





Abstract
Smokers are at increased risk of cardiovascular events. However, the exact mechanisms through which smoking influences cardiovascular disease resulting in accelerated atherosclerosis and vascular calcification are unknown. The aim of this study was to investigate effects of nicotine on initiation of vascular smooth muscle cell (VSMC) calcification and to elucidate underlying mechanisms.
We assessed vascular calcification of 62 carotid lesions of both smoking and non-smoking patients using ex vivo micro-computed tomography (µCT) scanning. Calcification was present more often in carotid plaques of smokers (n = 22 of 30, 73.3%) compared to non-smokers (n = 11 of 32, 34.3%; P < 0.001), confirming higher atherosclerotic burden. The difference was particularly profound for microcalcifications, which was 17-fold higher in smokers compared to non-smokers. In vitro, nicotine-induced human primary VSMC calcification, and increased osteogenic gene expression (Runx2, Osx, BSP, and OPN) and extracellular vesicle (EV) secretion. The pro-calcifying effects of nicotine were mediated by Ca2+-dependent Nox5. SiRNA knock-down of Nox5 inhibited nicotine-induced EV release and calcification. Moreover, pre-treatment of hVSMCs with vitamin K2 ameliorated nicotine-induced intracellular oxidative stress, EV secretion, and calcification. Using nicotinic acetylcholine receptor (nAChR) blockers α-bungarotoxin and hexamethonium bromide, we found that the effects of nicotine on intracellular Ca2+ and oxidative stress were mediated by α7 and α3 nAChR. Finally, we showed that Nox5 expression was higher in carotid arteries of smokers and correlated with calcification levels in these vessels.
In this study, we provide evidence that nicotine induces Nox5-mediated pro-calcific processes as novel mechanism of increased atherosclerotic calcification. We identified that activation of α7 and α3 nAChR by nicotine increases intracellular Ca2+ and initiates calcification of hVSMCs through increased Nox5 activity, leading to oxidative stress-mediated EV release. Identifying the role of Nox5-induced oxidative stress opens novel avenues for diagnosis and treatment of smoking-induced cardiovascular disease.
Translational perspective
Our findings reveal that smoking causes vascular calcification through VSMC Nox-5-mediated pro-calcific processes. We elucidate the molecular effects of nicotine on VSMC phenotype and provide novel insights into mechanisms of nicotine-induced vascular calcification. Nicotine binding to the nAChR results in increased intracellular calcium and secretion of extracellular vesicles, providing potential targets for pharmacological intervention. The use of vitamin K2, besides being a cofactor for the carboxylation of matrix Gla-protein, is an effective antioxidant ameliorating nicotine-induced oxidative stress and extracellular vesicle release, and has the potential to open new avenues for the treatment of vascular disease.
1. Introduction
Cigarette smoking is a major cause of coronary heart disease and cardiovascular diseases (CVDs). According to the 2014 Surgeon General’s Report on smoking and health, smoking is responsible for approximately one of every four deaths from CVD.1 Smoking has been shown to be associated with increased cardiovascular morbidity and mortality in a dose-dependent manner.2,3 Because of its detrimental effect, smoking remains the most important modifiable risk factor for CVD outcome.4
Vascular calcification is a strong and independent risk factor of CVD and used as a parameter to detect atherosclerotic burden in patients.5 Vascular calcification is the deposition of hydroxyapatite crystals in the vasculature which can be categorized by size.6 Microcalcification is an active dynamic process caused by vesicle-mediated mineralization, apoptotic bodies, and osteogenic differentiation.7,8 It precedes macrocalcification and is considered detrimental.9 Vascular calcification is commonly observed in atherosclerosis, aortic aneurysm, chronic kidney disease, and diabetes.10,11 Arterial intimal calcification is associated with arterial obstruction and atherosclerotic plaque rupture, whereas arterial medial calcification is associated with arterial stiffness, systolic hypertension, and aortic aneurysm.12 Vascular calcification is an active process driven by vascular smooth muscle cells (VSMCs), which are the major cell type in the vessel wall and are located in the tunica media.13 Vascular calcification arises from differentiation of VSMC, VSMC apoptosis, and mineralization of extracellular vesicles (EVs).12,14–16 Lineage-tracing studies reveal that VSMCs undergo phenotypic switching and give rise to a variety of cells, including osteogenic-like cells, which contribute to vascular calcification.17,18 Thus, VSMCs are well recognized as key player in atherogenesis and atherosclerotic plaque calcification.19
Cigarette smoking is strongly associated with (sub-)clinical atherosclerosis including coronary artery, peripheral artery, and aortic calcification.20–23 A growing body of evidence suggests that nicotine, a major constituent of cigarette smoke,24accounts for impaired vascular function and vascular calcification.25 In this study, we aimed to unravel the mechanistic link between nicotine- and VSMC-induced vascular calcification. We investigate the effect of smoking on calcification in human carotid plaques and explore the effect of nicotine on VSMC processes that lead to vascular calcification in vitro.
2. Methods
2.1 Quantification of microcalcifications in patients
Human carotid artery lesion specimen from 30 patients, previously collected during autopsies at the University Hospital Maastricht, Maastricht, the Netherlands, via the Maastricht Pathology Tissue Collection, was used.26 The collection, storage, and processing of tissue and patient data are in accordance with the ‘Code for Proper Use of Secondary Human Tissue in the Netherlands’ and conform to the principles outlined in the Declaration of Helsinki.26
Micro-computed tomography (µCT) images were captured using a µCT scanner (Tomoscope DUO, CT-Imaging, Erlangen, Germany). Sources of the µCT were operated with voltage of 65 kV and current of 0.5 mA. About 720 projections with 1032 x 1012 pixels were acquired during one revolution with duration of 90 seconds. Volumetric images were reconstructed using a Feldkamp Reconstruction at voxel size 35 µm using a medium sharp reconstruction kernel (T60). Volumetric images were visualized and analysed using the Imalytics Preclinical Software.27 Calcifications were interactively segmented and counted using a fixed threshold (100 HU) and connected component analysis.27 In order to show differences in calcification according to calcification size, segmented components were grouped into small, medium, and large classes based on their volumes (< 0.01 mm³, < 0.1 mm³, and > 0.1 mm³). Due to the lack of previous studies using this design and set-up, these cut-offs were chosen based on the scanners resolution, specimen size, and distribution of calcification size. Volumes were determined based on the segmentation size, that is proportional to the number of segmented voxels. Calcification amount per voxel was assumed to be proportional to the voxel intensity, and the amount of calcification per plaque was computed as the sum of the corresponding voxels.
