



Abstract
Aims
Thoracic aortic dissection (TAD) is a highly fatal disease lacking effective pharmacologic interventions in clinical practice. Emerging evidence indicates that the natriuretic peptide receptor C (NPR-C) plays a crucial role in the regulation of cardiovascular diseases. However, the precise involvement of NPR-C in TAD remains elusive. In this study, the role and molecular mechanisms of NPR-C in the pathogenesis of TAD were investigated.
Methods and Results
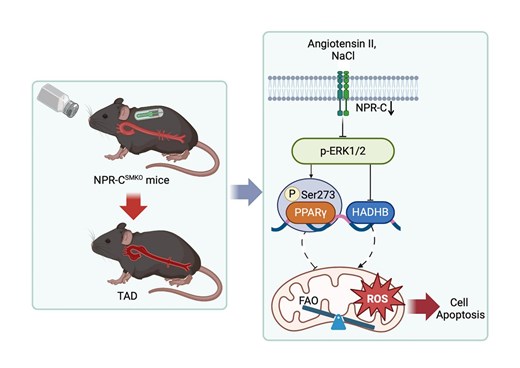
Through integrated analyses of human TAD transcriptome and single-cell sequencing data sets, we identified that NPR-C was downregulated in the aortas of acute TAD patients and in beta-aminopropionitrile (BAPN)-treated mice. Intriguingly, vascular smooth muscle cell (VSMC)-specific NPR-C knockout (NPR-CSMKO) mice, rather than endothelial cell-specific NPR-C knockout mice, developed TAD after treated with angiotensin II (Ang II) plus high salt diet (HSD), but not Ang II alone. Loss of NPR-C function promoted extracellular matrix degeneration, VSMCs apoptosis and inflammation. RNA-sequencing analysis revealed that mitochondrial fatty acid oxidation (FAO) genes were significantly downregulated in the thoracic aortas of NPR-CSMKO mice treated with Ang II plus HSD. Notably, the expression of HADHB, a subunit of mitochondrial trifunctional protein (MTP) responsible for FAO, was obviously decreased in NPR-CSMKO mice treated with Ang II plus HSD. Mechanistically, knockdown of NPR-C activated ERK1/2 pathway, which decreased the expression and activity of peroxisome proliferator-activated receptor γ (PPARγ) and inhibited HADHB expression. Furthermore, NPR-C agonist, C-ANP4-23, mitigated the progression of TAD in BAPN-treated mice. Activation of MTP by spermidine (SPD) effectively prevented TAD formation in NPR-CSMKO mice treated with Ang II plus HSD.
Conclusions
Our data highlight a critical role of HSD in triggering TAD and a previously unrecognized role of NPR-C that protects against TAD through regulating mitochondrial homeostasis. Therefore, NPR-C activation and SPD supplementation could be new prevention and treatment strategies for TAD.
 Accepted manuscripts
Accepted manuscripts


{kind=link}