





Abstract
Despite improvements in patient identification and management, heart failure (HF) remains a major public health burden and an important clinical challenge. A variety of animal and human studies have provided evidence suggesting a central role of calcium/calmodulin-dependent protein kinase II (CaMKII) in the development of pathological cardiac remodelling and HF. Here, we describe a new potent, selective, and orally available CaMKII inhibitor.
Chemical optimization led to the identification of RA306 as a selective CaMKII inhibitor. This compound was found potent on the cardiac CaMKII isoforms delta and gamma (IC50 in the 10 nM range), with pharmacokinetic properties allowing oral administration in animal models of HF. RA306 was administered to diseased mice carrying a mutation in alpha-actin that is responsible for dilated cardiomyopathy (DCM) in humans. In two separate studies, RA306 was orally administered at 30 mg/kg either for 2 weeks (twice a day) or for 2 months (once a day). Echocardiography monitoring showed that RA306 significantly improved cardiac function (ejection fraction and cardiac output) as compared to vehicle. These disease modifying effects of RA306 were associated with inhibition of cardiac phosphorylation of phospholamban (PLN) at threonine-17, indicating reduced cardiac CaMKII activity.
This work supports the feasibility of identifying potent orally available CaMKII inhibitors suitable for clinical use to treat heart disease.
1. Introduction
Heart failure (HF) is a leading cause of death and hospitalization that represents a significant economic burden worldwide.1 The prevalence of HF and associated costs are expected to double over the next 20 years due to the ageing of the population and because of improved treatment of ischaemic and valvular heart disease.2,3 Despite advances in HF therapies, which mainly act by reducing workload on the heart, the prognosis for patients remains poor, highlighting the need for new therapeutic options. calcium/calmodulin-dependent protein kinase II (CaMKII) has emerged as a promising target for drug discovery based on both animal and human pre-clinical data.
CaMKII is a serine/threonine kinase expressed as hetero-multimers of 12 subunits encoded by four different genes (α, β, γ, and δ).4 The α and β isoforms are predominant in the brain where they play a role in learning and memory processes,5,6 whereas the δ and γ isoforms, which are more ubiquitous, represent the main cardiac isoforms. CaMKII regulates key proteins involved in Ca2+ handling, excitation-contraction coupling, and cardiomyocyte hypertrophy, including the ryanodine receptor 2 (RyR2), PLN, L-Type Ca2+ channel (LTCC), Na+ and K+ channels, and histone deacetylases (HDAC).7,8 Up-regulation of cardiac CaMKII expression and activity was reported in HF subjects with reduced ejection fraction, familial hypertrophic cardiomyopathy, atrial fibrillation, and diabetic cardiomyopathy.9–16 The role of CaMKII in the pathophysiology of cardiac diseases was first established in CaMKII over-expressing transgenic (TG) mice that develop severe HF with ventricular arrhythmias.17–19 In contrast, CaMKII knock out (KO) mice or animals treated with a CaMKII chemical inhibitor were shown to be protected from cardiac dysfunction, adverse cardiac remodelling, cardiac arrhythmias, and mortality in HF models, with a redundant pathological role of CaMKII δ and γ isoforms in the progression of HF.20–28 Furthermore, treatment of cardiac tissue obtained from HF patients with CaMKII chemical inhibitors increased contractility and reduced RyR2-mediated Ca2+ leaks.11
Despite these data that suggest a key role of CaMKII in the pathophysiology of HF and cardiac arrhythmias, there are still no CaMKII inhibitors in clinical trials, highlighting the difficulty in identifying potent, selective, and orally available compounds suitable for chronic cardiovascular indications.
We initiated an internal chemical optimization programme following re-scaffolding of the previously described pyridopyrimidines series of ATP-competitive CaMKII inhibitors.29 Pre-defined criteria were applied to guide chemical optimization, including: potency in the nanomolar (nM) range on recombinant CaMKIIδ, high kinase selectivity to avoid off-target side effects, good selectivity (>10 fold) towards the α and β isoforms or low brain permeation (<10-fold heart exposure) to avoid central nervous system (CNS) side effects, good selectivity towards ion channels, sub-micromolar activity on cardiomyocytes, and good oral bioavailability with a sustained inhibition of CaMKII in the myocardium (>12 h).
The present study describes the lead compound RA306, and reports its effects in diseased alpha-actin mutant mice.
2. Methods
All detailed information is provided in the Supplementary material online.
2.1 CaMKII enzymatic assays
CaMKII inhibition was measured by capillary electrophoresis mobility shift assay technology from Caliper LifeSciences (PerkinElmer) using purified human CaMKII protein isoforms: alpha (Carna Bioscience), beta (Millipore), gamma (Millipore), and delta (Millipore).
2.2 Evaluation of RA306 activity in cardiomyocytes
Cor4U® cells from Ncardia and adult rat and mouse cardiomyocytes were treated with RA306 (30 nM–10 µM) for 30 min prior to isoproterenol (Iso) challenge at EC80 concentration (1 µM for each species) for 15 min. In a second set of experiments, Cor4U® were pre-treated with RA306 (3 and 10 µM) prior to an oxidative challenge with H2O2 (H1009, Sigma) at 1 mM. After lysis, samples were subjected to SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Phosphorylation of PLN on Thr17 (sc-17024-R, Santa Cruz) and Ser16 (A010-12AP, Badrilla) were normalized to Total-PLN (#14562, Cell Signaling Technologies).
2.3 Selectivity profiling
Kinase profiling was performed by Eurofins on 304 human recombinant kinases, representative of the full human kinome (Supplementary material online, Table S1), using P33 radioactive assays at Km ATP concentrations and RA306 at 1 µM. When a kinase was inhibited by more than 50%, a dose-response evaluation of RA306 was performed at eight concentrations in duplicates with IC50 estimation by curve fitting. Extended profiling was performed by CEREP at 10 µM of RA306 on 107 targets (listed in Supplementary material online, Table S2) using binding, enzyme, and uptake catalogue assays. RA306 was retested at 1 µM in the sigma (1/2) (h) binding assay and in dose-response in the 15-lipoxygenase-2 (h) enzymatic assay. RA306 and KN93 profiling on human ion channels was performed by patch-clamp in dose-response experiments on CHO recombinant cell line (hERG), Human Embryonic Kidney recombinant cell line (Nav1.5, Kv4.3) or human induced-pluripotent stem cell-derived cardiomyocytes (Cav1.2).