2.2 VSMC culture and treatments
Human primary VSMCs were isolated from a non-diseased region of aortic tissue obtained from patients undergoing aortic aneurysm surgical repair. Tissue was considered as a waste material from the Vascular Surgery Unit of University Hospital Maastricht, Maastricht, the Netherlands, and University Hospital RWTH Aachen, Aachen, Germany. The collection, storage, and processing of tissue and patient data are in accordance with the ‘Code for Proper Use of Secondary Human Tissue in the Netherlands’, the Ethics Committee of the Faculty of Medicine, University Hospital RWTH Aachen, and the Declaration of Helsinki. VSMCs were cultured in DMEM (Gibco) supplemented with 20% FBS (Gibco) and 1% penicillin/streptomycin (PS) (Gibco). VSMCs were passaged when 90% confluent and used for experiments at passage 3–10. VSMCs were stimulated with nicotine (150 nM, 1 µM, 1 mM, 5 mM, 10 mM, Sigma, N3876) or vehicle control (ethanol). Inhibitor treatments were carried out prior to nicotine stimulation: GKT136901 (1 µM, 5 µM, Cayman chemical, 17764), α-bungarotoxin (1 µM, 10 µM, Life Technologies, B1601), N-acetyl-cysteine (0.5 µM, Sigma, A7250), Vitamin K (Menaquinone-7, 10 µM, Nattopharma ASA), Stattic (1 µM, Abcam, ab120952), SU6656-SrcFK inhibitor (2 µM, Sigma, S9692), GSK2656157-pERK inhibitor (2 µM, ApexBio, B2175), and BAPTA-AM (1 µM, Molecular probes #d1207). SiRNA transfection (Nox5: S103243856, Qiagen; p22phox: L-011020-02-0005, Horizon Discovery) was carried out using the Neon Transfection System (Thermo Fisher Scientific) and kit following the manufacturer’s protocol for human aortic smooth muscle cells.
2.3 Statistical analysis
Data are shown as mean ± SD and were obtained in three or more independent experiments. Normality of all data was tested using the Shapiro–Wilk test. If data were not normally distributed, Mann–Whitney and Kruskal–Wallis tests were used; otherwise, statistical significance was tested with t-test, one-sample test, and one-way ANOVA with Bonferroni post hoc for experiments with two and more groups, respectively. Statistical analysis was carried out using GraphPad Prism 8.2.0. All clinical data were analysed on a per lesion basis, and the statistical test did not account for the correlation of multiple lesions within patients. Clinical data computations were performed using SPSS (IBM Corp., Armonk, NY, USA). The exact test used for each data set is mentioned in figure legends. * denotes P < 0.05, ** P < 0.01, *** P < 0.001.
Extended methods are provided in Supplementary material online.
3. Results
3.1 Smoking increases the incidence of calcification in human atherosclerotic plaques
Smoking has been shown to correlate with higher incidence of vascular calcification.28 We assessed calcifications in 30 carotid artery atherosclerotic plaques of 15 smokers and compared to 32 plaques of 15 non-smokers using µCT scanning (Figure 1A and B). We divided the observed calcifications into three groups based on size and quantified their number and volume. Results show that, overall, calcification was present in 73.3% smokers and 34.4% non-smokers (P = 0.002, Table 1). Smoking was associated with a significantly higher prevalence of plaque calcifications of all size categories (microcalcifications were present in 21 lesions (70.0%) of smokers vs. eight lesions (25.0%) of non-smokers, P < 0.001; intermediate calcifications in 20 lesions (77.7%) vs. seven lesions (21.9%), P < 0.001; macrocalcifications in 18 lesions (60.0%) vs. four lesions (12.5%), P < 0.001; Table 1). Additionally, smokers had significantly more calcifications of all sizes (Figure 1C–E). The total volume of microcalcifications and intermediate calcifications, but not macrocalcifications, was also higher in smokers than in non-smokers (Figure 1F–H). These results suggest that smoking predisposes the formation of microcalcifications as well as calcification progression resulting in increased numbers of intermediate calcification and macrocalcification.
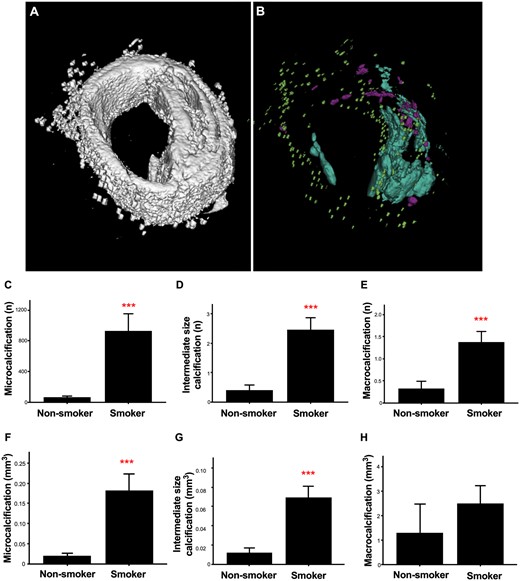
Smoking increases the incidence and amount of calcification in human atherosclerotic plaques. (A) Representative µCT image of carotid plaque from a non-smoker. Carotid plaque including soft tissue and (B) the same plaque without soft tissue displaying compartmentation of microcalcification in green, intermediate size calcification in magenta, and macrocalcification in turquoise. (C) Number of microcalcifications, (D) intermediate size calcifications, and (E) macrocalcifications in carotid artery atherosclerotic plaques from smokers and non-smokers quantified by µCT. (F) Volume of microcalcifications, (G) intermediate size calcifications, and (H) from the same analysis. Statistical significance was tested using t-test, n = 30 plaques for smokers, n = 32 plaques for non-smokers. All graphs show mean and SEM.
Characteristics of patients, whose lesions were used for the study
| Clinical data | Smoking | Non-smoking | P-value |
|---|---|---|---|
| n = 30 | n = 32 | ||
| Age (years) | 74.5 ± 11.5 | 75.2 ± 10.4 | NS |
| Male (n, %) | 14 (46.7) | 17 (53.1) | NS |
| Diabetes (n, %) | 14 (46.7) | 14 (43.8) | NS |
| Glucose (mmol/L) | 8.8 ± 3.6 | 9.1 ± 3.4 | NS |
| CRP (mg/L) | 155.1 ± 149 | 113.4 ± 112.2 | NS |
| Calcification (n, %) | 22 (73.3) | 11 (34.4) | 0.002 |
| Overall NOC (n) | 921.5 ± 1305.5 | 54.3 ± 146.2 | 0.001 |
| Microcalcification | |||
| Present (n, %) | 21 (70.0) | 8 (25.0) | <0.001 |
| NOC (n) | 917.7 ± 1303.7 | 53.7 ± 145.8 | 0.001 |
| Volume (mm2) | 0.180 ± 0.235 | 0.018 ± 0.045 | 0.001 |
| Total calcification (AU) | 207.8 ± 268.7 | 22.5 ± 55.3 | 0.001 |
| Intermediate size calcification | |||
| Present (n, %) | 20 (66.7) | 7 (21.9) | <0.001 |
| NOC (n) | 2.43 ± 2.40 | 0.38 ± 1.10 | <0.001 |
| Volume (mm2) | 0.069 ± 0.068 | 0.011 ± 0.034 | <0.001 |
| Total calcification (AU) | 89.55 ± 91.97 | 15.79 ± 45.59 | <0.001 |
| Macrocalcification | |||
| Present (n, %) | 18 (60.0) | 4 (12.5) | <0.001 |
| NOC (n) | 1.37 ± 1.38 | 0.31 ± 1.00 | 0.001 |
| Volume (mm2) | 2.47 ± 4.07 | 1.28 ± 6.77 | NS |
| Total calcification (AU) | 4610.8 ± 9766.4 | 2360.9 ± 12 654.4 | NS |
| Clinical data | Smoking | Non-smoking | P-value |
|---|---|---|---|
| n = 30 | n = 32 | ||
| Age (years) | 74.5 ± 11.5 | 75.2 ± 10.4 | NS |
| Male (n, %) | 14 (46.7) | 17 (53.1) | NS |
| Diabetes (n, %) | 14 (46.7) | 14 (43.8) | NS |
| Glucose (mmol/L) | 8.8 ± 3.6 | 9.1 ± 3.4 | NS |
| CRP (mg/L) | 155.1 ± 149 | 113.4 ± 112.2 | NS |
| Calcification (n, %) | 22 (73.3) | 11 (34.4) | 0.002 |
| Overall NOC (n) | 921.5 ± 1305.5 | 54.3 ± 146.2 | 0.001 |
| Microcalcification | |||
| Present (n, %) | 21 (70.0) | 8 (25.0) | <0.001 |
| NOC (n) | 917.7 ± 1303.7 | 53.7 ± 145.8 | 0.001 |
| Volume (mm2) | 0.180 ± 0.235 | 0.018 ± 0.045 | 0.001 |
| Total calcification (AU) | 207.8 ± 268.7 | 22.5 ± 55.3 | 0.001 |
| Intermediate size calcification | |||
| Present (n, %) | 20 (66.7) | 7 (21.9) | <0.001 |
| NOC (n) | 2.43 ± 2.40 | 0.38 ± 1.10 | <0.001 |
| Volume (mm2) | 0.069 ± 0.068 | 0.011 ± 0.034 | <0.001 |
| Total calcification (AU) | 89.55 ± 91.97 | 15.79 ± 45.59 | <0.001 |
| Macrocalcification | |||
| Present (n, %) | 18 (60.0) | 4 (12.5) | <0.001 |
| NOC (n) | 1.37 ± 1.38 | 0.31 ± 1.00 | 0.001 |
| Volume (mm2) | 2.47 ± 4.07 | 1.28 ± 6.77 | NS |
| Total calcification (AU) | 4610.8 ± 9766.4 | 2360.9 ± 12 654.4 | NS |
The data are presented as mean ± SD or n (%). Data were analysed on a per lesion basis, and the statistical test did not account for the correlation of multiple lesions within patients.
