





Abstract
Prolonged endothelin (ET) receptor signalling causes vasoconstriction and can lead to hypertension, vascular smooth muscle hypertrophy, and hyperplasia. Usually, G protein-coupled receptor signalling is negatively regulated by G protein-coupled receptor kinases (GRKs), preventing prolonged or inappropriate signalling. This study investigated whether GRKs regulate ET receptor contractile signalling in adult Wistar rat mesenteric arterial smooth muscle cells (MSMCs).
ET-1-stimulated phospholipase C (PLC) activity and changes in [Ca2+]i were assessed using confocal microscopy in rat MSMCs transfected with the pleckstrin-homology domain of PLCδ1 (eGFP-PH) and loaded with Fura-Red. ET-1 applications (30 s) stimulated transient concentration-dependent eGFP-PH translocations from plasma membrane to cytoplasm and graded [Ca2+]i increases. ET-1-mediated PLC signalling was blocked by the type A endothelin receptor (ETAR) antagonist, BQ123. To characterize ETAR desensitization, cells were stimulated with a maximally effective concentration of ET-1 (50 nM, 30 s) followed by a variable washout period and a second identical application of ET-1. This brief exposure to ET-1 markedly decreased ETAR responsiveness to re-challenge, and reversal was incomplete even after increasing the time period between agonist challenges to 60 min. To assess GRK involvement in ETAR desensitization, MSMCs were co-transfected with eGFP-PH and catalytically inactive D110A,K220RGRK2, D110A,K220RGRK3, K215RGRK5, or K215RGRK6 constructs. D110A,K220RGRK2 expression significantly attenuated ETAR desensitization, whereas other constructs were ineffective. Small interfering RNA-targeted GRK2 depletion equally attenuated ETAR desensitization. Finally, immunocyotchemical data showed that ETAR activation recruited endogenous GRK2 from cytoplasm to membrane.
These studies identify GRK2 as a key regulator of ETAR responsiveness in resistance arteries, highlighting the potential importance of this GRK isoenzyme in regulating vasoconstrictor signalling pathways implicated in vascular disease.
1. Introduction
Endothelin (ET)-1 is a potent vasoconstrictor controlling vascular tone with an established role in vascular smooth muscle hypertrophy1,2 and hyperplasia,3,4 fibrosis and inflammatory responses, leading to vascular remodelling and hypertension.4,5 ET-1 plays a crucial role in a number of experimental models of hypertension,5,6 and increased plasma ET-1 levels have been reported in hypertensive patients.7 Endothelins can signal through two receptor subtypes ETA and ETB, however in arterial smooth muscle ETA receptors (ETAR) mediate constrictor responses.8 ETARs couple through Gαq/11 to activate phospholipase C (PLC), generating inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), and leading to the release of intracellular Ca2+ stores and the activation of protein kinase C.2,8 ETAR-mediated intracellular Ca2+ increases underlie a major component of ET-1-induced vasoconstriction. ET-1 also promotes PKC-induced inhibition of voltage-gated K+ channels to further enhance vasoconstriction.9
Previous studies have shown that antagonizing ETAR signalling can reverse hypertrophy and hypertension in animal models,10 indicating a potentially important role for ETAR over-activation in the development of vascular disease. Over-stimulation of ETAR signalling clearly has adverse effects on vascular smooth muscle cells, and understanding the mechanisms that regulate Gαq/11 signalling is of potential importance in understanding and treating vascular diseases. Continual or repeated stimulation of receptors by agonists usually leads to reduced responsiveness to further agonist challenge.11 This process, known as receptor desensitization, has been shown to protect cells from the adverse effects of over-stimulation or inappropriate signalling. Phosphorylation of key serine and/or threonine residues within the third intracellular loop and/or C-terminal tail of the G protein-coupled receptor is thought to be the primary event initiating desensitization and is often mediated by one or more members of a family of seven serine/threonine kinases, the G protein-coupled receptor kinases (GRKs).11 Phosphorylation by GRKs enhances receptor affinity for the non-visual arrestins 2 and 3, which can sterically suppress further interaction between receptor and G proteins.12
Although previous studies have shown that recombinantly expressed ETARs are phosphorylated and desensitized in a GRK-dependent manner in model cells, such as HEK293 cells,13 little evidence is currently available to indicate which, if any, GRKs regulate endogenous ETAR signalling in vascular smooth muscle. This is an important issue as recombinant receptors are often regulated very differently to those expressed endogenously. Indeed, a growing number of studies are revealing that precise definition of the GRKs involved in receptor regulation can only be truly defined in a system endogenously expressing the receptor of interest and through manipulation of endogenous GRK populations.11,14–16 Here, we have combined confocal imaging techniques and specific inhibition of endogenous GRK isoenzymic activities to examine the interaction between GRKs and the native ETAR population in isolated, cultured mesenteric smooth muscle cells (MSMCs), a widely studied model of systemic resistance arteries.9,17
2. Methods
2.1 Isolation and culture of mesenteric arterial smooth muscle cells
Adult male Wistar rats were sacrificed by stunning and cervical dislocation, a method approved under the UK Animals (Scientific Procedures) Act 1986. Smooth muscle cells were isolated from small branches of mesenteric artery by enzymatic dissociation as previously described.17 Smooth muscle cells were separated by gentle trituration in 231 medium (Cascade Biologics, Nottingham, UK), supplemented with smooth muscle growth supplement, 100 IU−1 penicillin, 100 µg/mL streptomycin, and 2.5 µg/mL amphotercin B. Cells plated onto coverslips were maintained at 37°C, 5% CO2 in humidified conditions. MSMCs were characterized by immunocytochemistry and immunoblotting for the presence of smooth muscle-specific α-actin and calponin (data not shown). The investigation conforms to Guide for Care and Use of Laboratory Animals US (NIH Publication No. 85-23, revised 1996).