2.4 Pharmacokinetic profiling
After an extraction step, RA306 concentrations were determined in plasma, brain, and heart tissues by LC/MS/MS method using an API4000 instrument (Sciex) in positive electrospray ionization mode and Analyst software (version1.6.2). The pharmacokinetic analysis was performed using a non-compartmental model and WinNonLin software (Phoenix, version 6.4).
2.5 Quantification by simple WesternTM technology of Phospho-Thr17-PLN, Phospho-Ser16-PLN, and total-PLN in freeze-clamped mouse heart samples
After lysis and homogenization of freeze-clamped tissues, samples were diluted to normalize total protein concentration at 0.2 mg/mL. Simple WesternTM technology was performed using anti-total PLN 1:25 (#8495, Cell Signaling Technology), anti-pThr17-PLN 1:160 (#sc-17024, Santa Cruz Biotechnology), and anti-pSer16-PLN 1:50 (#A010-12AP, Badrilla).
2.6 In vivo pharmacology studies
Experiments were performed in full compliance with standards for the care and use of laboratory animals, according to French and European Community (Directive2010/63/EU) legislation. At the end of each study, mice were euthanized using profound anaesthesia with isoflurane 4% followed by exsanguination and heart removal.
In both studies, cardiac function was assessed by transthoracic echocardiography using Vevo 1100 (VisualSonics) in isoflurane 1.5%-anaesthetized animals.
In the first study, 8–10-month-old alpha-actin TG mice were treated orally twice daily for 2 weeks either with RA306 (30 mg/kg; n = 11) or vehicle (Captisol 10%; n = 13). Control age-matched wild-type C57BL6 mice (n = 13) received vehicle only.
Echocardiography was performed before and 2 weeks after the onset of treatments and 2 h after the last administration. The last echocardiography was followed by heart clamp freezing for quantification of phospho-Thr17-PLN/total PLN by Simple WesternTM technology.
In the second study, 8-month-old alpha-actin TG mice were treated orally once daily for 2 months either with RA306 (30 mg/kg; n = 9) or vehicle (Captisol 10%; n = 9). Control age-matched wild type C57BL6 mice (n = 15) received vehicle only.
Echocardiography was performed before, 2 weeks and 2 months after the onset of treatments and within 4 h after the last administration.
At the end of the study, after euthanasia, hearts were collected, weighted, and processed for pathological examination.
2.7 Statistical analysis
Detailed statistical analysis is described in Supplementary material online, Methods.
3. Results
3.1 RA306 inhibits human CaMKII in an ATP-competitive manner
RA306 is a low molecular weight compound (MW = 571.7 Da) having a thienopyrimidine core scaffold (Figure 1A). Its in vitro potency was assessed on the four human CaMKII isoforms with enzymatic caliper assays. At Michaelis–Menten constant (Km) ATP concentration, RA306 potently inhibited CaMKII delta (IC50 = 15 nM), gamma (IC50 = 25 nM), and alpha (IC50 = 61 nM) isoforms, with a drop of potency on CaMKII beta (IC50 = 420 nM) (Figure 1B). A shift of IC50 was observed in each assay at 10× Km ATP concentration, confirming the ATP-competitive mechanism of action of RA306 (Figure 1C and D).
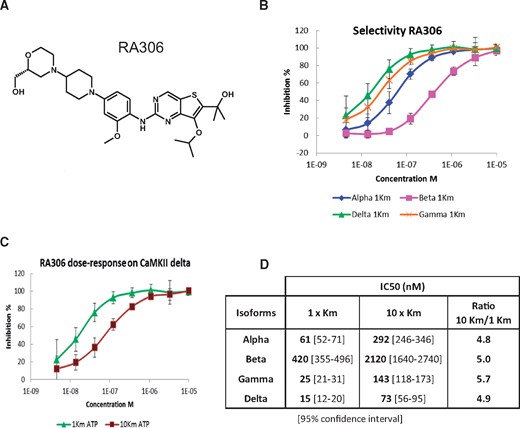
RA306 enzymatic activity. (A) Chemical structure of RA306. (B) Dose-response effect of RA306 at 1× Km ATP for CaMKII delta (green), gamma (orange), alpha (blue), and beta (pink) isoforms. (C) Dose-response effect of RA306 on CaMKII delta at 30 µM ATP (purple) and 300 µM ATP (green) corresponding to 1× and 10× Km. Data are represented as means ± SD in B and C (N = 6 experiments). (D) Table of RA306 IC50 estimated for each CaMKII isoform at 1× and 10× Km ATP.
3.2 RA306 selectivity profile
Most small-molecule kinase inhibitors inhibit multiple kinases and this contribute to off-target toxic side effects.30 At Km ATP concentration, RA306 potently inhibited CaMKII delta and gamma isoforms in a panel of 304 radioactive kinase assays, with an estimated IC50 below 3 nM and a shift of activity towards the beta isoform as observed in caliper assays (Table 1). On the 301 kinases tested outside of CaMKII delta, gamma, and delta (alpha not tested), only nine were inhibited by RA306 with IC50 below the threshold value of 300 nM corresponding to a selectivity ratio of at least 100-fold as compared to CaMKII delta IC50 (Table 1).