AU, arbitrary units; NOC, number of calcifications.
Characteristics of patients, whose lesions were used for the study
| Clinical data | Smoking | Non-smoking | P-value |
|---|---|---|---|
| n = 30 | n = 32 | ||
| Age (years) | 74.5 ± 11.5 | 75.2 ± 10.4 | NS |
| Male (n, %) | 14 (46.7) | 17 (53.1) | NS |
| Diabetes (n, %) | 14 (46.7) | 14 (43.8) | NS |
| Glucose (mmol/L) | 8.8 ± 3.6 | 9.1 ± 3.4 | NS |
| CRP (mg/L) | 155.1 ± 149 | 113.4 ± 112.2 | NS |
| Calcification (n, %) | 22 (73.3) | 11 (34.4) | 0.002 |
| Overall NOC (n) | 921.5 ± 1305.5 | 54.3 ± 146.2 | 0.001 |
| Microcalcification | |||
| Present (n, %) | 21 (70.0) | 8 (25.0) | <0.001 |
| NOC (n) | 917.7 ± 1303.7 | 53.7 ± 145.8 | 0.001 |
| Volume (mm2) | 0.180 ± 0.235 | 0.018 ± 0.045 | 0.001 |
| Total calcification (AU) | 207.8 ± 268.7 | 22.5 ± 55.3 | 0.001 |
| Intermediate size calcification | |||
| Present (n, %) | 20 (66.7) | 7 (21.9) | <0.001 |
| NOC (n) | 2.43 ± 2.40 | 0.38 ± 1.10 | <0.001 |
| Volume (mm2) | 0.069 ± 0.068 | 0.011 ± 0.034 | <0.001 |
| Total calcification (AU) | 89.55 ± 91.97 | 15.79 ± 45.59 | <0.001 |
| Macrocalcification | |||
| Present (n, %) | 18 (60.0) | 4 (12.5) | <0.001 |
| NOC (n) | 1.37 ± 1.38 | 0.31 ± 1.00 | 0.001 |
| Volume (mm2) | 2.47 ± 4.07 | 1.28 ± 6.77 | NS |
| Total calcification (AU) | 4610.8 ± 9766.4 | 2360.9 ± 12 654.4 | NS |
| Clinical data | Smoking | Non-smoking | P-value |
|---|---|---|---|
| n = 30 | n = 32 | ||
| Age (years) | 74.5 ± 11.5 | 75.2 ± 10.4 | NS |
| Male (n, %) | 14 (46.7) | 17 (53.1) | NS |
| Diabetes (n, %) | 14 (46.7) | 14 (43.8) | NS |
| Glucose (mmol/L) | 8.8 ± 3.6 | 9.1 ± 3.4 | NS |
| CRP (mg/L) | 155.1 ± 149 | 113.4 ± 112.2 | NS |
| Calcification (n, %) | 22 (73.3) | 11 (34.4) | 0.002 |
| Overall NOC (n) | 921.5 ± 1305.5 | 54.3 ± 146.2 | 0.001 |
| Microcalcification | |||
| Present (n, %) | 21 (70.0) | 8 (25.0) | <0.001 |
| NOC (n) | 917.7 ± 1303.7 | 53.7 ± 145.8 | 0.001 |
| Volume (mm2) | 0.180 ± 0.235 | 0.018 ± 0.045 | 0.001 |
| Total calcification (AU) | 207.8 ± 268.7 | 22.5 ± 55.3 | 0.001 |
| Intermediate size calcification | |||
| Present (n, %) | 20 (66.7) | 7 (21.9) | <0.001 |
| NOC (n) | 2.43 ± 2.40 | 0.38 ± 1.10 | <0.001 |
| Volume (mm2) | 0.069 ± 0.068 | 0.011 ± 0.034 | <0.001 |
| Total calcification (AU) | 89.55 ± 91.97 | 15.79 ± 45.59 | <0.001 |
| Macrocalcification | |||
| Present (n, %) | 18 (60.0) | 4 (12.5) | <0.001 |
| NOC (n) | 1.37 ± 1.38 | 0.31 ± 1.00 | 0.001 |
| Volume (mm2) | 2.47 ± 4.07 | 1.28 ± 6.77 | NS |
| Total calcification (AU) | 4610.8 ± 9766.4 | 2360.9 ± 12 654.4 | NS |
The data are presented as mean ± SD or n (%). Data were analysed on a per lesion basis, and the statistical test did not account for the correlation of multiple lesions within patients.
AU, arbitrary units; NOC, number of calcifications.
3.2 Nicotine increases VSMC calcification in vitro
To investigate the mechanisms underlying increased calcification observed with smoking, we investigated effects of nicotine, a major component of cigarette smoke, on human primary VSMC calcification in vitro. Nicotine increased calcification of VSMCs (Figure 2A), as well as other hallmarks of active mineralization processes. Expression of some osteogenic markers was increased: transcription factors Runx2 and Osx and downstream targets OCN, BSP, and OPN (Figure 2B–F); however, others did not show a consistent pattern of upregulation (Supplementary material online, Figure S1A–D). Increased osteogenic gene expression was accompanied by a decrease in expression of some contractile markers: SM22α mRNA (Figure 2G) and αSMA, CNN1, p-MLC on protein level (Figure 2H–K). Expression of other contractile markers did not show a consistent pattern of regulation (Supplementary material online, Figure S1E–I). We also examined expression of markers characteristic of the dedifferentiated (synthetic) phenotype (KLF4, S100A4) and found it not to be changed by nicotine (Supplementary material online, Figure S1J and K).