2.2 Immunocytochemistry
MSMCs were cultured for 72 h before fixation and permeabilization in 100% methanol (10 min at −20°C). GRK expression was identified through addition of specific anti-rabbit polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) against GRK2, GRK3, GRK5, and GRK6. Protein expression was visualized with the addition of fluorescein isothiocyanate-conjugated secondary antibodies, before viewing on an Olympus FV500 laser scanning confocal IX70 inverted microscope.
2.3 Small interfering RNA knockdown of endogenous GRK2 levels and western blotting
Expression of GRKs and dominant-negative GRKs (see below) was determined using standard immunoblotting protocols as described previously14 using specific anti-rabbit polyclonal antibodies against GRK2, GRK3, GRK5, and GRK6. MSMCs (1 × 106) were transfected with 10 or 100 nM negative-control or anti-GRK2 (5′-GCAGGUACCUCCAGAUCUCtt-3′) (Applied Biosystems, UK) small interfering (si)RNAs. Cells were lysed after 48 h and GRK expression assessed by immunoblotting (see above). GRK expression was quantified using the GeneGnome image analysis system (Syngene, Cambridge, UK).
2.4 Manipulating cellular GRK activity
GRK2/3 constructs were mutated in both the N-terminal RGS-like domain (D110A) (to prevent binding to Gαq/11 proteins11) and the catalytic-domain (K220R) to allow potential receptor/GRK phosphorylation-dependent interactions to be examined.11,19 In MSMCs transfected with D110A,K220RGRK2 or D110A,K220RGRK3 constructs, normal PLC signalling was unaffected. In contrast, expression of K220RGRK2 or K220RGRK3 markedly attenuated PLC signalling (data not shown), most likely through the previously reported phosphorylation-independent inhibition of Gαq/11 transduction through an interaction with the GRK2/3 RGS-homology (RH) domain.19
GRK activity was manipulated using the dominant-negative constructs D110A,K220RGRK2, D110A,K220RGRK3, K215RGRK5, or K215RGRK6 (0.5 µg), negative-control plasmid (pcDNA3), or through addition of anti-GRK2, or negative-control siRNAs (10 nM). Cells were transfected using LipofectAMINE2000 (Invitrogen, Paisley, UK) according to the manufacturer's instructions.
2.5 Single cell confocal imaging
MSMCs were transfected (24–48 h after isolation) with the pleckstrin-homology domain of PLCδ1 tagged with enhanced green fluorescent protein (eGFP-PH, 0.5 µg), a previously extensively characterized IP3 biosensor.18 Cells were maintained at 37°C by a Peltier unit and continually perfused with a modified Krebs-Henseleit buffer (in mM: NaCl 134, KCl 6, MgCl2 1, glucose 10, HEPES 10, CaCl2 1.3, pH 7.4). Real-time images were taken using an Olympus FV500 laser scanning confocal IX70 inverted microscope (oil immersion objective ×60). Cells transfected with eGFP-PH and loaded with Fura-Red were excited (Ex) at 488 nm and eGFP-PH and Fura-Red emissions (Em) were collected at 505–560 and >660 nm, respectively. ET-1 (50 nM) was applied via the perfusion line as indicated. Changes in cytosolic eGFP-PH fluorescence are represented as the fluorescence emission (F)/initial basal fluorescence (F0) (F/F0) and increases in intracellular Ca2+ ([Ca2+]i) are reported by Fura-Red as decreases in F/F0 fluorescence. Additional Ca2+ experiments in the absence of eGFP-PH were conducted using Fluo-4-AM (3 µM, 60 min) and imaged as for eGFP above.