RA306 selectivity profile
| Eurofins panel (304 kinases) | |||
| P33 assays | |||
| IC50 (nM) | IC50 ratioa | ATP (µM) | |
| CaMKIIγ (h) | <3 | ∼1 | 10 |
| CaMKIIδ (h) | <3 | 1 | 10 |
| CaMKIδ (h) | 12 | >4 | 15 |
| CaMKIIβ (h) | 17 | >6 | 15 |
| ACK1 (h) | 26 | >9 | 70 |
| MLK1 (h) | 89 | >30 | 45 |
| SIK (h) | 149 | >50 | 45 |
| PTK5 (h) | 216 | >72 | 155 |
| Pyk2 (h) | 249 | >83 | 90 |
| IRE1 (h) | 254 | >85 | 15 |
| ULK2 (h) | 255 | >85 | 15 |
| CaMKK2 (h) | 294 | >98 | 90 |
| Others | >300 | >100 | Km |
| CEREP panel (107 targets) | |||
| Binding, enzyme, and uptake assays | |||
| I%–10 µM | I%–1 µM | IC50 (nM) | |
| CaMK2α (h) | 101 | NA | NA |
| Sigma (1/2) (h) | 96 | 73 | NA |
| 15-Lipoxygenase-2 (h) | 96 | 73 | 270 |
| M3 (h) | 90 | NA | NA |
| IRK (h) | 74 | NA | NA |
| Others | <50 | NA | NA |
| Ion channels | |||
| Manual patch-clamp | |||
| RA306 IC50 (µM) | KN93 IC50 (µM) | IC50 ratiob | |
| ERG (h) | >30 | 0.08 | >375 |
| Kv4.3 (h) | >30 | 4 | >7.5 |
| Nav1.5 (h) | >30 | 2.7 | >11 |
| Cav1.2 (h) | 14 | 0.9 | 15 |
| Eurofins panel (304 kinases) | |||
| P33 assays | |||
| IC50 (nM) | IC50 ratioa | ATP (µM) | |
| CaMKIIγ (h) | <3 | ∼1 | 10 |
| CaMKIIδ (h) | <3 | 1 | 10 |
| CaMKIδ (h) | 12 | >4 | 15 |
| CaMKIIβ (h) | 17 | >6 | 15 |
| ACK1 (h) | 26 | >9 | 70 |
| MLK1 (h) | 89 | >30 | 45 |
| SIK (h) | 149 | >50 | 45 |
| PTK5 (h) | 216 | >72 | 155 |
| Pyk2 (h) | 249 | >83 | 90 |
| IRE1 (h) | 254 | >85 | 15 |
| ULK2 (h) | 255 | >85 | 15 |
| CaMKK2 (h) | 294 | >98 | 90 |
| Others | >300 | >100 | Km |
| CEREP panel (107 targets) | |||
| Binding, enzyme, and uptake assays | |||
| I%–10 µM | I%–1 µM | IC50 (nM) | |
| CaMK2α (h) | 101 | NA | NA |
| Sigma (1/2) (h) | 96 | 73 | NA |
| 15-Lipoxygenase-2 (h) | 96 | 73 | 270 |
| M3 (h) | 90 | NA | NA |
| IRK (h) | 74 | NA | NA |
| Others | <50 | NA | NA |
| Ion channels | |||
| Manual patch-clamp | |||
| RA306 IC50 (µM) | KN93 IC50 (µM) | IC50 ratiob | |
| ERG (h) | >30 | 0.08 | >375 |
| Kv4.3 (h) | >30 | 4 | >7.5 |
| Nav1.5 (h) | >30 | 2.7 | >11 |
| Cav1.2 (h) | 14 | 0.9 | 15 |
NA, not available.
Kinase IC50/CaMKIIδ IC50.
RA306 IC50/KN93 IC50.
RA306 selectivity profile
| Eurofins panel (304 kinases) | |||
| P33 assays | |||
| IC50 (nM) | IC50 ratioa | ATP (µM) | |
| CaMKIIγ (h) | <3 | ∼1 | 10 |
| CaMKIIδ (h) | <3 | 1 | 10 |
| CaMKIδ (h) | 12 | >4 | 15 |
| CaMKIIβ (h) | 17 | >6 | 15 |
| ACK1 (h) | 26 | >9 | 70 |
| MLK1 (h) | 89 | >30 | 45 |
| SIK (h) | 149 | >50 | 45 |
| PTK5 (h) | 216 | >72 | 155 |
| Pyk2 (h) | 249 | >83 | 90 |
| IRE1 (h) | 254 | >85 | 15 |
| ULK2 (h) | 255 | >85 | 15 |
| CaMKK2 (h) | 294 | >98 | 90 |
| Others | >300 | >100 | Km |
| CEREP panel (107 targets) | |||
| Binding, enzyme, and uptake assays | |||
| I%–10 µM | I%–1 µM | IC50 (nM) | |
| CaMK2α (h) | 101 | NA | NA |
| Sigma (1/2) (h) | 96 | 73 | NA |
| 15-Lipoxygenase-2 (h) | 96 | 73 | 270 |
| M3 (h) | 90 | NA | NA |
| IRK (h) | 74 | NA | NA |
| Others | <50 | NA | NA |
| Ion channels | |||
| Manual patch-clamp | |||
| RA306 IC50 (µM) | KN93 IC50 (µM) | IC50 ratiob | |
| ERG (h) | >30 | 0.08 | >375 |
| Kv4.3 (h) | >30 | 4 | >7.5 |
| Nav1.5 (h) | >30 | 2.7 | >11 |
| Cav1.2 (h) | 14 | 0.9 | 15 |
| Eurofins panel (304 kinases) | |||
| P33 assays | |||
| IC50 (nM) | IC50 ratioa | ATP (µM) | |
| CaMKIIγ (h) | <3 | ∼1 | 10 |
| CaMKIIδ (h) | <3 | 1 | 10 |
| CaMKIδ (h) | 12 | >4 | 15 |
| CaMKIIβ (h) | 17 | >6 | 15 |
| ACK1 (h) | 26 | >9 | 70 |
| MLK1 (h) | 89 | >30 | 45 |
| SIK (h) | 149 | >50 | 45 |
| PTK5 (h) | 216 | >72 | 155 |
| Pyk2 (h) | 249 | >83 | 90 |
| IRE1 (h) | 254 | >85 | 15 |
| ULK2 (h) | 255 | >85 | 15 |
| CaMKK2 (h) | 294 | >98 | 90 |
| Others | >300 | >100 | Km |
| CEREP panel (107 targets) | |||
| Binding, enzyme, and uptake assays | |||
| I%–10 µM | I%–1 µM | IC50 (nM) | |
| CaMK2α (h) | 101 | NA | NA |
| Sigma (1/2) (h) | 96 | 73 | NA |
| 15-Lipoxygenase-2 (h) | 96 | 73 | 270 |
| M3 (h) | 90 | NA | NA |
| IRK (h) | 74 | NA | NA |
| Others | <50 | NA | NA |
| Ion channels | |||
| Manual patch-clamp | |||
| RA306 IC50 (µM) | KN93 IC50 (µM) | IC50 ratiob | |
| ERG (h) | >30 | 0.08 | >375 |
| Kv4.3 (h) | >30 | 4 | >7.5 |
| Nav1.5 (h) | >30 | 2.7 | >11 |
| Cav1.2 (h) | 14 | 0.9 | 15 |
NA, not available.