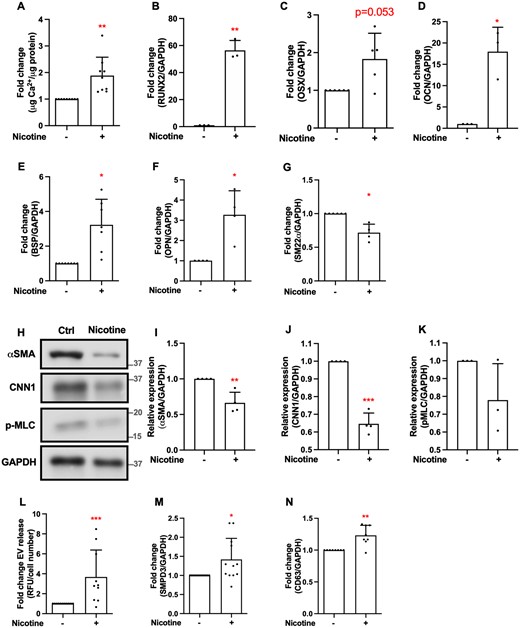
Nicotine increases VSMC calcification in vitro. (A) Calcification was induced with elevated Ca2+ concentrations (5.4 mM) in DMEM with 2.5% FBS in the presence of nicotine (1 mM, 5–7 days) and quantified using an o-cresolphthalein colorimetric assay. Cells were seeded in 48-well plates at 15 000 cells/well. Data from n = 9 independent experiments. Statistical significance was tested with one-sample test. (B–F) qPCR analysis of osteogenic genes RUNX2, Osx, OCN, BSP, OPN, and (G) contractile marker SM22α in VSMCs treated with nicotine (1 µM or 1 mM, 7 days in DMEM with 20% FBS). Statistical significance was tested with one-sample test. Data from n = 3 (B, D), n = 5 (C), n = 7 (E), n = 4 (F, G) independent experiments. In case of borderline significance, more samples were added to rule in/rule out the genes that are involved in nicotine-induced changes in VSMCs. (H–K) Western blotting and quantification of contractile markers αSMA, CNN1, p-MLC in VSMCs treated with nicotine (1 mM, 7 days in DMEM with 20% FBS). Labels show protein standard (kDa). Data from n = 4 (I, J) and n = 3 (K) independent experiments. Statistical significance was tested with one-sample test. (L) VSMCs treated with nicotine (1 mM, 24 h) in DMEM with 0.5% FBS secrete more EVs than VSMCs treated with vehicle control. EVs were captured with anti-CD63-coulped beads, detected with a fluorescently labelled anti-CD81 antibody and quantified using flow cytometry. Cells were seeded in 48-well plates at 15 000 cells/well. Data from n = 3 independent experiments in triplicate or quadruplicate. Statistical significance was tested with one-sample test. (M and N) qPCR analysis of EV-related genes; CD63 and SMPD3 mRNA expression treated with nicotine (1 mM, 7 days in DMEM with 20% FBS). Data from n = 12 (M) and n = 8 (N) independent experiments. Statistical significance was tested with one-sample test.
Calcifying EVs play an important role in the initiation and development of vascular calcification.29 We found that EV release was increased by nicotine (Figure 2L, Supplementary material online, Figure S1L), as was expression of EV-related genes (Figure 2M and N). Surprisingly, we observed that VSMCs treated with nicotine showed decreased proliferation rates compared to control (Supplementary material online, Figure S2A). Nicotine at the concentration and time point used for these assays did not affect VSMC viability at the time points the cells were collected for analysis (Supplementary material online, Figure S2B and C). Taken together, these results suggest dedifferentiation of VSMCs and activation of some calcification-promoting mechanisms as a result of exposure to nicotine.
3.3 Nicotine induces ROS production in VSMCs
As nicotine is a known inducer of oxidative stress30,31 and increased reactive oxygen species (ROS) are an important risk factor for atherosclerosis and calcification,24,32 we investigated the effect of nicotine on ROS production in VSMCs. We found that nicotine induced ROS production in VSMCs (Figure 3A). Further to that, nicotine-induced ROS and EV production could be blocked by antioxidant vitamin K2 (Figure 3B and C), and this leads to decreased calcification (Figure 3D). The same effect was observed for antioxidant N-acetylcysteine (NAC, Figure 3E). This suggests that the calcification-promoting effects of nicotine are mediated by increased ROS production.
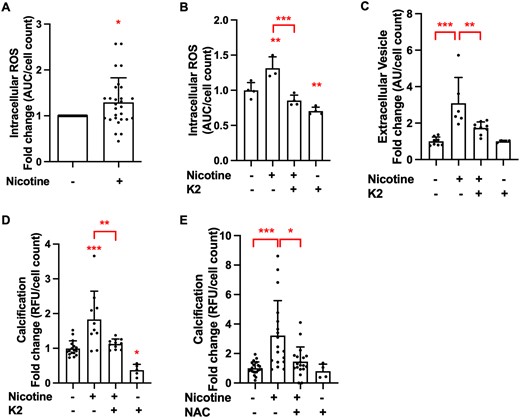
Nicotine-induced ROS production leads to VSMC calcification. (A) Stimulation of nicotine (1 mM, 40 min) induces intracellular ROS of VSMCs compared to vehicle control. ROS production was measured using a fluorescent assay with DCFDA. Cells were seeded in 96-well black plates at 8000 cells/well. Data from n = 7 independent experiments in quadruplicate. Statistical significance was tested with one-sample test. Pre-treatment of VSMCs with vitamin K (10 µM, 1 h) prior to stimulation with nicotine reduces (B) intracellular ROS (1 mM nicotine, 40 min, 8000 cells/well), data from n = 3 to 4 independent experiments (C) EV secretion (1 mM nicotine in DMEM with 0.5% FBS, 24 h, 15 000 cells/well, and (D) VSMC calcification (1 mM nicotine in DMEM with 2.5% FBS and 3.6 mM Ca2+, 3–5 days, 15 000 cells/well). Data from n = 3 independent experiments in duplicate or triplicate (C) and n = 3 to 5 independent experiments in duplicate, triplicate, or quadruplicate (D). Data of K2 control n = 1 experiment in quadruplicate (C) and n = 2 independent experiments in duplicate and triplicate (D). Statistical significance was tested with ANOVA (B–D). (E) Pre-treatment with NAC (0.5 µM, 1 h) prior to nicotine stimulation reduces VSMC calcification induced by DMEM with 2.5% FBS and 3.6 mM Ca2+ (3–5 days). Cells were seeded in 48-well plates at 15 000 cells/well. Calcification was detected using Fetuin A-Alexa-546. Data from n = 5 independent experiments, each in triplicate or quadruplicate. Data of NAC control n = 1 experiment in quadruplicate. Statistical significance was tested with Kruskal–Wallis test.