To assess GRK2 effects on ET-1-stimulated DAG/PKC signalling, MSMCs were transfected with eGFP-PKCα20 or eGFP-PKCε20 (0.5 µg), and anti-GRK2 siRNA as above. After 48 h, cells were loaded with Fura-Red (3 µM) for 50 min before commencing imaging experiments.
2.6 Data and statistical analysis
Data presented are from cells obtained from at least three separate preparations and are expressed as means ± SEM. Data were analysed using one-way ANOVA as indicated, with appropriate post hoc testing (GraphPad Prism, San Diego, CA, USA).
3. Results
3.1 ETAR desensitization and re-sensitization
ET-1 activation of PLC signalling was assessed in MSMCs transfected with the eGFP-PH biosensor and loaded with the Ca2+-sensitive dye Fura-Red to allow simultaneous measurement of changes in IP3 and [Ca2+]i.18 Continual ET-1 (50 nM) challenge produced transient [Ca2+]i increases, which rapidly returned to basal within 100 s (Figure 1A). Short applications (30 s) of ET-1 stimulated transient concentration-dependent translocations of eGFP-PH from the plasma membrane to the cytoplasm and increases in [Ca2+]i, with maximal effects at concentrations of ET-1 of ≥50 nM (data not shown). The ETAR antagonist BQ123 (1 µM) blocked ET-1-stimulated signals, suggesting that in our MSMC preparations ETARs are primarily responsible for PLC signalling (data not shown). To assess the time courses of ETAR desensitization and re-sensitization MSMCs were challenged with a short desensitizing pulse of ET-1 (50 nM, for 30 s, termed R1) followed by a wash period (of 10, 20, 30, or 60 min) and a second ET-1 challenge (50 nM, 30 s, termed R2). Initial ET-1 challenge (R1) increased eGFP-PH translocation and [Ca2+]i with responses returning to basal within 5 min of agonist washout (Figure 1B). A second ET-1 challenge applied 10 min after R1 caused less translocation of eGFP-PH and a reduced [Ca2+]i response (Figure 1B). The reduction in the R2/R1 ratio can be interpreted as an indicator of ETAR desensitization.14,19 Increasing the washout period provided evidence of a slow and incomplete re-sensitization, with R2 for the IP3 response still 70% less than R1 after 60 min (Figure 1C, D).
![Kinetics of ETAR desensitization and re-sensitization. (A) Representative trace showing desensitization of ETAR [Ca2+]i signals following ET-1 challenge (50 nM, 3 min) in Fluo4-loaded MSMCs. Data are representative of 16 cells from three or more separate animals. MSMCs were transfected with 0.5 µg eGFP-PH and loaded with Fura-Red and subjected to the standard R1/R2 desensitization protocol: cells were stimulated with ET-1 (50 nM, 30 sec) (R1), followed by a variable period of 10, 20, or 30 min before further ET-1 (50 nM, 30 s) exposure (R2). Representative traces from single cells with either a 10 min (B) or 30 min (C) wash period are shown. ETAR desensitization was determined as the relative change in R2 response compared with R1 for both eGFP-PH (solid traces) and Fura-Red (broken traces). (D) Cumulative data shown as means ± SEM from 14–15 cells from three or more different animals.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/85/3/10.1093_cvr_cvp310/1/m_cvp31001.gif?Expires=1750483879&Signature=gR7Pw3ww50kynlyAmRwuSxtvY6JYK9WRxySRnoAIJxI-jGkLk44EX4bh~lC5NshjoFvsHHqnVm-wmoe5jyzNbowkYDc~5uJaVLdkWv9CNR-ccRbgIGFjEWCKiD5ZpMawv1VDcH7QV6AomYgnOP8EaIGjnes7whtxbl0ygvb8yuMlqP4Ujcb35hCC2LOoBqAV9nc3~OaBb~Ys-T20gnBcy4ROqNAuYImspSv941Jgp1hGQSvb0ywwe3SCuT1RI3C3LEoffa7QMfaOlJjo1CTlsavck8PYudqmHg2lmxIFKbn0WxDtsb9eAjuE1TZHVxjptTi1fSCOAr7VUAhEHfQZ5Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Kinetics of ETAR desensitization and re-sensitization. (A) Representative trace showing desensitization of ETAR [Ca2+]i signals following ET-1 challenge (50 nM, 3 min) in Fluo4-loaded MSMCs. Data are representative of 16 cells from three or more separate animals. MSMCs were transfected with 0.5 µg eGFP-PH and loaded with Fura-Red and subjected to the standard R1/R2 desensitization protocol: cells were stimulated with ET-1 (50 nM, 30 sec) (R1), followed by a variable period of 10, 20, or 30 min before further ET-1 (50 nM, 30 s) exposure (R2). Representative traces from single cells with either a 10 min (B) or 30 min (C) wash period are shown. ETAR desensitization was determined as the relative change in R2 response compared with R1 for both eGFP-PH (solid traces) and Fura-Red (broken traces). (D) Cumulative data shown as means ± SEM from 14–15 cells from three or more different animals.
