





Abstract
After myocardial infarction (MI), injured cardiomyocytes recruit neutrophils and monocytes/macrophages to myocardium, which in turn initiates inflammatory and reparative cascades, respectively. Either insufficient or excessive inflammation impairs cardiac healing. As an endogenous inhibitor of neutrophil adhesion, EDIL3 plays a crucial role in inflammatory regulation. However, the role of EDIL3 in MI remains obscure. We aimed to define the role of EDIL3 in cardiac remodelling after MI.
Serum EDIL3 levels in MI patients were negatively associated with MI biomarkers. Consistently, WT mice after MI showed low levels of cardiac EDIL3. Compared with WT mice, Edil3−/− mice showed improvement of post-MI adverse remodelling, as they exhibited lower mortality, better cardiac function, shorter scar length, and smaller LV cavity. Accordingly, infarcted hearts of Edil3−/− mice contained fewer cellular debris and lower amounts of fibrosis content, with decreased collagen I/III expression and the percentage of α-smooth muscle actin myofibroblasts. Mechanistically, EDIL3 deficiency did not affect the recruitment of monocytes or T cells, but enhanced neutrophil recruitment and following expansion of pro-inflammatory Mertk−MHC-IIlo-int (myeloid-epithelial-reproductive tyrosine kinase/major histocompatibility complex II) macrophages. The injection of neutrophil-specific C-X-C motif chemokine receptor 2 antagonist eliminated the differences in macrophage polarization and cardiac function between WT and Edil3−/− mice after MI. Neutrophil extracellular traps (NETs), which were more abundant in the hearts of Edil3−/− mice, contributed to Mertk−MHC-IIlo-int polarization via Toll-like receptor 9 pathway. The inhibition of NET formation by treatment of neutrophil elastase inhibitor or DNase I impaired macrophage polarization, increased cellular debris and aggravated cardiac adverse remodelling, thus removed the differences of cardiac function between WT and Edil3−/− mice. Totally, EDIL3 plays an important role in NET-primed macrophage polarization and cardiac remodelling during MI.
We not only reveal that EDIL3 deficiency ameliorates adverse cardiac healing via NET-mediated pro-inflammatory macrophage polarization but also discover a new crosstalk between neutrophil and macrophage after MI.
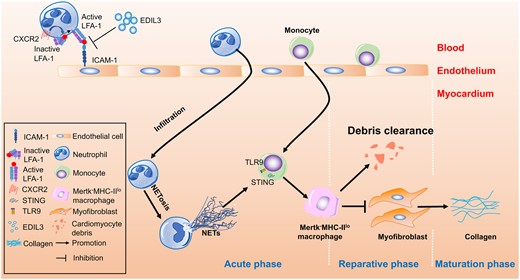
1. Introduction
Accumulating evidence has shown that the healing of myocardial infarction (MI) involves a set of delicately regulated inflammatory responses.1 After MI, injured cardiomyocytes release damage-associated molecular patterns (DAMPs), cytokines, and chemokines, resulting in massive recruitment of neutrophils and monocytes/macrophages to the myocardium. These neutrophils and monocytes contribute to the clearance of debris and dead cells, and the activation of reparative pathways. Moreover, recruited monocytes give rise to pro-inflammatory or reparative macrophages. Pro-inflammatory macrophages produce cytokines, release matrix metalloproteinase (MMP) to promote extracellular matrix destruction, and clear cellular debris, while reparative macrophages promote fibroblast transformation into myofibroblasts and enhance collagen deposition, thereby resulting in the formation of a scar of cross-linked collagen to protect left ventricle (LV) from cardiac rupture.2–4 However, excessive collagen deposition leads to adverse ventricular remodelling, including ventricular dilation and cardiac contractile dysfunction.5
Neutrophil recruitment depends on a well-coordinated adhesive cascade that includes selectin-mediated neutrophil capture and rolling, as well as integrin-mediated firm adhesion and transmigration.6 Three endogenous regulators, pentraxin (PTX)-3, growth-differentiation factor (GDF)-15, and EGF-like repeats and discoidin I-like domain 3 (EDIL3, also known as Del-1, encoded by the Edil3 gene), reportedly inhibit neutrophil recruitment and regulate inflammation.7 PTX-3 attenuates neutrophil rolling on vessels by binding to P-selectin8 and plays a protective role in an MI mouse model.9 GDF-15 represses the recruitment of polymorphonuclear leukocytes by interfering with the conformational activation of β2 integrins, thus protecting post-MI hearts from fatal cardiac rupture.10 As the third endogenous leukocyte–endothelial adhesion inhibitor, EDIL3 inhibits the interaction between LFA-1 (also known as αLβ2 and CD11a/CD18) integrin and endothelial counter-receptors, such as intercellular adhesion molecules (ICAMs), suppressing neutrophil recruitment during inflammatory bone loss.11 However, while PTX-3 and GDF-15 in MI are relatively well studied and may be candidate prognostic biomarkers for cardiovascular diseases, the role of EDIL3 in MI remains obscure.