3.4 Nox5 mediates pro-calcific effects of nicotine
To reveal the source of increased ROS, we examined the expression of p22phox, Nox1, Nox2, Nox4, and Nox5, enzymes implicated in oxidative stress in vascular disease.33 We confirmed that Nox2 is not expressed in aortic VSMCs (Supplementary material online, Figure S3A), and expression of Nox1 and Nox4 was not changed by nicotine treatment (Figure 4A, B, E, G). Interestingly, expression of p22phox was decreased by nicotine (Figure 4C), while expression of Nox5 was increased, however only at mRNA level and not protein (Figure 4D–F). To investigate this further, we used pan-Nox inhibitor GKT136901 and found that it significantly blocked nicotine-induced ROS production, suggesting a potential role for Nox enzymes in these processes (Figure 4H). Pan-Nox inhibitor GKT136901 was not sufficient to reduce nicotine-induced calcification (Figure 4I). Nox5 activity is known to be mediated by Ca2+ binding to this enzyme33, and we have previously shown that increased intracellular Ca2+ mediates increased ROS production by Nox5.34 Therefore, we wanted to investigate whether nicotine induces changes in intracellular Ca2+. First, we showed that nicotine did induce an increase in intracellular Ca2+ (Figure 4J). Further to that, intracellular Ca2+ chelator BAPTA-AM attenuated nicotine-induced ROS production in VSMCs (Figure 4K). To confirm that this effect is mediated by Nox5, we performed siRNA knockdown of Nox5 (Figure 4L and M). Results show that Nox5 siRNA significantly lowered nicotine-induced ROS production (Figure 4N), EV release (Figure 4O), and calcification (Figure 4P). In order to examine the possible contribution of other Nox enzymes to nicotine-mediated calcification, we carried out p22phox siRNA knockdown and demonstrated that it did not induce a significant change in this process (Supplementary material online, Figure S3B and C).
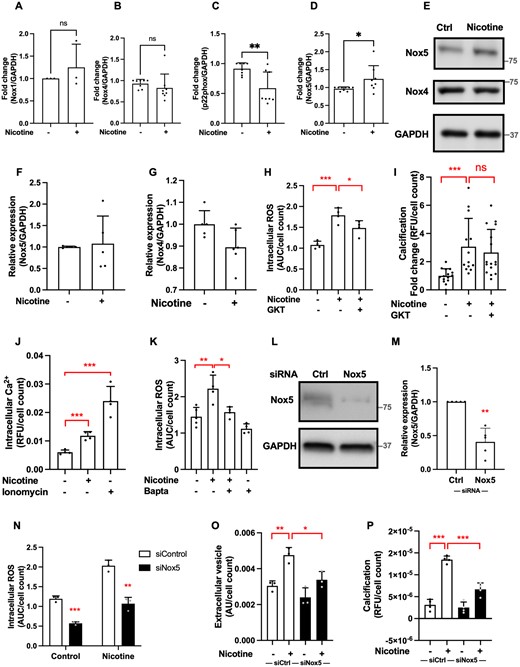
Nox5 mediates pro-calcific effects of nicotine. (A–D) qPCR analysis of NOX subunit mRNA expression in VSMCs treated with nicotine (1 mM, 7 days in DMEM with 20% FBS). Data from n = 3 independent experiments in single (A) and n = 4 independent experiments in duplicate or triplicate (B–D). Statistical significance was tested with t-test. (E–G) Western blotting and quantification of Nox4 and Nox5 protein in VSMCs treated with nicotine (1 mM, 7 days in DMEM with 20% FBS). Labels show protein standard (kDa). Data from n = 4 (F) independent experiments in single or duplicate and n = 4 (G) independent experiments in single or triplicate. Statistical significance was tested with t-test. (H) Pre-treatment of VSMCs with GKT136901 (1 µM, 1 h) prior nicotine stimulation (1 mM, 40 min, 8000 cells/well) reduces nicotine-induced intracellular ROS. Data from one experiment in quadruplicate, representative of three independent experiments. Statistical significance was tested with ANOVA. (I) VSMC calcification was induced with calcification medium (2.5% FBS, 3.6 mM Ca2+) in the presence of nicotine (1 mM), with or without pre-treatment with GKT (1 µM, 1 h), for 3–5 days. Cells were seeded in 48-well plates at 15 000 cells/well. Calcification was detected using Fetuin A-Alexa-546. Data from four independent experiments, each in triplicate or quadruplicate. Statistical significance was tested with Kruskal–Wallis test. (J) Nicotine (10 mM, 5 min) induces an increase in intracellular Ca2+ (measured using Fluo-4-AM). Cells were seeded in 96-well plates at 8000 cells/well. Data from one experiment in quadruplicate, representative of three independent experiments. Statistical significance was tested with t-test. (K) Pre-treatment of VSMCs with BAPTA-AM (1 µM, 1 h) reduces nicotine-induced intracellular Ca2+ (10 mM nicotine, 5 min, 8000 cells/well). Data from one experiment in quadruplicate, representative of three independent experiments. Statistical significance was tested with ANOVA. (L–M) Western blot showing that siRNA knock-down (24 h) decreased Nox5 protein expression. Labels show protein standard (kDa). Data from n = 4 independent experiments in single or duplicate. Statistical significance was tested with one-sample test (N) SiRNA knock-down of Nox5 (24 h) decreased VSMC intracellular ROS (data from one experiment in triplicate, representative of three independent experiments), (O) EV secretion (data from one experiment in triplicate, representative of three independent experiments), and (P) VSMC calcification (2.5% FBS, 3.6 mM Ca2+, 3–5 days). Data from one experiment in quadruplicate, representative of three independent experiments. Statistical significance was tested with ANOVA(N–P).
In order to examine alternative mechanisms leading to oxidative stress-mediated calcification and phenotype changes of VSMCs, we investigated the SrcFK/JAK-STAT3/ERK pathways (Supplementary material online, Figure S4A).35 While we observed a trend towards increased STAT3 phosphorylation (P = 0.051) after nicotine treatment (Supplementary material online, Figure S4B and C), inhibition of STAT3 phosphorylation with Stattic did not consistently change EV release and calcification (Supplementary material online, Figure S4D–E). Additionally, inhibition of SrcFK with SU6656 and ERK phosphorylation with GSK2656157 did not change nicotine-induced EV release or calcification (Supplementary material online, Figure S4D–E). Taken together, these results suggest that these pathways do not play a role in mediating nicotine-induced EV release and calcification.
3.5 Nicotine mediates its effects through interaction with α3 and α7 nicotinic acetylcholine receptors
Subsequently, we investigated which nicotinic acetylcholine receptor (nAchR) subunit mediates the effects of nicotine in VSMCs. We used two nAchR inhibitors, hexamethonium bromide and α-bungarotoxin, which inhibit α3 and α7-9 nAchR subunits, respectively.36,37 VSMCs do not express α8 and α9 subunits.36 Both hexamethonium bromide and α-bungarotoxin reduced nicotine-induced intracellular ROS production (Figure 5A). While hexamethonium bromide significantly decreased VSMC calcification, α-bungarotoxin did so only slightly (Figure 5B). However, α-bungarotoxin reduced nicotine-induced raise in intracellular Ca2+ (Figure 5C), but not EV release (Figure 5D) or expression of Nox5 and EV-related genes (Figure 5E–G). These data suggest that both α3 and α7 nAchR are involved in mediating effects of nicotine in VSMCs.