3.2 GRK expression in MSMCs
Immnuoblotting data showed that GRKs 2, 5, 6 are expressed in rat MSMCs, however, in agreement with the previous findings16 we were unable to detect GRK3 expression (see Supplementary material online, Figure S1). Immunocytochemical evidence highlights an anticipated cytoplasmic distribution of GRK2 (Figure 6A), whereas GRK5 expression appears to be primarily nuclear (Figure 6B and C).
3.3 Effects of inhibiting endogenous GRK activities on ETAR signalling
To determine which endogenous GRKs modulate ETAR signalling, we inhibited individual GRK isoenzymes by over-expressing catalytically inactive, dominant-negative GRK mutants. This approach has been successfully used in a number of studies to bring about highly selective GRK isoenzymic inhibition.14,15 MSMCs were co-transfected with eGFP-PH (0.5 µg) and pcDNA3 (control), D110A,K220RGRK2,19D110A,K220RGRK3, K215RGRK5 or K215RGRK6 (0.5 µg).21 MSMCs transfected with dominant-negative GRKs were subjected to the standard desensitization protocol (R1/R2) with 10 min washout between ET-1 additions. Control experiments, co-transfecting monomeric red fluorescent protein and eGFP-tagged dominant-negative GRKs indicated >90% co-transfection of cells (data not shown). In MSMCs transfected with pcDNA3 (Figure 2A, B, G), D110A,K220RGRK3 (Figure 2D, G), K215RGRK5 (Figure 2E, G), or K215RGRK6 (Figure 2F, G), the reduction in R2 compared with R1 was consistently ≥80% for eGFP-PH and ≥60% for [Ca2+]i signals, indicating similar reductions in ETAR responsiveness to those observed in untransfected cells. However, in the presence of D110A,K220RGRK2, the R2 IP3 response was decreased by ∼40% relative to R1, whereas the reduction in R2 for the ET-1-stimulated Ca2+ signal was almost completely ablated (Figure 2A, C, G). These data indicate that inhibition of endogenous GRK2 activity results in a highly significant attenuation of ET-1-induced ETAR desensitization.
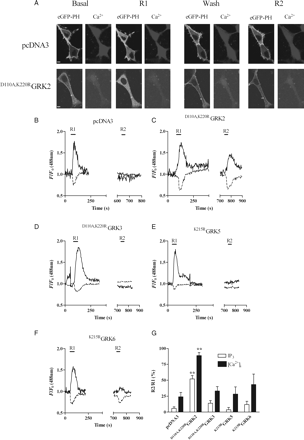
Inhibition of GRK2 reduces the extent of ETAR desensitization. MSMCs were co-transfected with 0.5 µg eGFP-PH and either pcDNA3 (control), D110A,K220RGRK2, D110A,K220RGRK3, K215RGRK5, or K215RGRK6 (0.5 µg). Cells were loaded with Fura-Red and subjected to the standard R1/R2 desensitization protocol (see Methods). Representative images showing both IP3 and Ca2+ changes in MSMCs expressing pcDNA3 (control) or D110A,K220RGRK2 at both the R1 and R2 stimulation points are shown in (A). Representative traces are from cells transfected with pcDNA3 (B), D110A,K220RGRK2 (C), D110A,K220RGRK3 (D), K215RGRK5 (E), or K215RGRK6 (F). ETAR desensitization was determined as the relative change in R2 response compared with R1 for both eGFP-PH (black traces) and Fura-Red (broken traces). Cumulative data (G) are expressed as means ± SEM for the percentage change in R2 relative to R1; n = 7–17 cells for each time-point, from at least eight separate experiments from three or more different animals. Statistical significance is indicated as **P < 0.01 vs. pcDNA3 (one-way ANOVA and Dunnett's post hoc test).
