





Microbial invasion of the amniotic fluid (AF) cavity stimulates an inflammatory response that involves activin-A, a pleiotropic mediator member of the TGFβ superfamily involved in connective tissue remodeling. The role of AF follistatin, a natural inhibitor of activin-A, in inflammation-induced preterm birth (PTB), has yet to be determined.
The objective of the study was to investigate the relationships between AF activin-A and follistatin in physiological gestation and in pregnancies complicated by PTB and to evaluate a possible role played by the activin-A-follistatin balance in processes leading to PTB and preterm premature rupture of membranes (PPROM).
The AF levels of total activin-A and follistatin were immunoassayed in 168 women with a normal pregnancy outcome or PTB with and without intraamniotic inflammation or PPROM. The impact of the activin-A-follistatin imbalance on PTB terminal effector pathways (prostaglandins [prostaglandin E2, prostaglandin F2α] and matrix metalloproteinases [MMP-1, MMP-2, MMP-3, and MMP-9]) was investigated in an amniochorion explant system challenged with lipopolysaccharide (LPS) to mimic inflammation.
AF follistatin and the activin-A to follistatin ratio varied with gestational age, both decreasing toward term (P < .001). Activin-A was up-regulated in AF infection (>2-fold elevation in activin-A to follistatin ratio) correlating directly with severity of inflammation (both P < .001). Activin-A increased prostaglandins, MMP-1, and MMP-9 released by amniochorion (P < .05) to LPS-equivalent levels. Follistatin effectively blunted the prostaglandin response to activin-A and LPS and that of MMPs after activin-A but not after LPS challenge.
Activin-A and follistatin are part of the complex inflammatory response of the gestational sac to infection and modulate effector pathways leading to PTB. The activin-A to follistatin ratio may play a role in determining the clinical phenotype of PTB as preterm labor or PPROM.
Activin-A and folistatin are part of the inflammatory response of amniotic cavity to infection. Their ratio modulates pathways leading to preterm labor or preterm premature rupture of membranes.
Activin-A is a dimeric protein, part of the TGF-β superfamily (1). Activin-A was originally identified as a reproductive hormone, inducer of FSH release from the pituitary gland. Since then, activin-A has been credited as regulator of multiple physiological and pathological processes associated with cell growth, differentiation, immunity, and extracellular matrix remodeling (2). Recently the important immunomodulatory cytokine-like function of activin-A has emerged as a critical link between inflammation, tissue damage, and repair (2).
Regulation of activin-A bioactivity involves the inhibitory factor follistatin, a glycoprotein that binds to activin with high affinity (3). Several follistatin isoforms resulting from either alternative splicing or cleavage have been described in human tissues and fluids (4). The imbalance between activin-A and follistatin has been identified as a driver of inflammation severity and tissue phenotypic shift in many pathological processes including asthma, pulmonary fibrosis, keloid scar formation, inflammatory bowel disease, carcinogenesis, and sepsis among others (5–9). Although the direction in which the activin-A-follistatin balance fine-tunes the immune response depends on various system-specific factors, animal models of infection point to its critical role for host survival (10). Because the release of activin-A precedes the release of other cytokines involved in the inflammatory cascade, the modulation of the activin-A-follistatin system may be a potential for therapeutic target in diseases with an important inflammatory component (10–12).
Preterm birth (PTB) is a heterogeneous clinical entity with many phenotypical presentations including advanced cervical dilatation, uterine bleeding, spontaneous onset of uterine contractions, or preterm premature rupture of membranes (PPROM) in the presence or absence of intrauterine or maternal systemic infection. Differentiation among clinical phenotypes is often difficult based on clinical grounds alone. At the molecular level, prostaglandins (PGs) E2 and F2α and matrix metalloproteases (MMPs) are essential components in terminal common pathways leading to onset of myometrial contractions, membrane rupture, or both (13, 14). For example, localization and expression of the PGE2 receptor subtype is higher in human myometrium compared with the fetal membranes (15). This implies that PGE2 acts on the myometrium and may be a key mediator for spontaneous idiopathic PTB. The receptor for PGF2α is expressed in both the myometrium and fetal membranes, implying that that PGF2α can play a role in both myometrial contractions and PPROM (15). Furthermore, PGE2 mediates the selective increase in MMP-9 after lipopolysaccharide (LPS) stimulation (16), suggesting the involvement in PPROM in the setting of intraamniotic infection. If activin-A plays intermediary roles in these scenarios, remains unknown.