Kinase IC50/CaMKIIδ IC50.
RA306 IC50/KN93 IC50.
RA306 selectivity was further assessed toward a CEREP panel of 107 pharmacological targets including GPCRs, enzymes, transporters, and ion channels. RA306 was found active with an inhibition above 50% at 10 µM on only five of these targets including CaMKII alpha (other CaMKII isoforms not tested) (Table 1). Despite a 90% inhibition at 10 µM in the M3 GPCR binding assay, RA306 was found inactive when tested in CEREP M3 functional assays (not shown). Full dose-response curve was performed on the 15-lipoxgenase-2 assay allowing determination of an IC50 of 270 nM for RA306.
RA306 activity was also tested by patch clamp on four key cardiac ion channels (Table 1). As compared to KN93 that inhibited the four channels with IC50 from 80 nM to 4 µM, RA306 only modestly inhibited Cav1.2 with an IC50 of 14 µM (vs. 0.9 µM for KN93).
All these results demonstrate a good selectivity profile of RA306 vs. kinases and non-kinase targets.
3.3 RA306 cellular activity in human and rodent cardiomyocytes
The phosphorylation site of PLN (Thr17)31,32 was previously shown to be specifically phosphorylated by CaMKII in myocardium after beta-adrenergic stimulation.20,33 We used Thr17-PLN phosphorylation as direct marker of CaMKII activity to assess the ability of RA306 to inhibit CaMKII in human iPSC-derived cardiomyocytes (Cor4U®) and isolated cardiomyocytes from adult mice and rats. After 15 min of stimulation with the beta-adrenergic agonist isoproterenol, markedly increased Thr17-PLN phosphorylation was detected by immunoblotting with a phospho-Thr17-PLN specific antibody (Figure 2A). Isoproterenol-induced phosphorylation of Thr17-PLN, normalized to total PLN, was fully inhibited by RA306 in a dose-dependent manner, with IC50s of 140 nM in rat, 205 nM in mouse, and 220 nM in human cardiomyocytes (Figure 2A, B, and Supplementary material online, Figure S1).
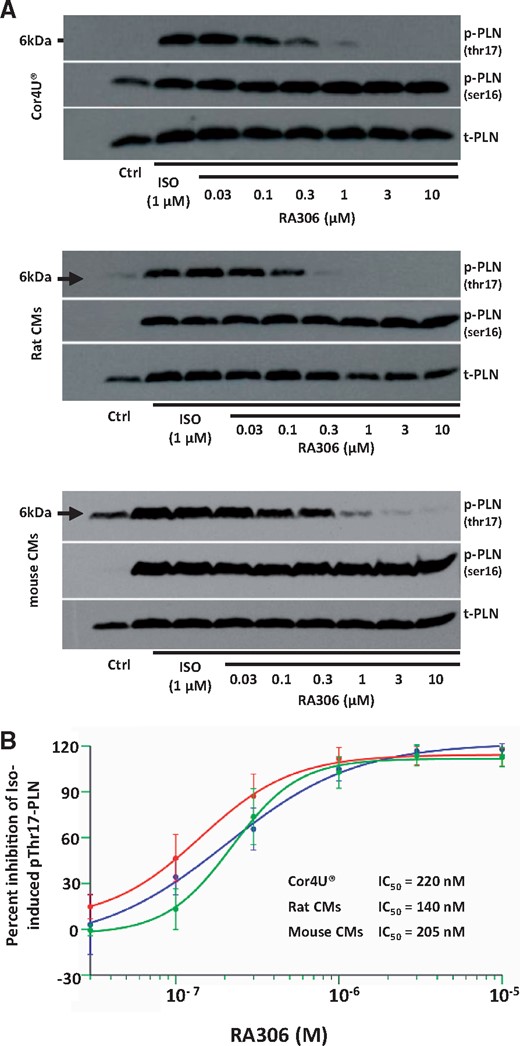
Effect of RA306 on Thr17-PLN phosphorylation in cardiomyocytes. (A) Representative western blots obtained in human iPS-derived cardiomyocytes (Cor4U®, upper panel), adult rat cardiomyocytes (middle panel), and mouse cardiomyocytes (lower panel). (B) Concentration-response curves of RA306 obtained after densitometric integration of each band followed by normalization to total-PLN. Results are expressed as percent inhibition of isoproterenol-induced Thr17-PLN and Ser16-PLN phosphorylation. IC50 were estimated with SAS 9.2 using a four-parameter logistic model. N = 5 experiments in Cor4U®, rat cardiomyocytes, and mouse cardiomyocytes.
Isoproterenol-induced phosphorylation of Ser16-PLN, used as PKA control, was not inhibited by RA306, but even significantly increased, as shown for Cor4U® in Supplementary material online, Figure S2. The unrelated CaMKII inhibitor KN-93 also significantly increased iso-induced Ser16-PLN phosphorylation in Cor4U® at a concentration fully inhibiting Thr17-PLN phosphorylation (Supplementary material online, Figure S3). This suggests that this activator effect on Ser16-PLN phosphorylation is linked to CaMKII inhibition rather than to a direct activation of PKA by RA306, which is also consistent with the lack of RA306 activity on recombinant PKA (1% inhibition at 1 µM in Eurofins panel).
In complement to the classical mechanism of CaMKII activation by Ca2+/CaM and autophosphorylation, other calcium-independent mechanisms of CaMKII activation by oxidation and GlcNAcylation have been described.16,34 In agreement with its ATP competitive mechanism of action, RA306 fully inhibited Thr17-PLN phosphorylation induced by oxidation (H2O2) in Cor4U® when tested at 3 and 10 µM (Supplementary material online, Figure S4).