To confirm and extend our findings, MSMCs were transfected with siRNAs designed to target GRK2. Optimal depletion of endogenous GRK2 was achieved 48 h after siRNA transfection at concentrations of siRNA of ≥10 nM (Figure 3A, B). This effect was shown to be GRK2-specific as the anti-GRK2-siRNA did not affect GRK6 expression (Figure 3A, B). A negative-control siRNA had no effect on either GRK2 or GRK6 expression (Figure 3B). In addition, we co-transfected MSMCs with eGFP (0.5 µg) and either negative-control or anti-GRK2 siRNAs (10 or 100 nM) and 48 h later GRK2 expression was determined immunocytochemically. Our previous work indicates that co-transfection rates are >90%; therefore, we have assumed that all eGFP-expressing cells were also transfected with siRNA constructs. When compared with non-eGFP-expressing cells, the presence of eGFP and negative-control siRNA did not affect GRK2 expression (data not shown), however, transfection with anti-GRK2 siRNA caused a marked reduction (∼75%) in GRK2 immunoreactivity (Figure 3C, D), whereas GRK5 and 6 expression was unaffected (Figure 3D).
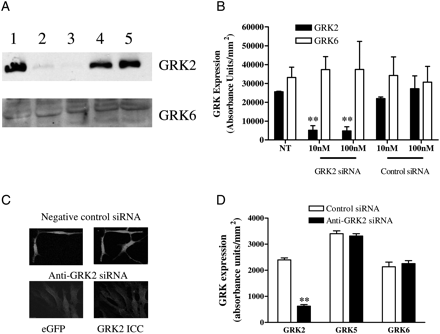
Anti-GRK2 siRNA causes endogenous GRK2 protein depletion. MSMCs were transfected with control or anti-GRK2 siRNAs (10 or 100 nM) using Amaxa nucleofection. After 48 h, cells were lysed and 40 µg of protein loaded for SDS–PAGE separation and immunoblotting. (A) Representative western blots of GRK2 and GRK6 levels are shown: untreated cells (lane 1), cells treated with 10 or 100 nM of anti-GRK2 siRNA (lanes 2 and 3, respectively), 10 or 100 nM negative-control siRNA (lanes 4 and 5, respectively). (B) Cumulative densitometric data show means ± SEM from three animals. (C) Representative images showing depletion of endogenous GRK2 in 4-day-old isolated MSMCs, 48 h after transfection with the same GRK2 siRNA validated in (A and B). On the left are eGFP-transfected cells and the right GRK2 immunoreactivity. (D) Cumulative immunocytochemical data showing that GRK2 siRNA treatment significantly decreased GRK2, but not GRK5 or GRK6 expression in MSMCs. Data are means ± SEM for 20–65 cells for each treatment from three separate animals. Statistical significance is indicated as **P < 0.01 vs. untreated cells (one-way ANOVA and Dunnett's post hoc test).
To examine the effect of siRNA-mediated GRK2 knockdown on ETAR desensitization, MSMCs were co-transfected with eGFP-PH (0.5 µg) and negative-control (10 nM) or anti-GRK2 (10 nM) siRNAs and subjected to the standard R1/R2 desensitization protocol. In the presence of negative-control siRNA, R2 responses were decreased by ≥80% for eGFP-PH and by ≥60% for [Ca2+]i signals compared with R1, consistent with the degree of receptor desensitization observed in untransfected cells (Figure 4A, B, D). In contrast, in cells transfected with anti-GRK2 siRNA, R2 and R1 responses were similar to those previously seen in D110A,K220RGRK2-transfected MSMCs, with a 50% decrease in eGFP-PH and ∼30% decrease in Ca2+ signals (Figure 4A, C, D). To assess whether quantitatively similar effects of manipulating cellular GRK2 are seen with respect to the DAG/PKC arm of the ETAR signalling pathway, we have assessed ET-1-stimulated translocation of the Ca2+ and DAG-sensitive PKCα and DAG-sensitive PKCε isoenzymes. Applying the standard desensitization protocol to eGFP-PKCα- (Figure 5A, C) or eGFP-PKCε (Figure 5B, C)-transfected MSMCs showed that a significantly greater recovery with respect to PKC recruitment responses was also observed when cellular GRK2 levels were selectively diminished.