Microbial invasion of the amniotic fluid (AF) cavity is an important cause of PTB and adverse neonatal outcomes. This process stimulates a robust innate inflammatory response that involves increased levels of free activin-A in the AF (17). Both activin-A and follistatin have been postulated to play a role in parturition based on their changes in systemic concentrations surrounding the time of labor (18–20). Our current study examined the levels of AF follistatin, which have not been previously investigated in relationship to severity of intraamniotic inflammation or PTB phenotype (PPROM). Furthermore, the role played by the activin-A-follistatin balance in events initiating PTB was not explored prior to this research. We hypothesized that pregnancies complicated by intraamniotic inflammation are characterized by an imbalance in AF activin-A-follistatin system, which in turn modulates key biological processes involved in PTB and PPROM. Our goals were as follows: 1) to quantify the components of the activin-A-follistatin system in vivo in AF of pregnancies complicated by intraamniotic infection and PTB and 2) to examine the consequences of activin-A-follistatin imbalance on the production of PGE2 and MMP-9, two important effectors in initiating uterine contractions and membrane rupture, respectively.
Materials and Methods
Patients and amniotic fluid collection
We investigated AF samples from 168 women pregnant with singletons who had a clinically indicated amniocentesis to rule out infection. Samples were retrieved by transabdominal amniocentesis for the following purposes: 1) second-trimester genetic karyotyping (gestational age [GA], median [interquartile range[: 19 [18–20] wk, n = 19); 2) third-trimester fetal lung maturity testing (GA: 36 [36–37] wk, n = 20); and 3) rule out AF infection in women who had preterm labor contractions refractory to tocolysis, PPROM, or advanced cervical dilatation (>3 cm) (GA 28 [25–31] wk, n = 129). Rule-out AF infection cases were further grouped by timing of birth (term or preterm), membrane status at amniocentesis (intact membranes or PPROM), and microbiological culture results of the AF (positive or negative). The workflow of subjects and biopecimens used in this study is presented in Supplemental Figure 1.
Exclusion criteria, definition of GA, preterm labor, and PPROM are presented in the Supplemental Materials and Methods. All women were recruited at Yale New Haven Hospital and were followed up prospectively until delivery. The Human Investigation Committee of Yale University approved the study protocol. All patients provided written informed consent.
Chemical and microbiological studies of the AF
After retrieval under sterile conditions, the AF was analyzed for glucose concentration, lactate dehydrogenase (LDH) activity, white blood cell (WBC) count, Gram stain, and standard culturing methods for aerobic and anaerobic bacteria, including Ureaplasma and Mycoplasma species. These results were available for clinical management. An AF glucose cutoff of 15 mg/d or less, an LDH level of 419 U/L or greater, a positive Gram stain, and/or culture result were considered suggestive of intraamniotic infection (21). The results were reported as final 5 days after culturing. The remaining AF was spun, aliquoted, and stored at −80°C.
Mass spectrometry of the AF
Due to known technical limitations of AF cultures in identifying infection, we assessed for the presence of intraamniotic inflammation using the mass-restricted (MR) score, a previously reported AF proteomic fingerprint generated using mass spectrometry (Supplemental Materials and Methods) (22).
Evaluation of histological chorioamnionitis
Tissues were available for all PTB cases and were submitted for histological examination by a perinatal pathologist. Each section was examined systematically for the presence or absence of inflammation and funisitis (neutrophil infiltration of the umbilical vessels walls or Wharton's jelly), as previously described (Supplemental Materials and Methods) (23–25).