3.4 Pharmacokinetic properties in mice
Oral bioavailability of RA306 was first evaluated in mice at 3 mg/kg intravenous (iv) and 10 mg/kg per os (po) (Figure 3). Following iv administration at 3 mg/kg, RA306 displayed a low clearance (0.49 L/h/kg), a large volume of distribution at steady state (3.3 L/kg), and a long-terminal elimination half-life (t1/2z) close to 5 h. Following oral administration at 10 mg/kg, maximal RA306 concentrations of 2.9 µg/mL and 3.7 µg/mL (∼7 µM) were reached at 2 h and 1 h post-dosing in plasma and heart, with long-terminal elimination half-life of 4.3 and 5.6 h, respectively and bioavailability close to 100%.
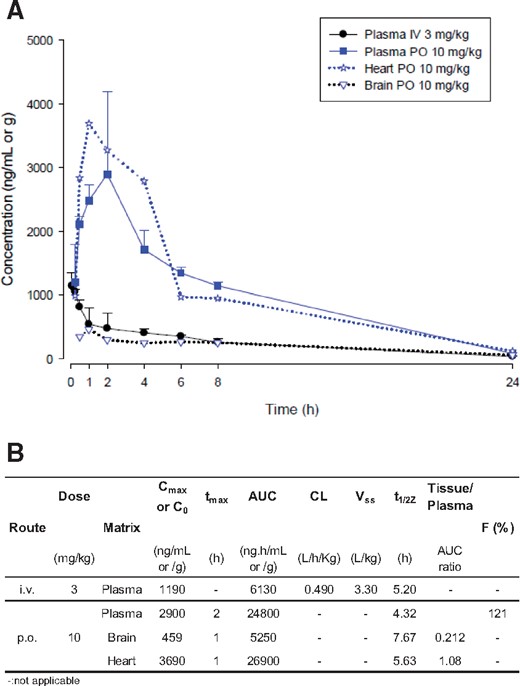
Mouse PK profile of RA306. (A) RA306 time course concentration after IV administration at 3 mg/kg in plasma (dark circle) or after per os administration at 10 mg/kg in plasma (blue square), heart (blue star), and brain (blue triangle). n = 3 male C57BL/6 mice per time point. (B) Determination of PK parameters of RA306 in male C57BL/6 mice after IV (3mg/kg) and PO administration (10 mg/kg) using a non-compartmental model and WinNonLin software (Phoenix, version 6.4).
3.5 RA306 inhibits CaMKII at the heart level following oral delivery
Based on these desirable pharmacokinetic properties, RA306 was evaluated for its ability to inhibit isoproterenol-induced Thr17-PLN phosphorylation in the heart following oral delivery at 10 and 30 mg/kg in mice. The robust increase in phospho-Thr17-PLN/Total PLN ratio induced by isoproterenol was prevented by oral treatment of mice with RA306 at a dose of 10 or 30 mg/kg, with almost complete inhibition at early time points post-treatment and over 50% inhibition maintained up to 4 or 16 h, respectively (Figure 4A and B). Parallel measurement of RA306 concentrations in heart samples (Figure 4C) indicated that a minimal cardiac concentration of ∼2.5 µg/g (∼5 µM) was necessary to inhibit isoproterenol-induced phosphorylation of Thr17-PLN by more than 50% (Figure 4D).
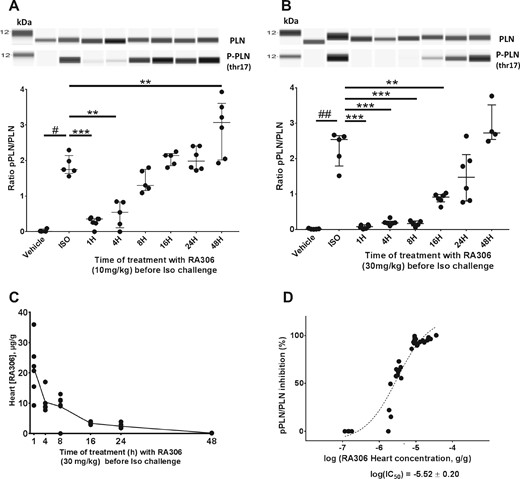
Cardiac pharmacodynamics effects of RA306 following oral delivery. Quantification of pThr17/Total PLN ratio by simple western on heart lysates from control mice, mice challenged by iv isoproterenol injection, or mice treated by 10 mg/kg (A) or 30 mg/kg (B) of RA306 administered orally at different time points before isoproterenol challenge. Representative lane views of simple western and quantification of pThr17/Total-PLN ratio with individual values (n = 4–7 per group), medians and interquartile ranges are shown. #P < 0.05; ##P<0.01: P-value obtained using a Wilcoxon test. **P < 0.01; ***P < 0.001: P-value obtained using the post hoc Wilcoxon analyses with Bonferroni–Holm adjustment for multiplicity after a Kruskall–Wallis test. (C) Cardiac concentrations of RA306 in mice corresponding to B. (D) Dose-response curve between cardiac concentration of RA306 and percentage inhibition of pThr17/total-PLN ratio. Full concentration-response curve was fitted using a logistic equation and IC50 estimated with SAS 9.2.
As observed in cardiomyocytes, RA306 significantly increased iso-induced Ser16-PLN phosphorylation following oral administration at 30 mg/kg (Supplementary material online, Figure S5). In order to fully exclude the possibility that RA306 could directly activate PKA at pharmacologically active dose, we analysed the level of VASP phosphorylation at the Ser157 PKA specific site35 in the same samples. As shown in Supplementary material online, Figure S5, RA306 had no significant effect on iso-induced Ser157-VASP phosphorylation, further suggesting that the increase of Ser16-PLN phosphorylation by RA306 is not due to a direct activation of PKA.
3.6 Disease modifying effects of RA306 in a genetic model of DCM
CaMKII was previously shown to be up-regulated in a mouse genetic model of systolic HF due to an alpha-actin mutation responsible for DCM in humans.24,36 KN-93 was demonstrated to prevent the decrease in left ventricular ejection fraction in this model when administered from 2 to 5 months of age.24 We tested RA306 in the same model but used older mice (8–10 months) with an advanced disease state to evaluate the curative effects of the compound.