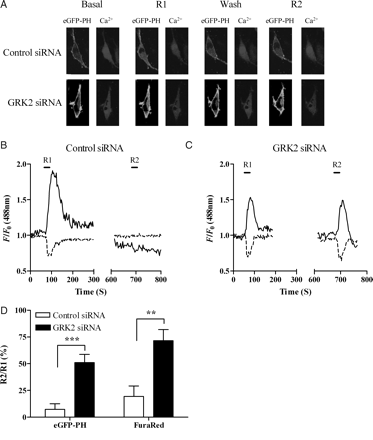
Depletion of endogenous GRK2 inhibits ETAR desensitization. MSMCs were co-transfected with 0.5 µg eGFP-PH and either negative-control or anti-GRK2 (10 nM) siRNAs. Cells were loaded with Fura-Red and subjected to the standard R1/R2 desensitization protocol (see Methods). (A) Representative images of changes in IP3 and Ca2+ in MSMCs expressing either control siRNA or anti-GRK2 siRNA at both the R1 and R2 stimulation points are shown. Representative traces from single cells transfected with control siRNA (B) or anti-GRK2 siRNA (C). ETAR desensitization was determined as the relative change in R2 response compared with R1 for both eGFP-PH (black traces) and Fura-Red (broken traces). Cumulative data (D) show means ± SEM from 7–9 cells from four or more animals. Statistical significance is indicated as **P < 0.01; ***P < 0.001 (one-way ANOVA, unpaired t-test).
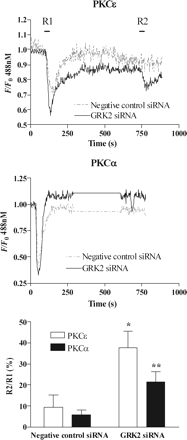
GRK2 depletion decreases ET1-induced desensitization of ETA receptor-stimulated PKC recruitment. MSMCs were co-transfected with 0.5 µg of either eGFP-PKCα or eGFP-PKCε and negative-control or anti-GRK2 (10 nM) siRNAs. Representative traces showing eGFP-PKCε (A) or eGFP-PKCα (B) translocations in MSMCs subjected to the standard R1/R2 desensitization protocol (see Methods) in the presence of negative-control (broken traces) or GRK2 siRNA (black traces). Cumulative data (C) are shown as means ± SEM from 8–20 cells from three to four animals. Statistical significance is indicated as *P < 0.05; **P < 0.01 (one-way ANOVA, Dunnett's post hoc test).
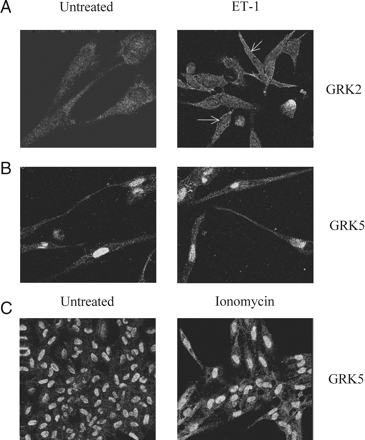
ET-1 stimulates recruitment of endogenous GRK2, but not GRK5 to the plasma membrane. MSMCs were challenged with ET-1 (50 nM) for 3 min, before fixation and processing for immunocytochemical detection of GRKs 2 and 5 (see Methods). Representative confocal images show the distribution of GRK2 and GRK5 in MSMCs. Left-hand panels are images of untreated cells, whereas the right-hand panels show: (A) GRK2 recruitment to the plasma membrane after ET-1 challenge (indicated by arrows); (B) Lack of movement of GRK5 after ET-1 challenge; (C) Redistribution of GRK5 from the nucleus to the cytoplasm after ionomycin (2 µM, 15 min) treatment. Data are representative of experiments from cells isolated from three separate animals.
3.4 ET-1-stimulated recruitment of endogenous GRKs
To investigate further GRK2-mediated regulation of ETAR signalling, we examined the redistribution of this GRK isoenzyme following ET-1 addition. The MSMCs were treated with ET-1 (50 nM) for 3 min, after which cells were fixed and processed to allow immunocytochemical detection of GRKs. Confocal images show GRK2 recruitment to the plasma membrane following ET-1 exposure (Figure 6A). The predominantly nuclear localization of GRK5 was unaltered following ET-1 challenge (Figure 6B), whereas addition of the Ca2+-ionophore ionomycin promoted GRK5 translocation to the cytoplasm (Figure 6C). We were unable to detect recruitment of GRK3 (data not shown), and due to its ‘constitutive’ plasma membrane localization GRK611 was not studied here.
3.5 Does protein kinase C have a role in ETAR desensitization
ETAR activation can recruit PKC isoenzymes to the plasma membrane20 raising the possibility that PKCs may also be involved in ETAR regulation. To assess whether PKC plays a role in ETAR desensitization, we used two complementary approaches: (i) pre-incubation of MSMCs with staurosporine (1 µM) or dimethyl sulphoxide (vehicle control) for 10 min prior to and throughout the standard R1/R2 desensitization protocol and (ii) down-regulation of PKC isoenzymes by a 24 h pre-treatment with phorbol 12,13-dibutyrate (PDBu, 1 µM). Neither acute treatment with staurosporine (see Supplementary material online, Figure S2C, E) nor chronic phorbol ester treatment (see Supplementary material online, Figure S2E) had any effect on ET-1-induced ETAR desensitization. In addition, the magnitude of effect on ETAR desensitization caused by the siRNA GRK2 knockdown strategy was unaltered in MSMCs additionally treated with either staurosporine or chronic PDBu (see Supplementary material online, Figure S2D, E).