Enzyme-linked immunosorbent assays
Immunoassays for activin-A, follistatin, and IL-6 were conducted in the AF. The explant medium was analyzed for the levels of PGE2 and PGF2α, the chemokine IL-8, and the following MMPs: MMP-1 (interstitial collagenase), MMP-2 (gelatinase A), MMP-3 (stromelysin-1), and MMP-9 (gelatinase B). These MMPs were chosen based on their wide range of preferential substrates (26). MMP-1 breaks down collagens I, II, and III. MMP-2 breaks down type IV collagen present in the basement membranes whereas MMP-9 degrades both types IV and V collagens, respectively. Lastly, MMP-3 degrades a wide range of collagens and extracellular matrix components such as proteoglycans, fibronectin, laminin, and elastin and activates other MMPs (26). All immunoassays were performed in accordance with the manufacturer's instructions after appropriate dilutions (Supplemental Materials and Methods).
Amniochorion explant experiments
Amniochorion was obtained from healthy women at term (n = 5, GA 38 [38–40] wk) undergoing elective cesarean delivery in the absence of labor. Using sterile surgical scissors, we collected full-thickness amniochorion from a site located at distance from the umbilical cord insertion and the point of iatrogenic rupture. Fresh fetal membranes were exposed for 24 hours to recombinant activin-A (R&D Systems; 50 ng/mL), recombinant follistatin (FST288, 310 ng/mL; R&D Systems), or both. Control wells received sterile water. The doses of activin-A and follistatin were equivalent to human AF and the previously reported biologically relevant ratio of 2.5 in the follistatin to activin-A molar ratio (27). LPS (50 ng/mL) was used to mimic inflammation in vitro. The explant medium was collected at 24 hours of incubation for the measurements of MMP-9 and PGE2. Details are presented in Supplemental Materials and Methods.
Statistical analysis
For our data analysis, we used one-way and two-way ANOVA, Spearman correlations, and χ2 tests. A value of P < .05 was considered significant throughout the analysis. Details are presented in Supplemental Materials and Methods.
Results
Clinical and laboratory characteristics of the study population
Demographic and outcome characteristics of the women at amniocentesis are presented in Supplemental Table 1. Women who had a genetic amniocentesis were older and all had a term delivery. Clinical chorioamnionitis and PPROM were encountered only in the group of women who had an amniocentesis to rule out infection. This group of women delivered babies of lower birth weight than the other two groups.
The demographic, clinical, laboratory, and outcome characteristics for the rule-out infection group are presented in Supplemental Table 2. Women with positive AF cultures were enrolled and delivered at earlier GAs and had shorter amniocentesis-to-delivery intervals, independent of membrane status. Women with infection had more often clinical chorioamnionitis and delivered babies of lower birth weight. Among groups who delivered preterm, there was no difference in the frequency of antenatal steroid administration preceding amniocentesis. Women presenting with intact membranes more often had been administered tocolytics before amniocentesis. The results of the biochemical and microbiological studies of the AF showed the group of women with positive cultures had lower AF glucose levels, higher AF LDH activity, higher WBC count, and higher frequency of a positive Gram stain. Histological examination of the placenta confirmed a higher frequency of severe inflammation in the groups of women with positive cultures.
Gestational age regulation of AF activin-A, follistatin, and their molar ratio
AF samples from the group of women with intact membranes who delivered at term were used along with the genetic and lung maturity samples to evaluate GA regulation of AF total activin-A, follistatin, and their relative ratio. We found that AF concentration of total activin-A did not vary significantly across gestation (Figure-1A, r = −0.050, P = .685). In contrast, AF levels of follistatin progressively decreased (Figure 1B, r = −0.462, P < .001). This resulted in a rise in the activin-A to follistatin molar ratio with advancing GA (Figure 1C, r = 0.375, P = .002). There was a direct correlation between total activin-A and follistatin levels (r = 0.347, P = .009), which was independent of GA, suggesting their concentration in AF may be coregulated.
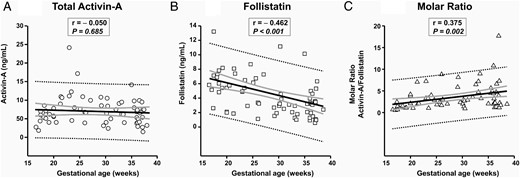
GA regulation of AF activin-A (A), follistatin (B), and activin-A to follistatin ratio (C) is shown.