In this study, we revealed the correlation between EDIL3 serum levels and the MI biomarkers in acute myocardial infarction (AMI) patients. In the MI mouse model, we compared the cardiac function, cardiac remodelling, and immunological microenvironment between WT and Edil3−/− mice. We also described the role of neutrophil extracellular trap (NET) for the improvement of cardiac function after MI and delineated the molecular mechanism of pro-inflammatory macrophage polarization in the presence of NETs. Therefore, we not only demonstrated the role of EDIL3 in cardiac remodelling after MI but also revealed new neutrophil-macrophage crosstalk after MI.
2. Methods
The detailed description of Section 2 is available in the Supplementary material online.
3. Study population and blood samples
A total of 58 AMI patients were recruited from West China Hospital between 1 September 2013 and 28 February 2014. The patients were diagnosed with AMI according to the following two criteria. First, the patient’s creatine kinase-MB (CK-MB) level was twice as high as that of the normal population or the concentration of cardiac troponin I was >0.1 ng/mL. Second, new pathological Q waves or ST-segment and T-wave changes were observed on electrocardiogram. The exclusion criteria included previous valve replacement, disseminated malignancy, inflammatory disease or autoimmune disease, and transplant patients. Based on the exclusion criteria, eight patients were excluded. These exclusions ensured that all included patients are with minimal inflammatory burden. Finally, 50 patients signed written informed consent and were studied in the follow-up. All the blood samples were collected within 24 h after MI. This study complied with the Declaration of Helsinki and was approved by the Ethics Committee of West China Hospital.
4. Animal model
Adult (8–12 weeks old) litter-matched male mice were used in all LAD ligation and I/R model. Edil3−/− mice, generously provided by Dr Thomas Quertermous (Stanford University), were generated by homologous recombination in C57BL/6 background.12 The Institute of Laboratory Animal Resources (Beijing, China) provided TLR9−/− mice, which were originally generated by OrientalBioService Inc. (Kyoto, Japan) through homologous recombination.13STING−/− mice (C57BL/6J-Tmem173gt/J), GFP mice [C57BL/6-Tg (CAG-EGFP)131Osb/LeySopJ mice] were purchased from the Jackson Laboratory. CD45.1 C57BL/6 mice were kindly provided by Dr Chen Chong (Sichuan University).
MI was induced by permanent ligation of the left anterior descending coronary artery (LAD).14 Sham-operated mice underwent the same chest-open procedure except for LAD ligation. Mice not surviving the first 24 h after surgery were excluded. Infarct size was visualized using Masson’s trichrome staining. For ischaemia/reperfusion,15 after 90 min of ischaemia, the occlusion of the left anterior coronary artery occlusion was released, and reperfusion for the indicated time. For preoperative and post-operative analgesia, subcutaneous injection of buprenorphine (0.08 mg/kg) was used. Mice were anaesthetized in an induction chamber with 4% isoflurane mixed with 0.5 L/min of 100% O2. During the surgery, 1.5% isoflurane with 0.5 L/min O2 was maintained for analgesia. After terminal studies at the indicated time points, animals were sacrificed under isoflurane inhalation followed by cervical dislocation.
In additional experiments, WT and Edil3−/− mice received intraperitoneal (i.p.) injection of C-X-C motif chemokine receptor 2 (CXCR2) antagonist14 (2 μL of 10 mg/mL SB225002 in 98 μL 0.9% saline per mouse, Sigma-Aldrich) or vehicle (2 μL DMSO in 98 μL 0.9% saline per mouse) 5 min before LAD occlusion and then every 24 h for 3 or 4 days.14 In the experiments of deoxyribonuclease I (DNase I) injection, WT and Edil3−/− mice were injected intravenously (i.v.) with 200 U DNase I (Sigma-Aldrich) or vehicle (0.9% saline) 5 min before LAD occlusion and then every 24 h for 4 days.16 In the experiments of neutrophil elastase inhibitor (NEi) injection, WT mice were i.p. injected with 50 μg NEi (2 μL of 25 mg/mL NEi in 98 μL 0.9% saline per mouse, GW311616) or vehicle (2 μL DMSO in 98 μL 0.9% saline per mouse) every 12 h for 3 days.17 Heparin (10U in 50 μL 0.9% saline) was administrated i.p. 10 min before ischaemia and immediately after release of ligature.
All animal experiments were approved by the Committee on the Ethics of Animal Experiments of Sichuan University. The experimental procedures were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication No 85-23).
5. Histology
Heart tissues were fixed with 4% paraformaldehyde for 48 h and embedded in paraffin. The heart sections (5 μm) were stained with Masson’s trichrome. The infarct size was determined as infarct circumference divided by total LV circumference × 100%, as described previously.18,19 The percentage of fibrosis area was calculated as the ratio of fibrosis area (blue staining) to the LV area in each microscopic field using 20× objective. For each section, five randomly chosen high-power fields were collected and analysed. Quantifications were performed by ImageJ software (National Institutes of Health). For the quantification of debris, which showed irregular, small, and eosin positive fragments around necrotic cells, heart sections were firstly stained with haematoxylin and eosin (H&E). Then, three random fields (with 40× objective) per sample were randomly selected to calculate debris content.