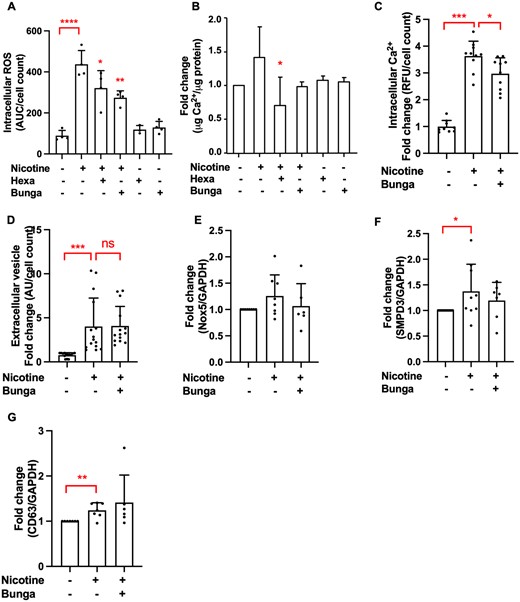
Nicotine mediates its effects through interaction with α3 and α7 nicotinic acetylcholine receptor. (A) Pre-treatment with α-bungarotoxin (1 µM, 1 h) and hexamethonium bromide (100 µM, 1 h) reduced nicotine-induced intracellular ROS (1 mM nicotine, 40 min, 8000 cells/well, data from one experiment in triplicate or quadruplicate, representative of three independent experiments) and (B) VSMC calcification (1 mM nicotine in DMEM with 2.5% FBS, 3.6 mM Ca2+, 3–5 days, 15 000 cells/well). Calcification was quantified using an o-cresolphthalein colorimetric assay. Data from n = 3 independent experiments, each in triplicate. Statistical significance was tested with ANOVA (A and B). (C) Pre-treatment with α-bungarotoxin (1 µM, 1 h) prior to nicotine stimulation (1 mM, 5 min) reduces intracellular Ca2+. Cells were seeded in 96-well plates at 8000 cells/well. Data from n = 3 independent experiments, each in triplicate or quadruplicate. Statistical significance was tested with ANOVA. (D) VSMCs were treated with nicotine (1 mM, 24 h) with or without pre-treatment with α-bungarotoxin (1 µM, 1 h) in DMEM with 0.5% FBS. EVs were captured with anti-CD63-coulped beads, detected with a fluorescently labelled anti-CD81 antibody and quantified using flow cytometry. Cells were seeded in 48-well plates at 15 000 cells/well. Data from n = 4 independent experiments, each in triplicate or quadruplicate. Statistical significance was tested with Kruskal–Wallis test. (E) qPCR analysis of Nox5 (data from n = 6 or 8 independent experiments) and (F–G) EV-related gene SMPD3 (data from n = 7 to 10 independent experiments) and CD63 (data from n = 6 or 7 independent experiments) expression in VSMCs treated with nicotine (1 mM, 7 days in DMEM with 20% FBS) with or without α-bungarotoxin pre-treatment (1 µM, 1 h). Statistical significance was tested with ANOVA (E) and Kruskal–Wallis test (F–G).
3.6 Nox5 expression correlates with the amount of calcification in human atherosclerotic plaques
Finally, to examine whether our findings have relevance in vivo, we examined expression of Nox5 as well as contractile marker CNN1, synthetic marker S100A4, and EV marker CD63 in human carotid arteries of 15 smokers and 15 non-smokers, whose arteries were previously examined by µCT (Figure 1). Immunohistochemistry showed markedly increased expression of Nox5 in smokers compared to non-smokers, while levels of CNN1, S100A4, and CD63 were not changed (Figure 6A–E). Interestingly, we found a positive correlation (r = 0.8187, P < 0.0001) between calcification (measured by µCT) and Nox5 expression (score) in both smokers and non-smokers (Figure 6E). Taken together, these data suggest that smoking is associated with increased Nox5 expression and that Nox5 promotes calcification in vivo.
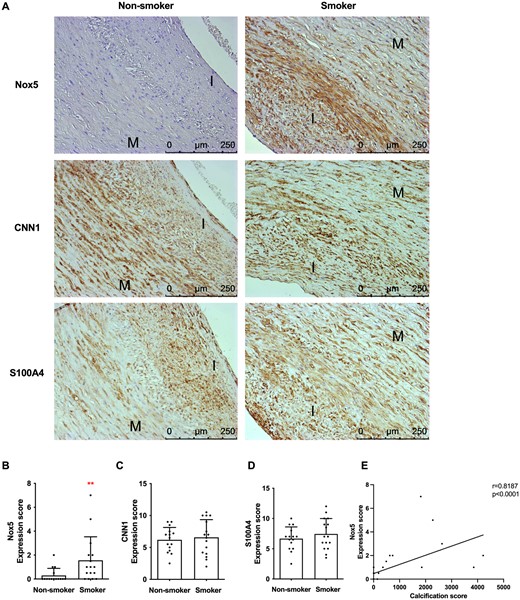
Smoking and calcification are associated with increased carotid artery Nox5 expression. (A) Immunohistochemical analysis of Nox5, CNN, and S100A4 expression in carotid artery samples from human donors. I and M indicate intima and media, respectively. Scale bars are 250 µm. Figure shows representative images from 32 carotid artery samples that were stained (15 smokers, 15 non-smokers). (B–D) Quantification of protein expression scores. Statistical significance was tested with Mann–Whitney U-test. Dots denote individual data points. Graphs (B–D) show n ≥ 15 plaques for smokers, n ≥ 15 plaques for non-smokers. (E) Nox5 expression positively correlates with calcification (P < 0.001, r = 0.8187 and P < 0.0001, r = 0.794, when outliers are removed). Nox5 expression scores were plotted against µCT total calcification scores for each patient. Graph shows individual data points (both smokers and non-smokers). Linear regression was plotted, and a two-tailed Pearson correlation test was carried out. Graph shows n = 14 plaques.
4. Discussion
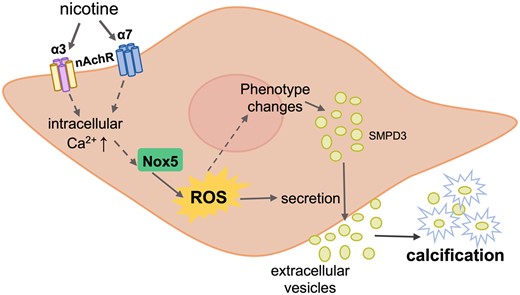
In this study, we demonstrated that smoking is associated with a significant increase in both micro- and macrocalcification in human carotid artery atherosclerotic plaques. We showed through in vitro studies that nicotine directly affects human VSMCs through α3 and α7 nAChR and induces all the major hallmarks of calcification. We uncovered a novel mechanism by which nicotine induces an increase in intracellular Ca2+, resulting in Nox5-mediated intracellular oxidative stress providing the perfect storm for VSMC calcification (Figure 7).