Thick black line, linear regression line; thick gray lines, 95% confidence intervals; dotted black lines, 95% prediction intervals. The Spearman correlations coefficients (r) and level of significance are presented on each graph.
Relationships of AF activin-A and follistatin with intraamniotic infection, intraamniotic inflammation, and membrane status
The results of the AF research analyses with absolute concentrations of activin-A and follistatin in the five groups of women who had amniocenteses to rule out infection are shown in Table 1. AF activin-A was increased in the groups with intraamniotic infection irrespective of membrane status (Figure 2A, P < .001). There was no significant difference in AF follistatin concentration among the groups (Figure 2B). The activin-A to follistatin ratio was increased in the groups of women with positive cultures driven primarily by the rise in activin-A immunoreactivity (Figure 2C, P < .001). When the patients were grouped based on severity of inflammation (MR score), the groups of women with severe inflammation (MR 3–4) showed increased median levels of activin-A (Figure 2D, P < .001). Follistatin levels appeared unaffected by either the presence or degree of AF inflammation (Figure 2E). The activin-A to follistatin ratio was increased in the women with severe inflammation but not in those with mild inflammation by the MR score (Figure 2F, P < .001). The level of significance was maintained after adjusting for GA at amniocentesis.
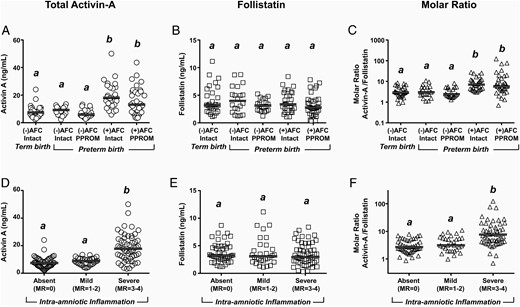
Scatterplots of AF activin-A (A and D), follistatin (B and E), and activin-A to follistatin ratio (C and F) are shown.
Cases are grouped by results of AF cultures (AFC; positive or negative) and membrane status (intact or PPROM) (A–C) or by severity of inflammation as reflected by MR score (D and E). The thick horizontal lines represent the group median. Groups marked with different letters are statistically different at P < .05. Data were analyzed using a one-way ANOVA on ranks followed by Dunn's post hoc tests.
Research Laboratory Results of Cases Who Had an Amniocentesis to Rule Out Intraamniotic Infection (n = 129)
| Variables | (−) AFC and Intact and TD n = 28 | (−) AFC and Intact and PTB n = 20 | (−) AFC and PPROM and PTB n = 20 | (+) AFC and Intact & PTB n = 28 | (+) AFC and PPROM and PTBn = 33 | P Value |
|---|---|---|---|---|---|---|
| Intraamniotic inflammation | ||||||
| Absent (MR = 0) | 18 | 13 | 12 | 0 | 1 | <.001 |
| Mild (MR = 1 or 2) | 10 | 7 | 8 | 0 | 5 | |
| Severe (MR = 3 or 4) | 0 | 0 | 0 | 28 | 27 | |
| IL-6, ng/mLa | 0.2 [0.1–0.2] | 0.7 [0.2–2.1] | 0.5 [0.2–1.0] | 85.4 [31.9–122.9] | 17.1 [9.0–42.5] | <.001 |
| Activin A, ng/mLa | 7.8 [5.3–10.4] | 9.2 [6.0–11.2] | 5.9 [4.6–9.0] | 18.1 [12.9–25.6] | 13.1 [7.7–22.7] | <.001 |
| Follistatin, ng/mLa | 3.2 [2.2–6.0] | 4.0 [2.1–6.2] | 3.1 [2.3–4.2] | 3.4 [2.3–5.5] | 2.8 [1.8–4.2] | .578 |
| Activin A to follistatin ratio† | 2.9 [1.9–5.1] | 3.0 [2.2–5.3] | 2.4 [1.9–5.0] | 7.0 [4.1–11.7] | 5.7 [3.6–16.2] | <.001 |