Immunohistochemistry was performed as described previously.20 After blocking endogenous peroxidase with 3% H2O2, the paraffin-embedded sections were blocked with goat serum and incubated overnight at 4°C with the primary rabbit monoclonal anti-α smooth muscle actin (α-SMA) (ab124964, Abcam), which is an antibody established by previous studies21,22 to be suitable for myofibroblast detection. The following day, the sections were incubated with the biotin-conjugated secondary antibody and streptavidin-HRP. With the appropriate DAB stain, the images of these sections were collected by Zeiss Imager Z2 equipped with an apochromat 20x/0.8 M27 objective and analysed by ImageJ software.
For NET staining,16,23 heart frozen sections were fixed by 4% paraformaldehyde for 15–20 min. Following incubation with 3% bovine serum albumin to block non-specific binding, sections were incubated with polyclonal goat anti-mouse myeloperoxidase (MPO) (AF3667, R&D) and polyclonal rabbit anti-citrullinated histone 3 (Cit-H3) (NB100-57135, Novus Biologicals) overnight at 4°C. On the following day, the sections were incubated with Alexa Fluor 647-conjugated donkey anti-goat IgG (A21447, Invitrogen) and FITC-conjugated donkey anti-rabbit IgG (A16036, Invitrogen) for 1 h at room temperature. Then, the sections were counterstained with DAPI (4,6-diamidino-2-phenylindole dihydrochloride) before being mounted and examined by confocal microscopy. Confocal microscopy was conducted using a NiKON A1RMP+ multiphoton confocal microscope equipped with a Plan Apo λ 40x/0.95 objective (NiKON). Images were collected using NIS-Elements AR (Advanced Research) and analysed offline using ImageJ (National Institutes of Health).
6. Flow cytometry
Mouse hearts were harvested, perfused with PBS, minced with scissors, and digested with collagenase I (500U/mL, Gibco), collagenase XI (0.1 mg/mL, Sigma-Aldrich), hyaluronidase type I-s (0.1 mg/mL, H3506, Sigma-Aldrich), and DNase I (0.1 mg/mL, Sigma-Aldrich) at 37°C for 45 min. Then, the single-cell suspension was passed through 70 μm strainer, centrifuged and resuspended in PBS. Flow cytometric data were acquired on ACEA flow cytometer, and analysed by NovoExpress software (ACEA, San Diego, CA, USA). For cell sorting, at least 10 000 events from each sample were acquired by MoFlo Astrios EQ (Beckman Coulter, Brea, CA, USA). Detailed methods and antibodies were described in the Supplementary material online.
7. Macrophage isolation and polarization in vitro
Bone marrow-derived monocytes (BMDMs) were prepared as previously described.24 Briefly, bone marrow cells were flushed from the femurs and tibia of WT, TLR9−/−, or STING−/− mice and subjected to red blood cell lysis. The cells were passed through a 40 μm strainer and cultured in RPMI 1640 medium supplemented with 10% foetal bovine serum, 1% penicillin-streptomycin and 10 ng/mL recombinant murine M-CSF (315-02, PeproTech) for 4 days. Monocytes were incubated with or without NETs, which were pre-treated with or without 5 μM NEi for 1 h. To confirm the components of NETs in macrophage polarization, WT monocytes were incubated with NETs, digested NETs (dNETs) (with 100 U/mL DNase), purified DNA from NETs or neutrophil elastase (NE) for 4 days. To study the molecular mechanism of NET-mediated macrophage polarization, monocytes isolated from WT, TLR9−/−, and STING−/− mice were incubated with NETs for 4 days. In another experiment, WT monocytes were incubated with Toll-like receptor 9 (TLR9) inhibitor (ODN 2088) or ODN negative control (tlrl-2088c) in the presence of NETs. Macrophage polarization was analysed by flow cytometry.
8. Statistical analysis
Clinical data were presented as the mean ± SEM or percentage. Mann–Whitney U test was used to analyse continuous data. The χ2 test was used for categorical data analysis. For survival analysis, the Kaplan–Meier method and the log-rank test were used. All the other data in this article were presented as the mean ± SEM. For comparisons between the two groups, P-values by unpaired t-test were reported if normality of the data can be confirmed by Shapiro–Wilk tests; otherwise, Mann–Whitney U test was used. For comparisons among multiple groups, one-way ANOVA followed by Tukey’s multiple comparison test were used to compare the difference, respectively. GraphPad Prism software was used to analyse these data. P-values < 0.05 were considered statistically significant.
9. Results
9.1 EDIL3 levels were decreased in severe AMI patients
The median of serum EDIL3 concentration among 50 recruited AMI patients was 6.3 ng/mL as determined by ELISA. Based on this median, the patients were stratified into a low EDIL3 group (n = 25) and a high EDIL3 group (n = 25). The levels of MI biomarkers, including cardiac troponin I (cTnI, P = 0.0406), CK-MB (P = 0.0123), and myoglobin (MGB, P = 0.0003) in the low EDIL3 group were significantly higher than those in the high EDIL3 group (Supplementary material online, Table S1). For serum lipids, EDIL3 levels showed no significant correlation with total cholesterol, high-density lipoproteins, or low-density lipoproteins, despite a negative correlation with TG (Supplementary material online, Figure S1A). However, for MI biomarkers, significant Spearman’s correlations were observed between the levels of EDIL3 and cTnI (r = −0.367; P = 0.0087), CK-MB (r = −0.415; P = 0.0028), and MGB (r = −0.550; P < 0.0001) (Supplementary material online, Figure S1B). Moreover, the EDIL3 levels in patients with ST-elevation MI (n = 33) were significantly lower (P = 0.0454) than those in non-ST-elevation MI patients (n = 17) (Supplementary material online, Figure S1C). These findings suggest EDIL3 levels are decreased in severe AMI patients.