RA306 was first orally administered to alpha-actin TG mice for 2 weeks at a dose of 30 mg/kg twice a day. This dosing regimen was chosen to maintain a sustained >50% inhibition of cardiac CaMKII based on PD results described in Figure 4. At baseline, alpha-actin TG mice from vehicle- and RA306-treated groups exhibited a significant reduction (P < 0.01) in ejection fraction (38.6 ± 1.64 and 34.2 ± 2.59 vs. 50.8 ± 1.47, respectively) and cardiac output (13.3 ± 0.99 and 13.2 ± 0.65 vs. 17.5 ± 0.96, respectively) as compared to age-matched wild type (WT) control mice. In TG mice, RA306 treatment for 2 weeks resulted in a significant increase (P < 0.05) in ejection fraction changes from baseline (EF) (from 34.2% at baseline to 44.8%) and cardiac output changes from baseline (from 13.22 ± 0.65 at baseline to 15.60 ± 0.84 mL/min) compared with the vehicle-treated TG group with no notable effect on heart rate. Neither EF nor CO were changed in the vehicle group (Figure 5A, B, and Supplementary material online, Figure S6). This effect of RA306 was associated with a significant inhibition of cardiac Thr17-PLN phosphorylation (Figure 5C), with no significant increase of Ser16-PLN phosphorylation by RA306 (Supplementary material online, Figure S7) as opposed to what was observed in the acute pharmacodynamics model.
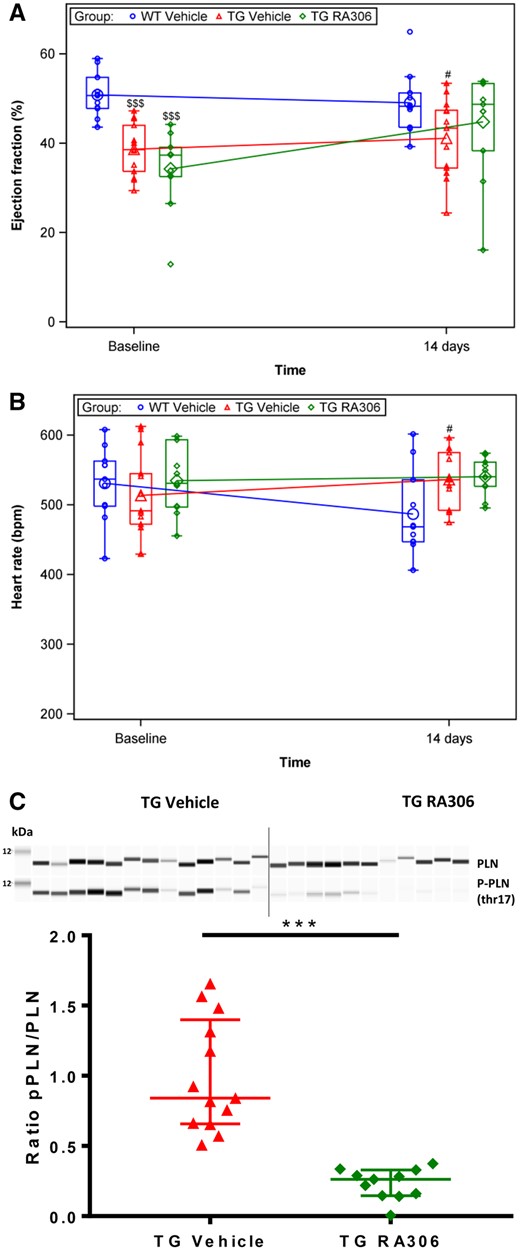
RA306 activity in alpha-actin TG mice (30 mg/kg twice a day, 2 weeks). Assessment of ejection fraction (A) and heart rate (B) by echocardiography performed in anaesthetized C57BL6 mice or alpha-actin TG mice before and after treatment with RA306 given by oral gavage at 30 mg/kg twice a day or its corresponding vehicle (Captisol 10% in water) for 14 days. Blue circle: vehicle administered to WT mice (n = 11), red triangle: vehicle administered to TG mice (n = 13), green diamond: RA306 administered to TG mice (n = 11). (C) Quantification of pThr17/total-PLN by Sally Sue technology in freeze-clamped heart samples from TG mice collected on day 14. Red triangle: vehicle-treated mice (n = 13), green diamond: RA306 treated mice (n=11). $$$P < 0.001: P-value to compare WT vs. TG groups at baseline were obtained using an ANOVA. #P < 0.05: P-value to compare WT vs. TG-vehicle group post-treatment was obtained using an ANOVA. ***P < 0.001: P-value to compare TG-vehicle to TG-RA306 groups post-treatment was obtained using an ANOVA (A and B) or Wilcoxon test (C). Individual values (small symbols), means (large symbols), and medians (Q1–Q3) (boxes) are reported in A and B. Individual values and medians (Q1–Q3) are shown in C.
Based on these results, a second study with a reduced dosing regimen (RA306 at 30 mg/kg/po, once a day) administered for 2 months was performed. As described in the first study, EF and CO were significantly reduced in alpha-actin TG mice as compared to WT control mice. After 2 weeks and 2 months treatment EF changes from baseline and CO changes from baseline were significantly increased (P < 0.05) in RA306-treated TG mice (Figure 6A and Supplementary material online, Figure S8) compared with vehicle-treated TG animals. These curative effects of RA306 were not associated with any modification of heart rate (Figure 6B) or LV internal diameter (Figure 6C).