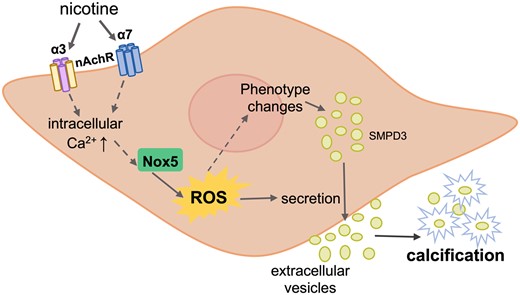
Mechanism of nicotine-induced VSMC calcification. Nicotine induces changes in VSMCs that initiate their calcification. First, nicotine binds to α3 and α7 nAchR on VSMCs, which rapidly raises intracellular Ca2+ levels. Nox5, whose activity is dependent on Ca2+, is then activated and subsequently increases production of intracellular ROS. Oxidative stress induces VSMC phenotypic switching and promotes transdifferentiation of VSMCs towards an osteogenic phenotype. Phenotypic switching of VMSC also results in higher secretion of EVs, which mineralize and contribute to calcification.
4.1 Smoking is associated with vascular microcalcification and vulnerable plaque phenotype
We show that calcification is significantly more present in smokers than in non-smokers. Our data support previous studies which reported a significant association between smoking and coronary artery as well as aortic calcification.20,38 While smoking has recently been shown to be an independent risk factor for carotid artery plaque calcification,39 calcifications have not been analysed with respect to their size. It has been shown that microcalcifications affect atherosclerotic plaque stability.40 We extend the current knowledge and show that smoking is associated with 17-fold increase in microcalcifications within atherosclerotic lesions. Since microcalcifications are biologically active and are an important feature of vulnerable plaques,9,40–42 our data suggest that smoking may be associated with a more vulnerable plaque phenotype. Indeed, studies have shown that cigarette smoke impacts the composition of the plaque, which contributes to plaque vulnerability and increased risk of plaque rupture.43,44
4.2 Nicotine induces phenotypic switching of VSMCs and initiates calcification
In this study, we elucidate the mechanism by which nicotine initiates vascular calcification in VSMCs. First, we show that nicotine downregulates VSMC contractile markers (SM22α, αSMA, CNN1, and p-MLC) and upregulates bone-specific markers (Runx2, Osx, OCN, BSP, and OPN). This is in line with previous studies showing that nicotine decreased expression of αSMA, SM22α, and increased expression of OPN in murine VSMCs45 and OPN and inflammatory cytokines in human VSMCs.46 Another study reported that nicotine differentiated VSMCs to a synthetic phenotype, demonstrated by a reduction in SM22α and h-caldesmon, and increased in myosin-II 10 and β-actin gene expression.47 Unexpectedly, the observed decrease in contractile markers in our experiments was not accompanied by a nicotine-induced increase in synthetic phenotype markers. This could be explained by the heterogeneity of VSMCs and sub-clonal population of VSMCs,17,48 which do not uniformly express specific markers at specific time points.48 More recent understanding points to the notion that VSMCs exhibit co-expression of genes rather than a two-end spectrum, contractile and synthetic phenotype, and that intrinsic and extrinsic factors as well as the developmental phase of the aorta should be taken into account.49,50
Phenotypic switching of VSMCs can be caused by various stimuli, including oxidized cholesterol, inflammatory cytokines, PDGF, and oxidative stress.51,52 We found that nicotine mainly acts via increasing oxidative stress, as it markedly induced intracellular ROS production and ROS-mediated nicotine-induced calcification. It is well documented that nicotine induces ROS generation both in animal studies53–55 and in murine and rat VSMCs (as cigarette smoke extract).45,56 However, our study is the first to show a causative link between ROS, nicotine, and VSMC calcification.
We also show that ROS production induced by nicotine leads to increased EV release from VSMCs. Both quantification using a CD63/CD81 bead capture assay and NTA suggest a role for exosomes; however, we cannot exclude a role for larger microvesicles, which were also present in VSMC culture supernatants. EVs are a well-known inducer of calcification, and it is known that phenotypic switching of VSMCs results in increased secretion of EVs.29,57 Pathological conditions induce release of EVs with distinct composition, which may affect cellular phenotypes.58 Indeed, release of EVs from osteogenic VSMCs promotes mineralization.57,59 Nicotine (as cigarette smoke extract) has been shown to affect the composition of EVs in lung cancer and bronchial epithelial cells,60,61 and higher levels of local site EVs were found in lungs of smokers.62 Interestingly, nicotine reduced monocyte-derived EV release.63 However, to our knowledge, there is no previous data on the effect of nicotine on EV release from VSMCs.
4.3 nAchR and Nox5 mediate pro-calcific processes induced by nicotine
We investigated the intracellular source of increased ROS, and, through pharmacological inhibition and knockdown experiments, we identified Nox5 as a key regulator of nicotine-induced ROS generation and calcification in human VSMCs. Nox enzymes are a major source of ROS in vascular diseases;64 however, the mechanism linking nicotine and oxidative stress in VSMCs is not well described in the literature. The effect of cigarette smoke on inducing vascular Nox activity has mainly been studied in endothelial cells.65,66 Cigarette smoke extract induced endothelial CXCL16-dependent leucocyte arrest via Nox5.67 Some data using rat and rabbit VSMCs suggest the involvement of Nox2 after nicotine induction.52,68 In our experiments, nicotine did not significantly change Nox1 or Nox4 expression, but decreased p22phox expression. However, Nox5 mRNA was increased by nicotine, and despite the lack of increased Nox5 protein expression in our study, we found that siRNA knock-down of Nox5, but not p22phox (the subunit required for Nox1, Nox2, and Nox4 activity), attenuated nicotine-induced ROS production, EV release, and calcification. This indicates that although Nox5 levels are not altered, activity of Nox5 is increased. Nox5 is a unique Nox, as it does not require other subunits for activity, but does require Ca2+ binding.33,69 We found that nicotine significantly raises intracellular Ca2+ within VSMCs, thereby contributing to increased Nox5 activity without increasing Nox5 expression. Our data are in line with a previous study showing that nicotine increases intracellular Ca2+ levels in human VSMCs.47
We also investigated an alternative mechanism leading to oxidative stress-mediated phenotypic switching of VSMCs and calcification, involving Janus kinase (JAK)/Signal transducers and activator of transcription (STAT) signalling, a critical pathway that regulates cell proliferation, differentiation, and apoptosis.70 Our data show that nicotine induces STAT3 phosphorylation in VSMCs. However, inhibiting STAT3, SrcFK, and ERK phosphorylation did not reduce nicotine-induced EV release and calcification, which was consistent with decreased proliferation rate of VSMCs treated with nicotine. This data merits caution as in vitro experimental time-points models more accurately interpret acute smoking than habitual smoking, which might involve activation of different signaling pathways.71 The STAT3/SrcFK pathway was shown to be only activated during 24-h nicotine exposure of rat VSMCs but not after 48-h nicotine exposure.71 The fact that we did not see nicotine-induced changes in EV release and VSMC calcification when inhibiting STAT3 after 7 days of treatment might be explained by a transient involvement of STAT3 phosphorylation at only the initial exposure to nicotine. As a result of this, STAT3 might not necessarily participate in the mechanisms of EV release and calcification of VSMCs. In line with our results, cholesteryl ester-transfer protein inhibitor treatment of rat VSMCs showed that STAT3 phosphorylation was unaffected by oxidative stress inhibitor NAC and Nox inhibitor GKT137831, suggesting that these two pathways are independent from each other.72 With the data obtained in our study, we conclude that nicotine-mediated VMSC calcification does not depend on SrcFK/JAK-STAT3/ERK pathways.