| Variables | (−) AFC and Intact and TD n = 28 | (−) AFC and Intact and PTB n = 20 | (−) AFC and PPROM and PTB n = 20 | (+) AFC and Intact & PTB n = 28 | (+) AFC and PPROM and PTBn = 33 | P Value |
|---|---|---|---|---|---|---|
| Intraamniotic inflammation | ||||||
| Absent (MR = 0) | 18 | 13 | 12 | 0 | 1 | <.001 |
| Mild (MR = 1 or 2) | 10 | 7 | 8 | 0 | 5 | |
| Severe (MR = 3 or 4) | 0 | 0 | 0 | 28 | 27 | |
| IL-6, ng/mLa | 0.2 [0.1–0.2] | 0.7 [0.2–2.1] | 0.5 [0.2–1.0] | 85.4 [31.9–122.9] | 17.1 [9.0–42.5] | <.001 |
| Activin A, ng/mLa | 7.8 [5.3–10.4] | 9.2 [6.0–11.2] | 5.9 [4.6–9.0] | 18.1 [12.9–25.6] | 13.1 [7.7–22.7] | <.001 |
| Follistatin, ng/mLa | 3.2 [2.2–6.0] | 4.0 [2.1–6.2] | 3.1 [2.3–4.2] | 3.4 [2.3–5.5] | 2.8 [1.8–4.2] | .578 |
| Activin A to follistatin ratio† | 2.9 [1.9–5.1] | 3.0 [2.2–5.3] | 2.4 [1.9–5.0] | 7.0 [4.1–11.7] | 5.7 [3.6–16.2] | <.001 |
Intraamniotic inflammation was assessed by proteomics analysis of AF and the MR score.
Data are presented as median [interquartile range] and analyzed by a Kruskal-Wallis one-way ANOVA.
Research Laboratory Results of Cases Who Had an Amniocentesis to Rule Out Intraamniotic Infection (n = 129)
| Variables | (−) AFC and Intact and TD n = 28 | (−) AFC and Intact and PTB n = 20 | (−) AFC and PPROM and PTB n = 20 | (+) AFC and Intact & PTB n = 28 | (+) AFC and PPROM and PTBn = 33 | P Value |
|---|---|---|---|---|---|---|
| Intraamniotic inflammation | ||||||
| Absent (MR = 0) | 18 | 13 | 12 | 0 | 1 | <.001 |
| Mild (MR = 1 or 2) | 10 | 7 | 8 | 0 | 5 | |
| Severe (MR = 3 or 4) | 0 | 0 | 0 | 28 | 27 | |
| IL-6, ng/mLa | 0.2 [0.1–0.2] | 0.7 [0.2–2.1] | 0.5 [0.2–1.0] | 85.4 [31.9–122.9] | 17.1 [9.0–42.5] | <.001 |
| Activin A, ng/mLa | 7.8 [5.3–10.4] | 9.2 [6.0–11.2] | 5.9 [4.6–9.0] | 18.1 [12.9–25.6] | 13.1 [7.7–22.7] | <.001 |
| Follistatin, ng/mLa | 3.2 [2.2–6.0] | 4.0 [2.1–6.2] | 3.1 [2.3–4.2] | 3.4 [2.3–5.5] | 2.8 [1.8–4.2] | .578 |
| Activin A to follistatin ratio† | 2.9 [1.9–5.1] | 3.0 [2.2–5.3] | 2.4 [1.9–5.0] | 7.0 [4.1–11.7] | 5.7 [3.6–16.2] | <.001 |
| Variables | (−) AFC and Intact and TD n = 28 | (−) AFC and Intact and PTB n = 20 | (−) AFC and PPROM and PTB n = 20 | (+) AFC and Intact & PTB n = 28 | (+) AFC and PPROM and PTBn = 33 | P Value |
|---|---|---|---|---|---|---|
| Intraamniotic inflammation | ||||||
| Absent (MR = 0) | 18 | 13 | 12 | 0 | 1 | <.001 |
| Mild (MR = 1 or 2) | 10 | 7 | 8 | 0 | 5 | |
| Severe (MR = 3 or 4) | 0 | 0 | 0 | 28 | 27 | |
| IL-6, ng/mLa | 0.2 [0.1–0.2] | 0.7 [0.2–2.1] | 0.5 [0.2–1.0] | 85.4 [31.9–122.9] | 17.1 [9.0–42.5] | <.001 |
| Activin A, ng/mLa | 7.8 [5.3–10.4] | 9.2 [6.0–11.2] | 5.9 [4.6–9.0] | 18.1 [12.9–25.6] | 13.1 [7.7–22.7] | <.001 |
| Follistatin, ng/mLa | 3.2 [2.2–6.0] | 4.0 [2.1–6.2] | 3.1 [2.3–4.2] | 3.4 [2.3–5.5] | 2.8 [1.8–4.2] | .578 |
| Activin A to follistatin ratio† | 2.9 [1.9–5.1] | 3.0 [2.2–5.3] | 2.4 [1.9–5.0] | 7.0 [4.1–11.7] | 5.7 [3.6–16.2] | <.001 |
Intraamniotic inflammation was assessed by proteomics analysis of AF and the MR score.
