spontaneous cell death in rat β cells with the most pronounced effect at 10 ng/ml follistatin (Figure 4D).
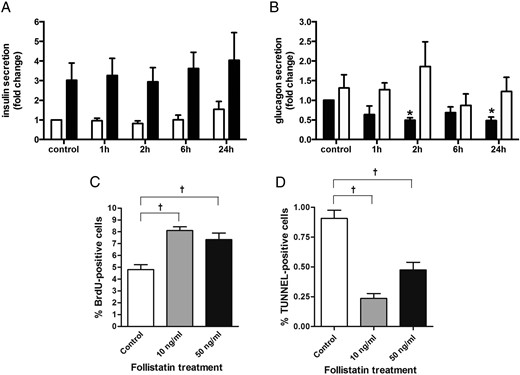
Follistatin treatment modulates glucagon secretion. Human islets were cultured for 0–24 hours on 804G matrix-coated dishes in the presence of 50 mM follistatin. (A) Insulin secretion: human islets were incubated for 60 minutes at 2.8 mM glucose (open bars) followed by 60 minutes at 16.7 mM glucose (closed bars) (n = 6). (B) Glucagon secretion: human islets were incubated for 60 minutes at 16.7 mM glucose (closed bars) followed by 60 minutes at 2.8 mM glucose (open bars) (n = 4). In sorted rat β cells, 24 hours of follistatin treatment at 10 or 50 ng/ml increases proliferation (C) and decreases cell death (D) (n = 4).*P < .05 compared to control as tested by ANOVA followed by Bonferroni post hoc test. †Significant by Student's t test. Data are presented as mean ± SEM.
Discussion
The present study demonstrates that: 1) in humans, follistatin is secreted from the splanchnic circulation. Because the in vitro data demonstrate follistatin secretion by hepatocytes, it seems that the liver is a major contributor to circulating follistatin levels, 2) both in vivo and in vitro data establish changes in the glucagon-to-insulin ratio as the key regulatory mechanism for hepatic follistatin production and 3) follistatin acutely regulates glucagon secretion from human islets of Langerhans within a time frame equivalent to the regulation observed in humans, whereas prolonged follistatin treatment decreases rat β-cell death and increases proliferation. Collectively, we demonstrate that follistatin is a liver-derived signal responsive to changes in the glucagon-to-insulin ratio that may interact with the endocrine pancreas.
Origin of circulating follistatin in man
Follistatin exists in two major isoforms of which the 288 isoform is considered a tissue-bound isoform, whereas the 315 isoform is considered to be circulating is plasma (31–33). Although the expression of follistatin in the liver has been described previously (34), the finding that circulating follistatin is liver-derived in humans is novel. We previously investigated the potential contribution of skeletal muscle to exercise-induced circulating follistatin in humans; however, we did not detect any arterial-to-venous difference across an exercising leg, and no regulation of the follistatin gene was detected in the skeletal muscle upon exercise (11). Thus, skeletal muscle does not seem to secrete follistatin to any significant extent. In cultured white adipose tissue, follistatin is secreted into the media, suggesting a potential contribution to systemic levels (35). However, while follistatin gene expression is markedly increased in the liver in response to exercise in mice (11), no increase was detected in tissues such as adipose tissue, muscle, heart, kidney, and spleen (11). Although follistatin may be secreted from the adipose tissue, it is not likely contributing to the exercise-induced increase in systemic follistatin. Based on existing data, the marked increase in circulating follistatin in response to exercise is most likely to the result of hepatic secretion, which places follistatin as one of the few identified hepatokines (36).
Regulation of hepatic follistatin secretion
The induction of plasma follistatin observed with high glucagon/low insulin resembles the exercise-induced follistatin increase. The hepatic secretion of follistatin seems to be controlled by the glucagon-to-insulin ratio: glucagon infusion (with high glucagon and high insulin) only increases plasma follistatin moderately, whereas glucagon infusion with a concomitant suppression of insulin increases plasma follistatin markedly. Interestingly, this phenomenon is also observed for FGF-21 (18), demonstrating a similar regulatory mechanism for these two hepatokines in vivo. In response to exercise, FGF-21 peaks 30 minutes after the end of the exercise and returns to baseline levels 2 hours after the exercise bout (18). Exercise-induced follistatin increases 1 hour after exercise and remains elevated up to 6 hours after exercise (11). Thus, although FGF-21 and follistatin share regulatory mechanism, they have distinct kinetic profiles. Although speculative, this raises the possibility that FGF-21 influences hepatic follistatin secretion via intrahepatic autocrine/paracrine signaling.