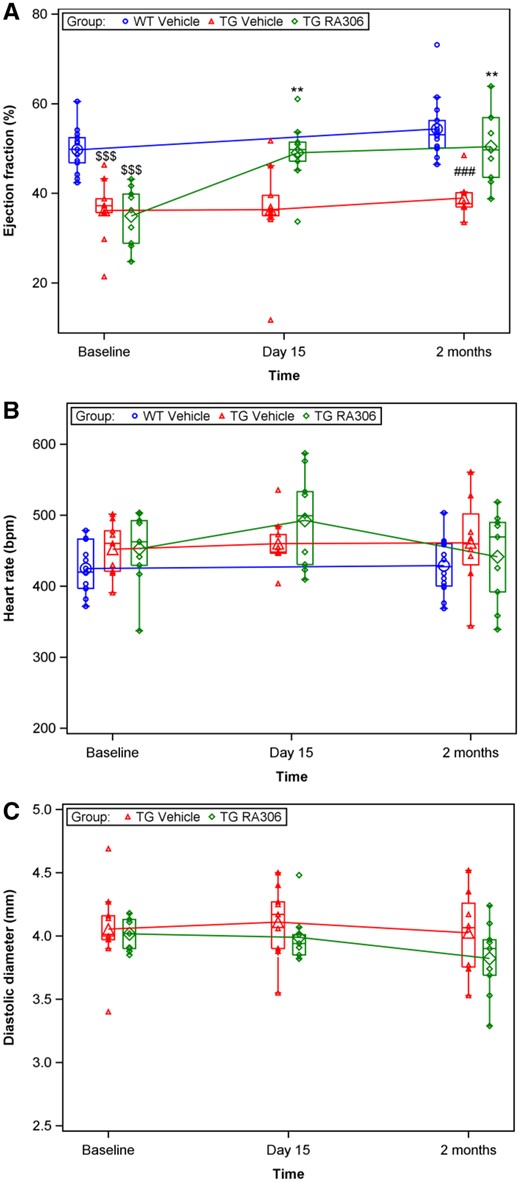
RA306 activity in alpha-actin TG mice (30 mg/kg once a day, 2 months). Assessment of ejection fraction (A), heart rate (B), and left ventricular diameter (C) by echocardiography performed in anaesthetized C57BL6 mice or alpha-actin TG mice before and after treatment with RA306 given by oral gavage at 30 mg/kg once a day or its corresponding vehicle (Captisol 10% in water) for 2 months. Blue circle: vehicle administered to WT mice (n = 15), red triangle: vehicle administered to TG mice (n = 9), and green diamond: RA306 administered to TG mice (n = 9). $$$P < 0.001: P-values to compare WT vs. TG-vehicle groups at baseline were obtained using an ANOVA. ###P < 0.001: P-value to compare WT vs. TG-vehicle groups post-treatment was obtained using an ANOVA. **P < 0.01: P-value to compare TG-vehicle to TG-RA306 groups post-treatment was obtained using an ANOVA. Individual values (small symbols), means (large symbols), and medians (Q1–Q3) (boxes) are depicted.
At terminal microscopic observation, minimal to mild focal/multifocal fibrosis of the myocardium was observed in alpha-actin TG mice treated with vehicle or RA306 (Supplementary material online, Figure S9). This was correlated with higher mean relative heart weight to body weight ratio in alpha-actin TG mice compared to WT, with no effect of RA306 on this parameter (Supplementary material online, Figure S10). No heart findings were observed in WT mice.
There was no significant myocardial inflammation (absence of inflammatory cell infiltrate readily visible on hemalun and eosin-stained sections) nor apoptotic cardiomyocytes (identified by their condensed and fragmented nuclei) in the heart of these animals at a microscopic level (Supplementary material online, Figure S9).
4. Discussion
The present report describes a novel, potent, ATP-competitive CaMKII inhibitor displaying a good selectivity profile, robust ability to inhibit CaMKII in myocardium following oral delivery, and in vivo efficacy demonstrated by disease modifying effects in alpha-actin TG mice.
RA306 is a potent inhibitor of human CaMKII delta and gamma, the two main cardiac CaMKII isoforms, with IC50s in the 10 nM range in the CALIPER assay and below 3 nM in the more sensitive P33 assay from Eurofins. This dual inhibition of CaMKII delta and gamma, exerting redundant pathological effects at the cardiac level, is expected to be beneficial in the context of HF.28 RA306 furthermore displays an ATP competitive mode of action, as illustrated by a shift in IC50 at 10× Km ATP as compared to 1× Km ATP, and as further supported by co-crystallization of a close analogue of equivalent potency, RA608, within the ATP binding site of human CaMKII delta (not shown). Such ATP-competitive mechanism of action, directly inhibiting the catalytic site of the enzyme, offers a significant mechanistic advantage over allosteric inhibitors like KN-93 that inhibit the initial activation step of CaMKII by Ca2+/CaM but are inactive on CaMKII once already activated.37
Inhibition of CaMKII alpha and beta isoforms at the brain level is expected to be potentially deleterious based on their role in synaptic plasticity and memory.5,6 However, the risk of affecting CAMKII beta activity in the brain is low with RA306. First, because RA306 displays an acceptable level of selectivity towards CaMKII beta (∼30 fold) thanks to one amino acid difference between the ATP binding site of CaMKII beta and delta isoforms, and second because its exposure is five-fold lower in the brain as compared to the heart in mice (Figure 3). Regarding the other isoform, CAMKII alpha, although no selectivity was achieved towards this isoform, the brain consequences should be limited again due to a limited central exposure. Therefore, altogether the risk of CaMKII-related CNS side effects is more likely limited with RA306 at active cardiac concentrations.
The shift of RA306 potency between hCaMKII delta recombinant assays and cardiomyocyte assays was expected for an ATP-competitive inhibitor due to higher intracellular ATP concentrations (mM range) as compared to the one used in recombinant assays (10 µM). RA306 nevertheless still displays a good cellular activity in cardiomyocytes from human, rat, and mice, with IC50s in the 200 nM range. Such cross-species inhibitory activity was anticipated based on the very high conservation of the CaMKII catalytic site (100% amino acid identity between human, rat, and mice for CaMKII delta).