Data are presented as median [interquartile range] and analyzed by a Kruskal-Wallis one-way ANOVA.
To better evaluate quantitative relationships between AF inflammation and the rise in activin-A, we analyzed its association with other AF inflammatory markers. We found significant direct correlations of activin-A with both AF WBC count (Figure 3A, r = 0.566, P < .001) and AF IL-6 concentration (Figure 3B, r = 0.585, P < .001), which upheld independent of GA. Interestingly, after GA correction, levels of follistatin correlated inversely with the AF WBC count (Figure 3C, r = −0.283, P = .001). The relationship of follistatin with AF IL-6 did not reach statistical significance (Figure 3D, r = −0.170, P = .060).
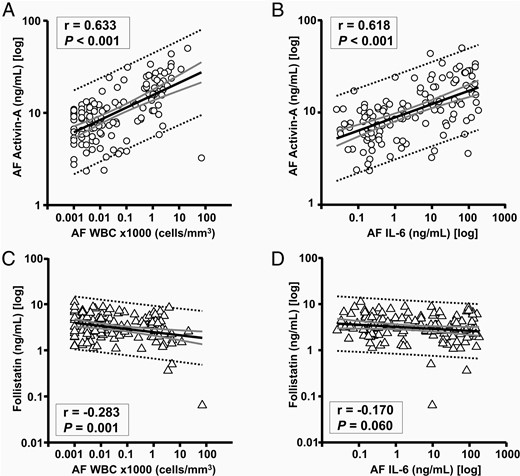
Relationships of AF activin-A with AF WBC count (A) and AF IL-6 (B) among women who had an amniocentesis to rule out infection (n = 129).
Relationships of AF follistatin with WBC count (C) and with AF IL-6 (D) are also shown. Thick black line, linear regression line; thick gray lines, 95% confidence intervals; dotted black lines, 95% prediction intervals. The correlations coefficients (r) and level of significance are presented on each graph after correction for gestational age at amniocentesis.
Effects of activin-A and follistatin imbalance on release of PGE2 and PGF2α from amniochorion explants
To examine the functional role of activin-A and follistatin and the potential consequences of the imbalance between the two factors, we challenged amniochorion explants with activin-A and follistatin using dose concentrations relevant for AF of women with inflammation-associated PTB. At 24 hours, fetal membranes had a basal release of PGE2 in significant excess compared with PGF2α (Figure-4A, P < .001). Follistatin alone had no significant effect over basal release of either PGE2 (Figure 4B) or PGF2α (Figure 4C). Conversely, activin-A stimulated the release of both PGs at levels comparable with explant-exposed endotoxin. This effect was significantly reversed by coincubation with follistatin.
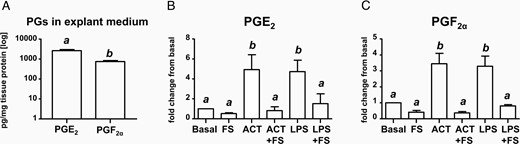
A, PGE2 and PGF2α concentration in explant medium after normalization for total tissue protein. Relative changes in PGE2 (B) and PGF2α (C) compared with basal levels (medium alone) at 24 hours of incubation. Full-thickness amniochorion tissues from women undergoing elective cesarean deliveries (n = 5) were challenged with activin-A (ACT), follistatin (FS), LPS (endotoxin), and combinations thereof. Bars marked with different letters are different statistically at P < .05. Data were analyzed using a one-way ANOVA on ranks followed by Dunn's post hoc tests.