In cultured human hepatocytes, glucagon stimulation rapidly promotes secretion of follistatin into the media. In addition follistatin is present in the media of (unstimulated) control cells after 8 hours, indicating a constant secretion of follistatin from hepatocytes. Glucagon and insulin have opposite effects on intracellular cAMP levels, and our data point toward intracellular cAMP as a key regulatory mechanism for hepatic follistatin secretion. Starvation of cells increases intracellular cAMP (37), and we found that glucose withdrawal from hepatocytes per se increased follistatin gene expression. Likewise, forskolin, an adenylate cyclase activator, stimulated follistatin expression in HepG2 cells, which was also demonstrated in rat granulosa cells (38). In agreement with this, the presence of a cAMP response element and AP-1 and AP-2 binding sites were demonstrated in the promoter region of the follistatin gene (38).
During exercise follistatin increases approximately 5-fold, whereas experimentally induced follistatin secretion through modulation of the glucagon-to-insulin ratio only increases approximately 4-fold, leaving room for other modulatory stimuli. Importantly, the kinetics of follistatin secretion are similar in the two conditions. Adrenalin acts on the liver and signals via cAMP and could stimulate follistatin secretion. However, conflicting results are reported; phenylephrine treatment of primary rat hepatocytes increased follistatin expression (12), whereas bolus administration of adrenalin in mice did not (11). Based on the present data, we speculate that, in addition to changes in glucagon-to-insulin ratio, adrenalin might also promote follistatin secretion.
In humans, circulating follistatin increases with prolonged fasting (39). Prolonged fasting and exercise are characterized by an increase in the glucagon-to-insulin ratio and elevated FFA (40). Thus, elevated FFA could confound our interpretation of the regulation of hepatic follistatin secretion. However, experimental elevation of FFA in healthy young subjects decreases circulating follistatin (ie, FFA per se does not stimulate follistatin secretion). Hence, the induction of follistatin observed with fasting (39) is likely explained by the increase glucagon and decrease insulin levels.
Effects of follistatin
Recently, Zhao et al provided novel data on follistatin's interaction with the β cells using an adeno associated virus delivery of follistatin to the β cells (19). Using this approach, it was demonstrated that β-cell–specific follistatin overexpression in db/db mice promotes β-cell proliferation and maintenance of pancreatic islet mass (19). In addition, it rescued hyperglycemia, relieved diabetic symptoms, and prolonged life span, indicating secondary whole-body effects of the treatment. Another study reported that long-term (72-hour) follistatin treatment increases expression of genes associated with a mature β-cell phenotype (20). In line with those reports, we find that long-term (24-hour) follistatin treatment induced β-cell survival by increasing proliferation and reducing cell death in isolated rat β cells. On the other hand, deletion of the follistatin 315 isoform in mice does not change β-cell mass or proliferation (41). In the present study, short-term follistatin stimulation is able to impair glucagon secretion in vitro, indicating that in states of energy abundance (high glucose) follistatin may decrease glucagon secretion from the islets of Langerhans. Such a condition could occur when food is available shortly (hours) after an exercise challenge where circulating follistatin is increased 5- to 7-fold. Consequently, follistatin could represent a feedback signal to the α cell to decrease glucagon secretion and, in turn, reduce hepatic glucose production. In contrast, long-term (48-hour) exposure of human islets to follistatin does not change glucagon mRNA levels (42). These data suggest that follistatin's effect on glucagon secretion is time dependent. Several studies have suggested the existence of a signal from the liver to the endocrine pancreas (43–45). Liver-specific insulin receptor knockout mice display increased β-cell mass explained by increased proliferation (45), whereas no α-cell proliferation is observed. By use of the parabiosis model, El Ouaamari et al found that the β-cell proliferation is promoted by a circulating factor (most likely a protein) derived from hepatocytes (45). Our data suggest that the liver may communicate to the endocrine pancreas via follistatin secretion potentially offering a mechanism for hepatokine-induced β-cell proliferation.
Follistatin is a known inhibitor of myostatin actions (1), and skeletal muscle overexpression of follistatin results in a hypermuscular phenotype (7). Importantly, treatment of mice (46) and ducks (47) with repeated injections of recombinant follistatin induces a hypertrophic response in skeletal muscle tissue. In addition, follistatin treatment induces skeletal muscle repair after injury and immobilization (46). As follistatin is induced during conditions of energy deprivation (eg, exercise, fasting) in vivo, it could be speculated that follistatin acts a as liver-to-muscle signal to spare the muscle from breakdown (eg, via myostatin inhibition during such conditions). The possible effects and regulation of exercise-induced follistatin is summarized in Figure 5.