9.2 EDIL3 deficiency improved cardiac function and ameliorated adverse remodelling after MI
To explore the role of EDIL3 after MI, we established an MI mouse model with permanent LAD ligation (referred to as LAD model). EDIL3 is a secretory protein, which is mainly expressed by endothelial cells and macrophages.25,26 Quantitative PCR (qPCR) and Western blots analysis showed that EDIL3 levels were significantly (P < 0.05) lower in the infarcted area than in the non-infarcted area on Days 1 and 4 after MI (Supplementary material online, Figure S2A and B), which suggest EDIL3 is down-regulated by MI injury.
To further study the role of EDIL3 after MI, we investigated the heart function of Edil3−/− mice, which was generously provided by the Quertermous lab at Stanford University.12 We confirmed the absence of EDIL3 expression in Edil3−/− mice and found that 8-week-old of WT and Edil3−/− mice showed similar cardiac function and heart weight/body weight ratio (Supplementary material online, Figure S3). Increased neutrophil-lymphocyte ratio (NLR) and platelet-lymphocyte ratio (PLR) are valuable predictors of adverse cardiovascular outcomes in clinical research.27,28 We did not observe significant correlation between EDIL3 and NLR or PLR in our recruited human patients (Supplementary material online, Figure S4), probably due to the small sample size and differences in patient age, gender, disease state, and medications. However, in LAD mouse model, Edil3−/− mice showed lower NLR value (post-MI day 1) and PLR value (post-MI days 7 and 9) than WT mice (Figure 1A), which was consistent with the finding that the mortality of Edil3−/− mice was lower than that of WT mice (17.6% vs. 39.3%; P = 0.0395) (Figure 1B). Echocardiographic analysis showed that Edil3−/− mice had a higher LVEF on Day 28 after MI than WT mice (27.7% ± 2.2% vs. 20.5% ± 2.5%; P < 0.05). The acceleration of cardiac dilatation in Edil3−/− mice was inhibited compared to WT, as evidenced by smaller values of LV internal diameter at diastole (LVIDd) (4.3 ± 0.3 mm vs. 5.5 ± 0.3 mm on Day 28; P < 0.05) and LV internal diameter at systole (LVIDs) (3.8 ± 0.4 mm vs. 5.2 ± 0.3 mm, P < 0.05) on post-MI day 28 (Figure 1C). Accordingly, the left ventricular posterior wall thickness at end-diastole (LVPWd) in Edil3−/− mice were larger than in WT mice on post-MI days 17 (LVPWd, 0.6 ± 0.09 mm vs. 0.43 ± 0.12 mm) and 28 (0.56 ± 0.13 mm vs. 0.32 ± 0.13 mm) (Figure 1C), which indicated EDIL3 deficiency improved cardiac function after MI. Consistent with the better cardiac function, on Day 28 after MI, Masson’s trichrome staining revealed that Edil3−/− mice had a smaller LV cavity size and shorter scar length than WT mice (Figure 1D). These results demonstrated that EDIL3 deficiency improved cardiac function, ameliorated adverse remodelling and reduced the risk of unfavourable outcome after MI. Bone marrow transplant experiment further confirmed the deficiency of non-haematopoietic-derived EDIL3 ameliorated cardiac adverse remodelling after MI (Supplementary material online, Figure S5A). Immunofluorescence histochemistry staining showed that almost all of the EDIL3-positive area was co-localized with CD31+ endothelial cells in the hearts (Supplementary material online, Figure S5B), indicating the endothelial-derived EDIL3 may play a main role in the hearts.