4. Discussion
ET-1, a potent vasoconstrictor important in the regulation of vascular tone, has also been reported to promote vascular hypertrophy and hyperplasia.1,3,4 In vascular smooth muscle, ETAR has been shown to be the dominant receptor subtype mediating ET-1 smooth muscle contraction.8 In agreement, we find that ET-1 acts through the ETAR to activate Gαq/11/PLC signalling and to elevate [Ca2+]i in our MSMC preparations2,20 (ultimately to modulate other pathways, including a number of K+ conductances9 and cause contraction). Despite the clear potential pathophysiological importance of ETAR, relatively little is currently known regarding the regulation of this receptor subtype in native vascular tissue. To address this, we have used simultaneous confocal imaging of IP3 (using eGFP-PH), [Ca2+]i (using the Ca2+-sensitive dye Fura-Red), and DAG/PKC (using eGFP-PKCs) to investigate ETAR regulation by ET-1 in smooth muscle cells within days of isolation from resistance arteries. MSMC culture conditions were chosen to enable us to examine ETAR regulation in cells maintaining as far as possible an in vivo phenotype. High levels of α-actin and calponin expression, combined with visual evidence of smooth muscle cell contractions elicited by ET-1 (and other contractile agonists) indicated the maintenance of a contractile phenotype in these cultures.
In agreement with the previous reports, for example in HEK293 cells,22 the initial increase in [Ca2+]i stimulated by ET-1 in MSMCs rapidly declined towards basal, even in the continued presence of agonist. Brief (30 s) exposure to ET-1 was sufficient to cause extensive and prolonged loss of ETAR responsiveness to subsequent ET-1 re-challenge with respect to both IP3 and Ca2+ signals. As expected, Ca2+ signals showed more rapid recovery than IP3 signals reflecting the greater amplification of the former signal in the ET-1-stimulated ETAR-PLC signalling pathway. Previous studies in arterial tissue have tended to use prolonged (>60 min) ET-1 exposures leading to marked reductions in arterial contractions on ET-1 re-challenge, indicating profound ETAR desensitization23 and most probably ETAR down-regulation.24
Data from studies in recombinant cell systems suggest that GRKs are able to regulate ETAR signalling.13 Indeed, when expressed in HEK293 cells, ETAR rapidly desensitized, and phosphorylation of the receptor was enhanced by recombinant over-expression of GRKs 2, 5, or 6.13 In addition, over-expression of recombinant GRK2 and GRK3 increased ETAR phosphorylation in CHO cells.25 Together these and other studies suggest that GRKs are able to cause ETAR phosphorylation, however, such studies are not necessarily predictive of how/if the receptor will be regulated by specific GRK isoenzymes in native ETAR expression systems. Therefore, a key objective here was to delineate for the first time the role that endogenous GRK isoenzymes play in ETAR regulation in resistance artery smooth muscle. Owing to the lack of effective and specific pharmacological GRK inhibitors, we applied previously validated molecular approaches to disrupt or decrease activity of specific endogenous GRK isoenzymes. Initially, we used dominant-negative (kinase-dead) GRK mutants to disrupt ETAR/GRK isoenzyme-specific interactions in an attempt to attenuate or prevent the reduction in ETAR responsiveness observed on re-addition of ET-1. The D110A,K220RGRK2 construct, which is mutated to prevent both kinase activity and Gαq/11-binding,19 markedly attenuated ETAR desensitization. In contrast, over-expression of D110A,K220RGRK3, K215RGRK5, or K215RGRK6 mutants had no effect on the extent or time course of recovery of ETAR responsiveness to ET-1.
