




Abstract
Aims
Neointimal hyperplasia (NIH) characterized by vascular smooth muscle cell (VSMC) dysfunctions plays a critical role in many vascular diseases including atherosclerosis and restenosis, which leads to serious ischemic complications and has limited therapeutic approaches. Our previous studies confirm a critical role for nuclear factor erythroid 2-related factor 3 (Nrf3) in VSMC differentiation. However, little is known about the functional implications of Nrf3 in NIH.
Methods and Results
Transcriptome dataset and human atherosclerotic samples were used to determine Nrf3 expression levels. Global (Nrf3-/-) and VSMC-specific (Nrf3ΔSMC) Nrf3 knockout mice were used to assess the role of Nrf3 in VSMC function and injury-induced NIH. Complementary molecular methods were performed to identify Nrf3 downstream targets and elucidate the regulatory role of Nrf3 in target gene regulation. Porcine carotid stenting model was used to validate the therapeutic effects of Nrf3 inhibition in vascular remodeling.
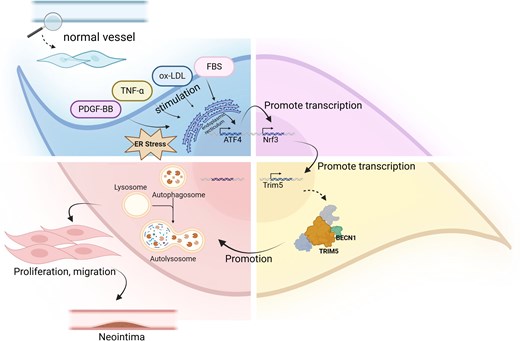
Transcriptomic data and immunostaining analysis showed increased levels of Nrf3, and a positive correlation between Nrf3 and NIH in the human atherosclerotic vessels. Various pathophysiological stimuli induced endoplasmic reticulum (ER) stress which enhanced Nrf3 expression via Activating Transcription Factor 4 (ATF4). Nrf3 overexpression promoted both human and mouse VSMC proliferation, migration, and inflammatory response, while opposite effects were observed when Nrf3 was deleted or knockdown. Nrf3-/- and Nrf3ΔSMC mice showed decreased VSMC accumulation and attenuated vascular injury-induced NIH. Mechanistically, tripartite motif-containing 5 (Trim5), a genetic risk locus for coronary artery disease, was identified as a functional downstream target of Nrf3 in VSMCs and in injured arteries. Nrf3 enhanced autophagy in VSMCs and injured arteries by upregulating Trim5 expression, which subsequently promoted dysfunctions of VSMCs, thereby increasing injury-induced NIH. Importantly, restoring either Nrf3 or Trim5 expression in Nrf3-/- mice rescued the arterial phenotypes observed in Nrf3-/- mice. Critically, the porcine carotid artery restenosis induced by ballooning and stenting was significantly reduced by suppressing Nrf3 expression through perivascular administration of the Nrf3 inhibitors.
Conclusions
We comprehensively demonstrate that Nrf3 is a novel modulator in VSMC dysfunctions and injury-induced NIH. Nrf3 exerts its pathological functions by transcriptional activation of Trim5 gene, which in turn triggers autophagy in VSMCs and injured arteries, promoting arterial remodeling. Inhibiting the Nrf3-Trim5 signal axis ameliorates injury-induced NIH, offering novel therapeutics for treating patients with NIH-related vascular diseases.
 Accepted manuscripts
Accepted manuscriptsAuthor notes
Chen Q, Sun S and Shi Z contributed equally to the study
Xiao Q and Zhang L contributed equally to the study as senior authors

{kind=link}