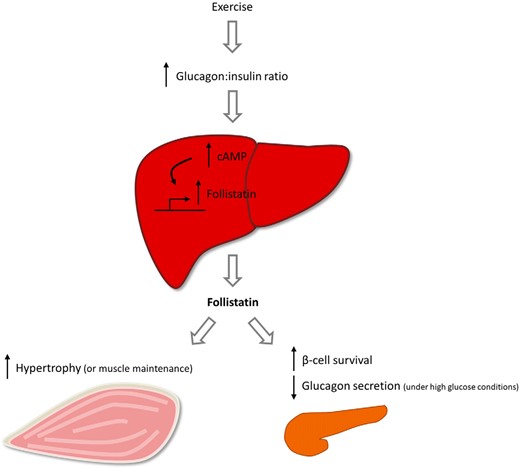
Schematic presentation of regulation and potential actions of follistatin. Liver-derived follistatin is regulated by the glucagon-to-insulin ratio during exercise, a mechanism dependent on cAMP in hepatocytes. Circulating follistatin may interact with the endocrine pancreas to increase β-cell survival and decrease glucagon secretion during energy abundance. Circulating follistatin may also interact with skeletal muscle to induce hypertrophy or to spare the muscle from breakdown under energy deprived conditions.
Follistatin in disease
In addition to being acutely regulated, circulating follistatin is increased in the patients with type 2 diabetes and correlate with markers of insulin resistance such as fasting glucose, glycated hemoglobin, and 2-hour glucose during an oral glucose tolerance test (9). As circulating follistatin is increased during states of energy deprivation such as prolonged fasting (39) and exercise, conditions associated with increased gluconeogenesis, it could be speculated that circulating follistatin acts in negative feedback loop to regulate hepatic gluconeogenesis. Surprisingly, deletion of the (circulating) follistatin 315 isoform in mice results in augmented gene expression of GLUT2, PEPCK, and G6P (48), consequently suggesting that circulating follistatin suppresses gluconeogenesis. Alternatively, follistatin may act to increase β-cell mass as a compensatory mechanism to combat increasing blood glucose. Circulating follistatin is also elevated in patients with NAFLD and NASH (10). Interestingly, follistatin 315-deletion in mice results in hepatic steatosis associated with increased hepatic gene expression of low-density lipoprotein receptor, lipoprotein lipase, acetyl-CoA carboxylase 1, and fatty acid synthase, suggestive of increased lipid uptake and de novo lipogenesis in these mice (48). In addition, mice with transgenic overexpression of follistatin (in skeletal muscle) show resistance to diet-induced hepatic steatosis (49). These data indicate that circulating follistatin reduce hepatic lipid uptake and synthesis and consequently suggest that circulating follistatin could counteract development of hepatic steatosis in NAFLD and NASH.
Conclusions
In conclusion, the liver is a major contributor to the circulating levels of follistatin in humans and its secretion is under control by the glucagon-to-insulin ratio. These data suggest that circulating follistatin may be a marker of the glucagon-to-insulin tone on the liver.
Acknowledgments
The Centre for Physical Activity Research (CFAS) is supported by a grant from Trygfonden. During the study period, the Centre of Inflammation and Metabolism (CIM) was supported by a grant from the Danish National Research Foundation (DNRF55). CIM and CFAS are members of and received funding from the Danish Center for Strategic Research in Type 2 Diabetes (grants 09-067009 and 09-075724). This study was further supported by grants from the Augustinus's and Aase og Ejnar Danielsen's. The Copenhagen Muscle Research Centre is supported by a grant from the Capital Region of Denmark. This work was also supported in part by grants from the German Federal Ministry of Education and Research to the German Centre for Diabetes Research (DZD e.V.; no. 01GI0925), the Deutsche Forschungsgemeinschaft GRK 1302/2 (to C.W.), the Swiss National Science Foundation (grant 31-135645), and Young Independent Investigator Grant from Swiss Society for Endocrinology and Diabetes. C.G.-G. was supported by the Swiss National Science Foundation (FNS 310030_138341). Human islets were kindly provided by the Cell Isolation and Transplant Centre of the University of Geneva, through JDRF award 31-2008-413 (ECIT Islet for Basic Research Program).
Disclosure Summary: The authors have declared that no conflict of interest exists.
Abbreviations
- a-hv
arterial-to-hepatic vein
- BMI
body mass index
- FFA
free fatty acid
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis.
References
Author notes
K.B. and P.P. contributed equally to the study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}