A potential criticism of the dominant-negative GRK over-expression approach is the possibility of ‘off-target’ effects or lack of sufficient specificity towards the GRK isoenzyme being targeted. This clearly was not the case here given the marked contrast between effects of the GRK2 and 3 constructs, nevertheless, to provide a complementary experimental approach we used specific siRNAs to deplete (by ≥75%) endogenous GRKs in MSMCs. In agreement with the D110A,K220RGRK2 experiments, siRNA-induced depletion of GRK2 also markedly attenuated the ET-1-induced ETAR desensitization with respect to IP3, Ca2+, and DAG/PKC signal readouts. Furthermore, redistribution of endogenous GRK2 from the cytoplasm to the membrane could be detected after a brief ET-1 stimulation, most likely indicating recruitment to agonist-occupied ETARs. Interestingly, despite previous reports of the plasma membrane association of GRK5, this isoenzyme is predominantly located within the nuclei of MSMCs. Nuclear accumulation of GRK5 has been reported previously in cardiomyocytes,26 presumably as a consequence of its nuclear localization sequence.27 Our study also represents a first report of the nuclear GRK5 localization in MSMCs, although the significance of this finding for GPCR signalling and vascular physiology remains to be established.
Collectively, these data indicate that GRK2 is the key endogenous GRK subtype initiating ETAR desensitization in MSMCs, with either GRK2 knockdown or disruption of the normal GRK2-ETAR interactions causing an ∼50% attenuation of ETAR desensitization. This partial blockade may arise, because the experimental strategy is incompletely effective, or may indicate that other, so far undefined mechanisms are involved in regulating ETAR responsiveness. In MSMCs, ETAR activation is known to recruit several PKC isoenzymes9,20 and PKC activation is known to phosphorylate ETAR, at least in a HEK293 cell background,13 suggesting a potential role for this protein kinase family in ETAR desensitization. In addition, PKCs are also known to phosphorylate GRK2 enhancing its membrane recruitment28 and ability to desensitize GPCRs.29 Unfortunately, most of the available PKC inhibitors are fluorescent molecules precluding their use with the translocating, fluorescent biosensors. Consequently, we used the complementary approaches of inhibiting PKC activity with either the broad-specificity PKC inhibitor staurosporine, or down-regulating conventional and novel PKC isoenzymes through chronic treatment of MSMCs with the phorbol ester, PDBu. Neither manipulation of MSMCs suggested that PKCs play a role in the ETAR desensitization process nor alter the ability of GRK2 to exert its effect on the ETAR. Nevertheless, it remains a possibility that PKC-mediated ETAR phosphorylation may have as yet undefined roles in directing the signalling outputs of this receptor subtype (e.g. ET-1-mediated inhibition of K+ channels to alter vasoconstrictor responses9,17). Nonetheless, since GRK2 inhibition appears only partially to prevent ETAR desensitization, it is possible that other mechanisms are involved, perhaps involving more distal regulatory processes such as β-arrestin binding,25 receptor internalization and/or receptor down-regulation.24
Accumulating evidence indicates that GRKs play an important role in the pathogenesis of vascular disease. In experimental systems, adenovirus-mediated over-expression of GRK2 in cultured rabbit aortic smooth muscle cells attenuated ET-1-induced proliferation,30 whereas a two- to three-fold over-expression of GRK2 in vascular smooth muscle significantly reduced functional (increases in mean arterial pressure) responses to angiotensin II in mice.31 These findings suggest a protective role for GRK2 expression, presumably by enhancing the desensitization of these pro-hypertensive signals. However, in apparent contradiction to these findings, GRK2 expression has been reported to be enhanced in both hypertensive patients32 and rat models of hypertension. A possible explanation for this discrepancy is that over-expression of GRK2 not only attenuates vasoconstrictor (e.g. angiotensin II) signalling, but also vasodilator signalling. Specifically, signalling through the β2-adrenoceptor is substantially reduced in vascular smooth muscle over-expressing GRK2.31 In addition, since GRK2 is able to suppress Gαq-signalling through its N-terminal RGS-like domain,11 it is conceivable that elevated GRK2 expression is initially an adaptive response to over-stimulation of Gαq-signalling pathways, preventing inappropriate vasoconstriction and resultant hypertension.33 It is therefore clear that GRK2 is linked to hypertensive adaptations, but its exact role in the development of hypertension merits further investigation. In summary, here we have provided important new evidence highlighting the role played by GRK2 in regulating ET-1/ETAR/Gαq-mediated vasoconstriction and suggest that this GRK isoenzyme might protect against ET-1-induced vascular dysfunction.1,3–6,34
Supplementary material
Supplementary Material is available at Cardiovascular Research online.
Funding
This work was generously supported by the British Heart Foundation [PG06/161/22136 and RG06/008/22062]. Funding to pay the Open Access publication charges for this article was provided by the British Heart Foundation.
Acknowledgements
We thank Tobias Meyer (Stanford University, USA) for generously providing the eGFP-PH biosensor, and Stephen Ferguson (University of Western Ontario, Canada) for the gift of eGFP-tagged PKC isoenzymes.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}