![EDIL3 deficiency improved cardiac function after MI. (A) NLRs and PLRs based on murine white blood cell counts in WT and Edil3−/− mice at different time points after MI. NLR, numbers of neutrophils/number of lymphocytes; PLR, numbers of platelets/number of lymphocytes. *P < 0.05 and **P < 0.01 vs. WT at the same time point, n = 4–7 (t-test). (B) Cumulative mortality within 28 days after MI in WT (n = 33) and Edil3−/− mice (n = 34) (log-rank test). (C) Representative echocardiograms of WT and Edil3−/− mice on Day 28 after MI (left). Echocardiographic data for LVEF, LVIDd, LVIDs, and LVPWd at the indicated time points after MI (right). *P < 0.05 and **P < 0.01 vs. WT at the same time point, n = 3–9 (t-test). (D) Representative Masson’s trichrome staining (collagen in blue) of cardiac tissue from WT and Edil3−/− mice on Day 28 after MI (left), scale bar = 1 mm. Quantitative analysis of LV cavity size and scar length (right) on Day 28 after MI in WT and Edil3−/− mice. *P < 0.05, n = 6–7 (t-test). (E) mRNA levels of collagen I, collagen III, MMP9, and TIMP1 at the indicated time points after MI, represented as the fold change compared with the sham-operated control. **P < 0.01 and ***P < 0.001 vs. WT at the same time point, n = 5–6 (t-test). (F) Collagen I protein expression in the scar tissues isolated from WT and Edil3−/− mice at post-MI day 4. *P < 0.05, n = 3 (t-test). (G) Immunohistochemical analyses of α-SMA in the infarcted hearts from WT and Edil3−/− mice at post-MI day 7 (left). Scale bar = 100 μm. Quantification of four fields per mouse (right). ***P < 0.001, n = 4 (t-test). (H) Representative collagen staining on Day 28 after MI (left). Scale bar = 100 μm. Quantitative analysis of the fibrosis content in the infarcted area, border area, and remote area on Day 28 after MI (right). *P < 0.05 and **P < 0.01 vs. WT at the same area, n = 7–8 (t-test). Here, five fields from each sample were randomly selected, and the fibrotic area was quantified in each field by the following formula: the fibrotic area= (blue collagen area/left ventricular area in the observed field) ×100%. (I) Representative H&E staining for debris (red arrows) in WT and Edil3−/− mice on Day 4 after MI [left, scale bar = 25 μm (upper) or 10 μm (lower)]. Quantification of the debris in three selected randomly fields from each sample (right). ***P < 0.001, n = 4–5 (t-test).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/118/9/10.1093_cvr_cvab269/2/m_cvab269f1.jpeg?Expires=1750238997&Signature=hXIMmqmVu6VQQoAyg4oYKAoTFJqQhoZrsB12aSPQKJJZcKnzEzEmp~GOiKzLMVvUgu19sUa~472WusyRv5Lgy-LcVe6rWY0L7tvABMTgK9YDvLtruCEtt78sTb10rJY8RfT5RSPL2RuO5-~wpSGA0snOqo-TMkRf2SUJGNmUNZHCfY9NMMHMxeiiaoyuTp5EvFNPTBPI7YVyB~3JGqipm-7KfJ9EZJUj0VpKxol7BC51a6J85ssN7iJ~BRTalr2J-Uuyx9wlwyLB6ubQcVk~ixzw9QiOIul-JueHV4pgK6Uu5L7nNNi75YWrRrWFwh8HrQnlDNaTXBQfys1mriWlSA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
EDIL3 deficiency improved cardiac function after MI. (A) NLRs and PLRs based on murine white blood cell counts in WT and Edil3−/− mice at different time points after MI. NLR, numbers of neutrophils/number of lymphocytes; PLR, numbers of platelets/number of lymphocytes. *P < 0.05 and **P < 0.01 vs. WT at the same time point, n = 4–7 (t-test). (B) Cumulative mortality within 28 days after MI in WT (n = 33) and Edil3−/− mice (n = 34) (log-rank test). (C) Representative echocardiograms of WT and Edil3−/− mice on Day 28 after MI (left). Echocardiographic data for LVEF, LVIDd, LVIDs, and LVPWd at the indicated time points after MI (right). *P < 0.05 and **P < 0.01 vs. WT at the same time point, n = 3–9 (t-test). (D) Representative Masson’s trichrome staining (collagen in blue) of cardiac tissue from WT and Edil3−/− mice on Day 28 after MI (left), scale bar = 1 mm. Quantitative analysis of LV cavity size and scar length (right) on Day 28 after MI in WT and Edil3−/− mice. *P < 0.05, n = 6–7 (t-test). (E) mRNA levels of collagen I, collagen III, MMP9, and TIMP1 at the indicated time points after MI, represented as the fold change compared with the sham-operated control. **P < 0.01 and ***P < 0.001 vs. WT at the same time point, n = 5–6 (t-test). (F) Collagen I protein expression in the scar tissues isolated from WT and Edil3−/− mice at post-MI day 4. *P < 0.05, n = 3 (t-test). (G) Immunohistochemical analyses of α-SMA in the infarcted hearts from WT and Edil3−/− mice at post-MI day 7 (left). Scale bar = 100 μm. Quantification of four fields per mouse (right). ***P < 0.001, n = 4 (t-test). (H) Representative collagen staining on Day 28 after MI (left). Scale bar = 100 μm. Quantitative analysis of the fibrosis content in the infarcted area, border area, and remote area on Day 28 after MI (right). *P < 0.05 and **P < 0.01 vs. WT at the same area, n = 7–8 (t-test). Here, five fields from each sample were randomly selected, and the fibrotic area was quantified in each field by the following formula: the fibrotic area= (blue collagen area/left ventricular area in the observed field) ×100%. (I) Representative H&E staining for debris (red arrows) in WT and Edil3−/− mice on Day 4 after MI [left, scale bar = 25 μm (upper) or 10 μm (lower)]. Quantification of the debris in three selected randomly fields from each sample (right). ***P < 0.001, n = 4–5 (t-test).
Since excessive collagen deposition leads to LV dilation, adverse remodelling and poor outcome,29 we analysed the expression of factors that affect collagen deposition. On Day 3 after MI, lower mRNA levels of collagen I/III and decreased protein expression of collagen I in Edil3−/− mice were confirmed by qPCR and Western blots, respectively (Figure 1E and F). The mRNA levels of MMP-9 in the hearts of Edil3−/− mice were three times higher than those of WT mice (Figure 1E). Furthermore, the area with positive staining of α-SMA, a marker of myofibroblast, in hearts of Edil3−/− mice were decreased on post-MI day 7 (Figure 1G). Accordingly, the collagen content in the MI areas and border zones were dramatically lower in Edil3−/− mice than in WT mice on Day 28 after MI (Figure 1H). Debris clearance is a necessary step for scar formation in cardiac healing after MI.30 Compared with WT mice, less amounts of debris were observed in Edil3−/− mice on post-MI day 4 (Figure 1I). These data indicated that EDIL3 deficiency facilitated debris clearance and protected hearts from adverse remodelling.