In this study, we show that nicotine affects VSMCs at least in part through binding of α3 and α7 nAchR. We show that inhibition of α3 and α7 nAchR results in decreased intracellular ROS. Inhibition of α3 nAchR reduces nicotine-induced VSMC calcification whereas inhibition of α7 nAchR reduces nicotine-induced intracellular Ca2+. Indeed, others have shown that nicotine increases intracellular Ca2+ of rat airway smooth muscle cells through α7 nAchR.73 However, the connection between α7 nAchR and Nox5-mediated oxidative stress in VSMCs has not been made before. Although a previous study has shown that α7 nAChR is the most abundantly expressed subunit in human VSMCs,74 we did not observe a significant reduction of nicotine-induced EV secretion when inhibiting α7 nAchR. Therefore, we conclude that other nAchRs may also play roles in VSMC phenotypic switching.
To confirm our in vitro findings, we show that carotid arteries of patients who smoked have higher Nox5 expression levels than arteries of non-smokers. Because we did not observe Nox5 expression consistently elevated in nicotine-induced VSMCs, the elevated expression in carotid arteries could be attributed to the length and amount of cigarette that smokers were exposed to, or other chemical constituents in the cigarette which also induce oxidative stress via Nox activity.75 More interestingly, Nox5 expression positively correlates with degree of calcification in these patients. Specifically, the correlation of Nox5 and calcification is the strongest at lower scores of calcifications. This observation may imply an involvement of Nox5 in initiating calcification after being induced by nicotine. Once calcification is present, it induces more changes in the vasculature that lead to more calcification.76–79 While the presence of Nox5 in the atherosclerotic plaques of human carotid arteries is known,80–82 a direct association between smoking and Nox5 expression has not been described. Amongst vascular diseases, smoking is known as one of the strongest life-style risk factors for development and calcification of abdominal aortic aneurysm (AAA).83–86 Interestingly, Nox5 expression was significantly increased in aortic aneurysm segments compared to matched non-dilated aortic specimens.87 Additionally, increased oxidative stress was significantly associated with smoking in the aortic aneurysm segments.87 Our in vivo data put forward Nox5 as a key mediator of vascular calcification caused by smoking.
4.4 Vitamin K ameliorates effects of nicotine on VSMCs
Given the role of oxidative stress in the pathogenesis of vascular calcification, antioxidant therapy may represent a viable therapeutic approach. We found that pre-treatment of VSMCs with vitamin K attenuates nicotine-induced intracellular ROS generation, reduces EV release, and subsequently inhibits calcification. However, we have not excluded that vitamin K could decrease calcification by its direct effect on γ-glutamylcarboxylation. Vitamin K is involved in multiple cellular processes and has important implications in cardiovascular and bone diseases.88 The primary role of vitamin K is to serve as a cofactor in the activation of coagulations factors and vitamin K-dependent calcification inhibitors such as matrix Gla-protein;89 however, vitamin K also exerts antioxidative properties, as several studies have demonstrated.90–94 It has also been shown that vitamin K can serve as an electron donor for the plasma membrane Nox of soya bean plant, which may implicate protection of plasma membranes against ROS.95 Nicotine administration in rats fed with a high-fat diet was shown to lower scavenging activity of antioxidant enzymes.96 It is therefore tempting to speculate that vitamin K may be a useful agent to counteract the development of nicotine-induced vascular pathologies.
Taken together, we propose a novel mechanism by which nicotine induces VSMC calcification, via a sequence of events involving a raise in intracellular Ca2+ via the α7 nAchR, increasing ROS production by Ca2+-dependent Nox5 and increasing EV release. We show that smoking is associated with increased vascular microcalcification in atherosclerotic plaques and that both smoking and degree of calcification correlated with increased Nox5 expression. Finally, we show that reducing oxidative stress using vitamin K ameliorates effects of nicotine on VSMCs. Nicotine is the primary agent in both cigarettes and e-cigarettes. Our findings add to the current literatures that vaping may not be a healthier choice when it comes to vascular health. Our study contributes to the understanding of how nicotine not only affects atherosclerotic plaque calcification but possibly also other VSMC-mediated vascular diseases, such as hypertension97,98 and aortic aneurysm.99,100
4.5 Limitations of the study
(i) Detailed characterization of the study cohort is not available. We have included all parameters we were able to access, but did not have information about further patient characteristics such as cardiovascular risk factors (e.g. family history, hypertension), laboratory values (LDL, HDL, cholesterol, triglycerides, hsCRP), and medication. (ii) The discrepancy between VSMC markers in vitro and in vivo observations could be attributed to the length and amount of cigarette that smokers were exposed to. In vivo data from vascular tissue of a smoker represent years of cigarette exposure, and possibly vascular repair and remodelling as compensation. In vitro data represent direct effect of nicotine and the immediate consequences that lead to VSMC pathology. We did not observe differences in S100A4 expression both on mRNA level in vitro and by immunohistochemistry in vivo. This is consistent with previous findings that S100A4, while associated with VSMC phenotype in certain conditions,101 does not increase with increased Nox5 activity. The tissue analysed by immunohistochemistry might represent various stages of atherosclerosis and various locations in the plaque, which we could not control for. It is possible that active synthetic differentiation occurs only at certain stages and locations of the vessel and thus any differences in expression are obscured, even though they may have been present. Additionally, the lack of correlation of S100A4 with Nox5 expression confirms our previous findings that overexpression of Nox5 or H2O2-induced oxidative stress downregulates expression of S100A4 in VSMCs. Therefore, we cannot unequivocally conclude the nature of S100A4 regulation because of the interaction between nicotine treatment and oxidative stress. Further to that, we saw no difference in CNN1 expression in smokers vs. non-smokers by immunohistochemistry, which could be attributed to similar reasons. (iii) We have not investigated the involvement of other members of the Nox family in mediating nicotine-induced effects on VSMCs. However, we have previously investigated the involvement of Nox1 and Nox4 in VSMC differentiation and calcification and found that they were not involved.34 While literature shows that Nox2 is involved in mediating oxidative stress in VSMCs,102,103 it has not been examined whether this leads to increased calcification.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
PP, MB, BW, NS, SH, and MF performed the experiments and analysed the data; PP, MB, MF, and LS wrote the manuscript; PP, MB, BW, BM, MF, and LS designed the study; FG, SR, KB, FK, NM, EN, EB, MJ, CR, BM, MF, and LS provided patient material and discussed the study; PP, MB, MF, and LS edited the manuscript, and all authors approved the final manuscript; PP and MB contributed equally to this work.
Acknowledgements
The authors would like to thank Cecile Maassen and Peter Leenders for excellent technical support.
Conflict of interest: L.S. is an unpaid consultant to immunodiagnostic systems (IDSs) and receives research grants from Nattopharma, Bayer, Boehringer Ingelheim, and Daiichi Sankyo. F.G. is the owner of Gremse-IT GmbH.
Funding
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 675111 and 722609. This study was partly funded by NWO ZonMw (MKMD 40-42600-98-13007) grant to C.R. and L.S.
Data availability
All data are incorporated into the article and its Supplementary material online.
References
CDC / U.S. Department of health and human services. Surgeon General’s Report: The Health Consequences of Smoking—50 Years of Progress 2014;8:458-512.
Author notes
Ploingarm Petsophonsakul and Mathias Burgmaier contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}