A pharmacokinetics/pharmacodynamics relationship was established for RA306 based on the inhibition of Thr17-PLN phosphorylation in the heart. Oral bioavailability of RA306, evaluated in mice, was associated with efficient inhibition of Thr17-PLN phosphorylation in the heart of diseased alpha-actin TG mice and the heart of WT mice challenged by isoproterenol. In vitro phosphorylation experiments coupled to site-directed mutagenesis previously showed that phosphorylation of amino acid Thr17 of PLN is specific to CaMKII.31,32 Phosphorylation of this site following isoproterenol-challenge was shown to be completely blunted in AC3-I TG mice that expressed a pseudo-substrate peptide inhibitor of CaMKII in cardiomyocytes.20 Therefore, inhibition of cardiac Thr17-PLN phosphorylation by RA306 can be considered to be a direct and specific marker of CaMKII inhibition. The cardiac concentration of RA306 (∼5 µM) required to achieve 50% inhibition of Thr17-PLN phosphorylation in isoproterenol-challenged WT mice was relatively high compared to cellular activity in cardiomyocytes. This could be due potentially to a high plasma protein binding (97 ± 0.2% and 96 ± 0.3% in mouse and human plasma respectively, not shown), or to activation of CaMKII at supra-physiological levels in response to isoproterenol-challenge.
RA306 increased iso-induced Ser16-PLN phosphorylation in cardiomyocytes and in the acute pharmacodynamics model. This effect is unlikely due to a direct activation of PKA since KN-93 displayed a similar effect in cardiomyocytes and VASP phosphorylation at the Ser157 PKA site was not increased by RA306 in the pharmacodynamics model. This might rather be due to a steric competition between CaMKII and PKA for their two adjacent phosphorylation sites, thus increasing the phosphorylation of PLN by PKA when CaMKII is inhibited. A similar effect was not observed in alpha-actin TG mice treated for 2 weeks by RA306 at 30 mg/kg, which could potentially be due to adaptive mechanisms decreasing the level of Ser16-PLN phosphorylation back to normal levels following a chronic inhibition of CaMKII by RA306. A lower level of PKA activity in alpha-actin TG mice as compared to the one induced by isoproterenol challenge could also minimize the effect of CaMKII inhibition on Ser16-PLN phosphorylation.
Although CaMKII inhibition is presumably the main driver of the beneficial effects of RA306 on cardiac function, it is possible that there are other potential contributing mechanisms. A limited number of protein kinases were inhibited by RA306 in the Eurofins panel, each exhibiting an IC50 ratio below 100 when compared with CaMKII delta. Amongst those protein kinases, MLK1 and SIK have been linked to cardiomyocyte hypertrophy in vitro,38,39 and TG inhibition of Pyk2 was shown to slow down pathological LV remodelling and HF progression in a mouse model of DCM due to cardiac expression of a constitutively active form of PKCε.40 Although inhibition of these kinases by RA306 is unlikely to occur in the heart at the pharmacological doses used, one cannot fully exclude that it could potentially contribute to the beneficial effect of RA306. In addition, two non-kinase targets (Sigma (1/2), 15-Lipoxygenase-2) were inhibited by RA306 in the CEREP panel but since their inhibition have not been reported to be associated with positive cardiac effects, it is unlikely that they could contribute to the therapeutic effects of RA306.
CaMKII is involved in HF progression and cardiac arrhythmia through pleiotropic mechanisms including cardiac fibrosis, hypertrophy, apoptosis, inflammation, and abnormal calcium handling.7,8,21,41 Prevention of HF progression by KN-93 in mutant alpha-actin TG mice was previously shown to be associated with a reduction of cardiomyocyte apoptosis after 3 months of chronic treatment.24 In our in vivo study, performed in mutant alpha-actin TG mice presenting an advanced disease state, no signs of apoptosis or inflammation were evidenced between WT and TG animals treated or not with RA306; therefore, the curative effects observed with RA306 on cardiac function following a 2-month treatment cannot be attributed to the inhibition of cardiac inflammation or cardiomyocyte apoptosis. The same conclusion applies for LV remodelling (cardiac hypertrophy and fibrosis) which was not affected by a 2-month treatment with RA306. Normalization of altered Ca2+ handling, a key pathological feature of HF,42,43 could be a plausible mechanism by which RA306 exerts its disease modifying effects following short- and long-term treatment. This hypothesis is supported by recent studies showing that RA608 (=RA123456), an analogue of RA306, potently inhibited RyR2-mediated SR Ca2+ sparks in diseased atrial myocytes from coronary artery disease patients with a subsequent increase in SR Ca2+ contents and post-rest contractile function.44
There is limited evidence supporting a beneficial effect of CaMKII inhibition in curative models of HF. Conditional knock-out of CaMKII delta and gamma induced in a HF mouse model with low ejection fraction did not improve cardiac function but prevented its further deterioration.23 Such disease stabilizing effects were reproduced with RA608 in the same HF mouse model.45 In contrast, RA306 caused a marked improvement in cardiac function in mutant alpha-actin TG mice. Whether these differences are model- (TAC vs. mutant alpha-actin TG mice), disease state- (early vs. late), or compound-related (RA608 vs. RA306) remains to be determined.
The present work characterized RA306 as the lead compound of a new class of potent orally active ATP-competitive CaMKII inhibitors. Other ATP-competitive CaMKII inhibitors AS105, GS-680, and SMP-114 inhibiting SR Ca2+ leaks in human-diseased cardiomyocytes were recently described,46–48 but no oral bioavailability were reported for AS105 and GS-680. In addition, SMP-114 suffers from a modest inhibitory potency in comparison with RA306 (i.e. IC50 = 30 µM for SMP-114 vs. 25 nM for RA306 on CaMKII gamma). Finally, RA306 displays a much better ion channel selectivity profile as compared to the KN93 allosteric inhibitor routinely used as a pharmacological tool in pre-clinical studies.
The identification of RA306 supports the feasibility of developing potent, orally available CaMKII inhibitors suitable for clinical use. Together with RA306 disease-modifying effects in a translational HF model linked to a sarcomeric mutation responsible of DCM in humans, these findings suggest that CaMKII inhibition may be beneficial for the treatment of heart diseases.
Time for primary review: 52 days
Acknowledgements
The authors acknowledge expert technical assistance of Alice Raffenne-Devillers, Maud Belot, Florence Coignée, Florence Gillot, Thibault Mahiet, Fatima Zikri, and Carine Bernard.
Conflict of interest: Authors are employed by Sanofi, a global biopharmaceutical company focused on human health.
Funding
This work was supported by the Sanofi R&D.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}