9.3 EDIL3 deficiency promoted neutrophil recruitment to myocardium after MI
Since EDIL3 is a leukocyte–endothelial adhesion inhibitor, we next analysed neutrophil recruitment in WT and Edil3−/− mice. Consistent with the known role of EDIL3 in myelopoiesis regulation,31 at steady state, decreased neutrophils were observed in the blood and hearts of Edil3−/− mice (Supplementary material online, Figure S6A and B), but probably are irrelevant for MI outcome, as WT and Edil3−/− mice show similar heart/body weight ratio (Supplementary material online, Figure S3). On Day 1 after MI, cardiac neutrophil levels peaked in both WT and Edil3−/− mice (Supplementary material online, Figure S6A). However, Edil3−/− mice tended to sustain this peak for 3 days after MI, while the neutrophil level started to decrease in WT mice. Meanwhile, on post-MI day 3, there is an 80% increase [from (0.20 ± 0.02)×106 to (0.36 ± 0.03)×106)] of blood neutrophil count in WT but only 33% increase [from (0.09 ± 0.01)×106 to (0.12 ± 0.03)×106)] of blood neutrophil in Edil3−/− mice (Supplementary material online, Figure S6B). Moreover, after adoptive transfer of the same number of GFP+ neutrophils into the blood of WT and Edil3−/− mice, more GFP+ neutrophils were observed in the hearts of Edil3−/− mice than in those of WT mice (Supplementary material online, Figure S6C), showing that EDIL3 deficiency enhances neutrophil recruitment. Neutrophils can be polarized to a proinflammatory N1 phenotype or an anti-inflammatory N2 phenotype.32 In the heart and blood, we did not observe a significant difference in neutrophil polarization between WT and Edil3−/− mice at different days after MI (Supplementary material online, Figure S6D and E). Therefore, instead of affecting neutrophil polarization, EDIL3 deficiency promoted the neutrophil recruitment from the blood to the myocardium after MI.
9.4 EDIL3 deficiency promoted the expansion of monocyte-derived macrophages and Mertk−MHC-IIlo-int macrophages
Macrophages are traditionally classified into M1 (CD206−F4/80+CD11b+) and M2 (CD206+F4/80+CD11b+) macrophages, roughly corresponding to pro-inflammatory and anti-inflammatory phenotypes, respectively. However, in recent years, it has been recognized that such a classification scheme is oversimplified.33 Indeed, in our results, the numbers of M1 and M2 were comparable between WT and Edil3−/− mice at different time points after MI (Supplementary material online, Figure S7). Instead of a simple M1/M2 classification system, a modified gating strategy34 was devised based on the expression of Ly6C and MHC-II to explore the sources of cardiac macrophage subsets in the hearts (Figure 2A). As per the previous report,34 within the gate of F4/80+CD11b+ cells, the CD11clo subsets in Ly6CloMHC-IIint-hi (R1) and Ly6CloMHC-IIlo cells (R2) were considered as resident macrophages. The other cells, especially Ly6C+MHC-IIlo-hi macrophages (R3) were considered as monocyte-derived macrophages. Consistent with the recent study,35 over 90% of Ly6CloMHC-IIint-hi (R1), Ly6CloMHC-IIlo cells (R2), and Ly6C+MHC-IIlo-hi macrophages (R3) were CD64+, which is constitutively expressed on macrophages. Ly6CloMHC-IIint-hiCD11clo and Ly6CloMHC-IIlo CD11clo macrophages showed higher expression of TIMD4 and LYVE1, which are the markers of resident macrophages, than Ly6C+MHC-IIlo-hi macrophages (R3). On the other hand, R3 macrophages showed higher expression of CCR2, which is the marker of recruited macrophages (Supplementary material online, Figure S8). Based on the above classification, all macrophage subsets were increased after MI and peaked at post-MI day 4 (Supplementary material online, Figure S9).