Effects of activin-A and follistatin imbalance on release of MMPs and IL-8 from amniochorion explants
In vitro, at 24 hours, fetal membranes released MMP-1 in significant excess compared with MMP-2, MMP-3, and MMP-9 (Figure-5A, P < .001). Endotoxin and activin-A elicited significant and comparable relative elevations in MMP-1 (Figure 5B) and MMP-9 (Figure 5C). Remarkably, although follistatin blunted the ability of activin-A to elicit release of MMP-1 and MMP-9, it was unable to counteract the stimulatory effect of LPS. Endotoxin but not activin-A induced a significant release of MMP-3 (Figure 5D) in the explant medium, which was not counteracted by the addition of follistatin. The levels of MMP-2 were not significantly affected by either activin-A or LPS (Figure 5E). Interestingly, the changes in PGs and MMPs described above in response to activin-A or follistatin were not accompanied by parallel changes in IL-8, which appeared affected solely by LPS (Figure 5D).

A, Concentration of measured MMPs in explant medium after normalization for total tissue protein. Relative changes in MMP-1 (B), MMP-9 (C), MMP-3 (D), MMP-2 (E), and the chemokine (IL-8) (F) compared with basal levels at 24 hours of incubation. Full-thickness amniochorion tissues from women undergoing elective cesarean deliveries (n = 5) were challenged with activin-A (ACT), follistatin (FS), LPS (endotoxin), and combinations thereof. Bars sharing a common letter are not different statistically (P > .05). Data were analyzed using a one-way ANOVA on ranks followed by Dunn's post hoc tests.
Discussion
The current study demonstrates that intraamniotic infection and inflammation results in the disturbance of activin-follistatin balance in AF, which in turn may increase PGs and the release of MMPs from fetal membranes, which are important mediators of uterine contractility and membrane rupture. Different from our prior investigation (17), the immunoassay used in the current study measured levels of total activin-A (free and bound), which was confirmed using spike and recovery experiments. The rationale was the need to determine the entire pool of activin-A and follistatin, theoretically able to interact with each other. Because follistatin and the relationship between activin-A and follistatin have never been assessed in relationship to PTB, our study is novel.
Activin-A is synthesized as a disulfide dimer of 402–426 amino acid precursors, which is processed to the mature bioactive protein of approximately 25 kDa by proteolysis (28). In addition, the bioavailability of activin-A is regulated by its ability to interact with follistatin, a single-chain glycoprotein that binds activin with high affinity and perturbs its access to cell surface receptors (29, 30). We previously evaluated free activin-A levels in human AF and determined its levels are decreased toward term (17). This is in contrast to the current observation that the level of total AF activin-A remains unchanged. Collectively and in the context of the concurrent decrease in follistatin observed in this study, these data imply that toward term, other factors involved in regulating free activin-A levels (such as proteolytic processing of the precursor) may also contribute.
In infection, the cellular origin(s) of AF activin-A and follistatin are yet to be established. Possible sources are fetal membranes and neutrophils with well-established roles on fighting infection (17, 31). Our current data showed that intraamniotic infection up-regulated activin-A directly related to the level of AF inflammation. Studies in humans and animal models of sepsis have shown that the release of activin-A occurs concomitantly with TNF-α, which is early in the inflammatory cascade preceding the rise in other cytokines such as IL-1β, IL-6, and C-reactive protein (10, 32). In mice, release of activin-A precedes the release of follistatin by approximately 2 hours, suggesting that in the acute phase of defense, activin-A may exercise its function unopposed. When stimulated by TNF-α, freshly isolated neutrophils release activin-A, implying that these cells are a source of prestored activin-A (33). A relevant question is, once released in the AF, what would be the role of activin-A in intraamniotic infection, especially when this polypeptide can carry both pro- and antiinflammatory roles (34)? Activin-A is part of the innate immune response to infection, and its release is linked to engagement of Toll-like receptor (TLR)-4 and TLR-2 (10, 35). Therefore, during infection, AF activin-A can stimulate fetal membranes' and immune cell synthesis and the release of proinflammatory cytokines (ie, IL-6, IL-1β, TNF-α). Based on our data, the overall balance would be turned toward a proinflammatory environment and potential damage because the AF follistatin levels remained unchanged. Interestingly, in vitro, activin-A inhibits LPS action on macrophages via suppressing TLR-4 expression (36). Whether during AF infection this antiinflammatory effect is exercised in human fetal membranes remains to be determined.