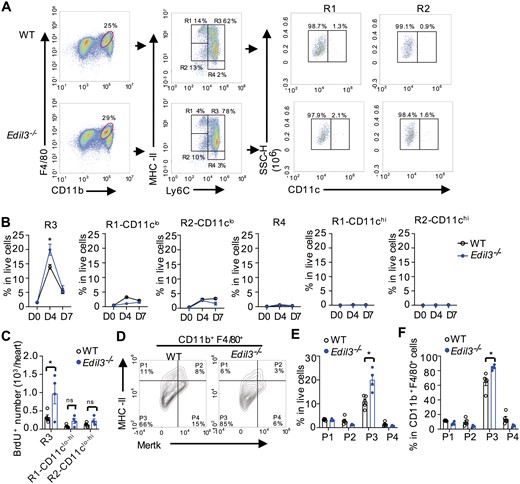
EDIL3 deficiency promoted the expansion of monocyte-derived macrophages and Mertk−MHC-IIlo-int macrophages. (A) Representative gating strategy for cardiac macrophages in WT and Edil3−/− mice at post-MI day 4. Cardiac cells gated on CD11b+F4/80+ were stratified by MHC-II, Ly6C and CD11c expression. R1, CD11b+F4/80+MHC-IIint-hiLy6Clo; R2, CD11b+F4/80+MHC-IIloLy6Clo; R3, CD11b+F4/80+MHC-IIlo-hiLy6Chi; R4, CD11b+F4/80+MHC-II-Ly6Chi. (B) Dynamics of cardiac macrophage subsets in WT and Edil3−/− mice. *P < 0.05 vs. WT at the same time point, n = 4–6 (t-test). (C) Ly6Chi monocytes were labelled with BrdU by i.p. injecting WT and Edil3−/− mice 48 h prior to harvest. Mice were sacrificed on Day 4 after MI, and BrdU+ cells were detected by flow cytometry. *P < 0.05, n = 4–5 (t-test). (D) Representative contour plots of cardiac macrophages in WT and Edil3−/− mice at post-MI day 4. Cardiac cells gated on CD11b+F4/80+ were stratified by MHC-II and Mertk expression. P1, CD11b+F4/80+Mertk−MHC-IIhi; P2, CD11b+F4/80+Mertk+MHC-IIhi; P3, CD11b+F4/80+Mertk−MHC-IIlo-int; P4, CD11b+F4/80+Mertk+MHC-IIlo-int. (E–F) Relative percentage of macrophage subsets in live cells (E) and in CD11b+F4/80+ macrophages (F) at post-MI day 4. *P < 0.05 and **P < 0.01 vs. WT at the same subset, n = 4–6 (t-test).
At steady state, the numbers of all cardiac macrophage subsets were comparable between WT and Edil3−/− mice. On post-MI day 4, the percentage of Ly6C+MHC-IIlo-hi macrophages (R3) was greater in Edil3−/− mice than in WT mice (19.9% ± 1.9% vs. 13.9% ± 0.8%, Figure 2B). Since EDIL3 deficiency restrained the conversion of Ly6Chi monocytes to Ly6Clo monocytes (Supplementary material online, Figure S10), we next want to confirm whether the increased Ly6C+MHC-IIlo-hi macrophages (R3) in Edil3−/− mice originated from monocytes. We labelled Ly6Chi monocytes, which are dominant monocytes at post-MI day 4, by injecting bromodeoxyuridine (BrdU) 48 h prior to harvest.34 Flow cytometric analysis revealed that more BrdU+Ly6C+MHC-IIlo-hi macrophages (R3) were observed in Edil3−/− mice than in WT mice (Figure 2C). These results suggested that the increased Ly6C+MHC-IIlo-hi macrophages (R3) due to EDIL3 deficiency were derived from monocytes after MI.
Mertk is mainly expressed on the macrophages and can be used as macrophage biomarker.34,36–38 We next used another gating strategy based on the expression of Mertk and MHC-II to further characterize cardiac macrophages (Figure 2D). At steady state (D0), Edil3−/− mice and WT mice showed identical numbers and ratios of Mertk−MHC-IIhi macrophages (P1), Mertk+MHC-IIhi macrophages (P2), Mertk−MHC-IIlo-int macrophages (P3), and Mertk+MHC-IIlo-int macrophages (P4) (Supplementary material online, Figure S11A). After MI, the amounts of all CD11b+F4/80+ macrophage subsets, which showed typical macrophage morphology using Giemsa staining (Supplementary material online, Figure S11B), were increased. Compared with WT mice, Edil3−/− mice showed a higher proportion of Mertk−MHC-IIlo-int macrophages (P3) on post-MI day 4 (WT, 64.6% ± 5.1% vs. Edil3−/−, 84.3% ± 1.7%, Figure 2D–F), while no significant difference was observed between the two groups at post-MI day 7 (Supplementary material online, Figure S12).
T lymphocytes, which were previously reported to affect cardiac wound healing by modulating monocyte/macrophage differentiation,39 are unlikely to be responsible for the differences in cardiac function between Edil3−/− and WT mice, because the numbers of Th, Tc, γδT, and Treg cells were comparable in the hearts of Edil3−/− and WT mice after MI (Supplementary material online, Figure S13). These data suggested that EDIL3 mainly mediated recruitment of neutrophils rather than other immune cells such as monocytes and T cells.