Different from our study, which focused on AF, blood follistatin has been reported to increase in septicemic patients (37). These results appear at odds with our findings in AF but are supported by the view that human pregnancy can be thought of as a compartmentalized state: the mother, the AF cavity, and the fetus (38). A successful pregnancy involves fine coordination of the maternal and fetal compartments. Thus, in maternal sepsis, follistatin could be up-regulated in maternal blood without transfer to the AF, especially if the follistatin source is not the placenta. An up-regulation of the AF follistatin levels in women with intraamniotic infection is also less probable based on recent findings that follistatin is predominantly expressed in the extravillous trophoblast with relatively weaker immunostaining in the syncytiotrophoblast layer of the villi (39). Nevertheless, our follistatin findings are supported by in vitro evidence suggesting that LPS had a limited effect of the follistatin protein expression in amniochorion (40). Furthermore, in vivo, histological chorioamnionitis did not change follistatin mRNA levels compared with controls (41).
Differences in maternal blood activin-A and follistatin of women with spontaneous labor have led to an interest on these two peptides as part of mechanisms triggering labor (20). As shown, activin-A stimulated the release from amniochorion of PGE2 and PGF2α at similar levels to LPS. The release of collagenase MMP-1 and the gelatinase MMP-9 was also augmented, confirming a possible role for activin-A in triggering both myometrial contractility and collagenolytic activity of the amniochorion. The results of our in vitro experiments imply that activin-A carries a selective stimulatory effect on MMPs because the release of MMP-2 or MMP-3 was not augmented. Previous research shows increases in AF concentrations of bioactive MMP-2 and MMP-3 in PPROM compared with a preterm labor group (42). These results underline again the complexity of the biochemical events leading to PPROM and the multitude of factors involved. Our data do not negate a role for MMP-2 and MMP-3 in PPROM but provided evidence that activin-A is most probably not responsible for their activation. Interestingly, follistatin reversed activin-A's effect on PGs, implying that the stimulation of PG production and/or the release from amniochorion is receptor mediated. The fact that follistatin was unable to inhibit the release of MMPs elicited by LPS supports the concept that that the pathway leading to PPROM in the context of inflammation initiates downstream and is distinct from that triggering uterine contractions.
Conclusions
Normal gestation associates with a relative excess of AF activin-A over follistatin, driven by a decrease in the AF follistatin level. In pregnancies complicated by intraamniotic infection and PTB, AF activin-A levels are elevated in relationship with the severity of inflammation. Because the presence and severity of the inflammatory process does not impact AF follistatin levels, this results in an excess of bioactive activin-A. Our study points to the possibility that the AF activin-follistatin balance may have a paracrine regulatory role on uterine and fetal membrane function and may participate in events leading to PTB.
Acknowledgments
We are indebted to the nurses, fellows, and residents in the Department of Obstetrics and Gynecology and Reproductive Sciences at Yale New Haven Hospital and to all the patients who participated in the study.
The funding source had no involvement in the study design, interpretation of the data, writing of the report, or the decision to submit the paper for publication.
The study was supported by National Institutes of Health/Eunice Kennedy Shriver National Institute of Child Health and Human Development Grant R01HD062007 (C.S.B. and I.A.B.).
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- AF
amniotic fluid
- GA
gestational age
- LDH
lactate dehydrogenase
- LPS
lipopolysaccharide
- MMP
matrix metalloproteinase
- MR
mass restriction
- PG
prostaglandin
- PPROM
premature rupture of membranes
- PTB
preterm birth
- TLR
Toll-like receptor
- WBC
white blood cell.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}