9.5 Targeting neutrophil recruitment eliminated the difference of macrophage polarization and cardiac function between WT and Edil3−/− mice
Neutrophils were reported to play a beneficial role in post-MI healing by affecting macrophage polarization.30 We wondered whether the increase of monocyte-derived Ly6C+MHC-IIlo-hi macrophages (R3) and Mertk−MHC-IIlo-int macrophages (P3) in Edil3−/− mice was due to enhanced neutrophil infiltration. CXCR2 is the main chemokine receptor for neutrophil recruitment by activating LFA-1.40 We found that SB225002,14 a CXCR2 antagonist, particularly repressed neutrophil recruitment, not monocyte recruitment after MI (Figure 3A and Supplementary material online, Figure S14A). Moreover, SB225002 injection decreased monocyte-derived macrophage (R3) number (Figure 3B) and increased LV cavity size and scar length (Supplementary material online, Figure S14B). Notably, the involvement of neutrophils in macrophage polarization is implicated by SB225002 treatment, which eliminated the differences in the proportions of Ly6C+MHC-IIlo-hi macrophages (R3) and Mertk−MHC-IIlo-int macrophages (P3) between WT and Edil3−/− mice on Day 4 after MI (Figure 3C and D), as well as their differences in cardiac function, LV cavity size, scar length, and fibrosis content (Figure 3E–G). A separate in vitro experiment excluded the direct effect of EDIL3 protein on macrophage polarization (Supplementary material online, Figure S14C). Therefore, our data implied that the contribution of EDIL3 deficiency in macrophage polarization and the improvement of cardiac function may be ascribed to the increased level of neutrophils.
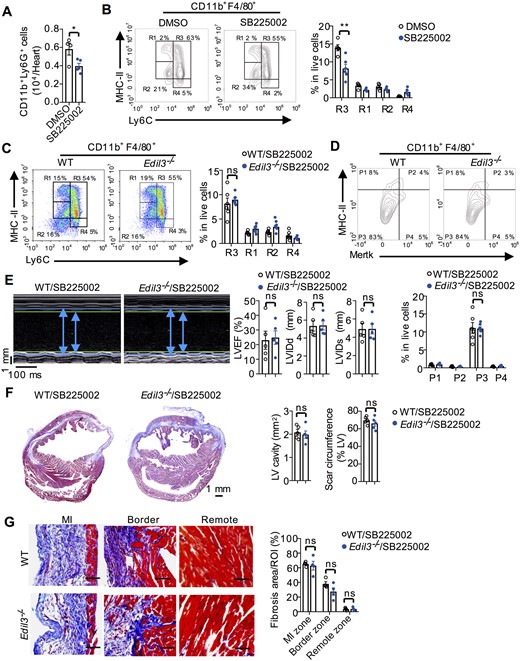
A CXCR2 antagonist eliminated the differences in macrophage polarization and cardiac function between WT and Edil3−/− mice. (A) Neutrophil numbers on Day 3 after MI in hearts of WT mice receiving the CXCR2 antagonist SB225002 or DMSO. P < 0.05, n = 4–5 (t-test). (B) Representative scatter plots of cardiac macrophages based on the expression of MHC-II and Ly6C on Day 4 after MI in WT mice receiving SB225002 or DMSO (left). Quantification of macrophage subsets in live cells (right). **P < 0.01 vs. WT at the same subset, n = 5 (t-test). (C–D) Flow cytometric quantification of cardiac macrophages based on MHC-II and Ly6C expression (C) or Mertk and MHC-II expression (D) on Day 4 after MI in WT and Edil3−/− mice receiving SB225002 (n = 6–7). (E) Echocardiographic measurement of LVEF, LVIDd, and LVIDs on Day 28 after MI in WT and Edil3−/− mice receiving SB225002 (n = 5). (F) Masson’s trichrome staining and quantitative analysis of LV cavity size and scar length on Day 28 after MI in WT and Edil3−/− mice receiving SB225002 (n = 5, scale bar = 1 mm). (G) Representative collagen staining (blue, scale bar = 100 μm) and quantitative analysis of the fibrosis content on Day 28 after MI in the hearts of WT mice and Edil3−/− mice receiving CXCR2 antagonist (n = 4–5).
9.6 EDIL3 deficiency increased the levels of NETs after MI
NETs, released by neutrophils, were observed in human sera and myocardial tissue.41 Consistent with these reports, we found that serum NET levels (MPO/DNA complex) in AMI patients correlated positively with MI biomarker cTnI and negatively with EDIL3 levels (Figure 4A). Accordingly, in MI mouse model, LAD ligation significantly increased serum NET levels (Figure 4B). By using the co-localization of Cit-H3 (a NET marker) and MPO to label NETs as per previous reports,16 we analysed total MPO+Cit-H3+ area and extracellular NETs (web-like structures) separately and found that NETs, especially extracellular NETs were present in the infarcted heart but not in sham heart (Figure 4C). High-mobility group box 1 (HMGB1), a DAMP released by ischaemic cardiomyocytes,42 induces NET formation (NETosis) via the TLR4-MYD88 pathway.43 After MI, HMGB1 levels in WT mice increased significantly.44 Anti-HMGB1 neutralizing antibody decreased the amount of Cit-H3+ neutrophils in the myocardium on day 3 after MI (from 0.79% ± 0.08% to 0.17% ± 0.04%, Figure 4D). Rossaint et al.45 reported that ICAM-1/β2 integrin signalling and simultaneous stimulation by CXCL4-CCL5-induced neutrophils to form NETs. This NETosis induction is suppressed by EDIL3, which can interfere the interaction of ICAM-1 with β2 integrins (Supplementary material online, Figure S15). Therefore, Edil3−/− mice showed higher serum and heart NETs levels than WT mice after MI (Figure 4B and C), which may be explained by the increased recruitment of neutrophils and NETosis (Supplementary material online, Figure S6A and Figure 4C). These results prompted us to determine whether NETs released from neutrophils in Edil3−/− mice contribute to cardiac improvement.
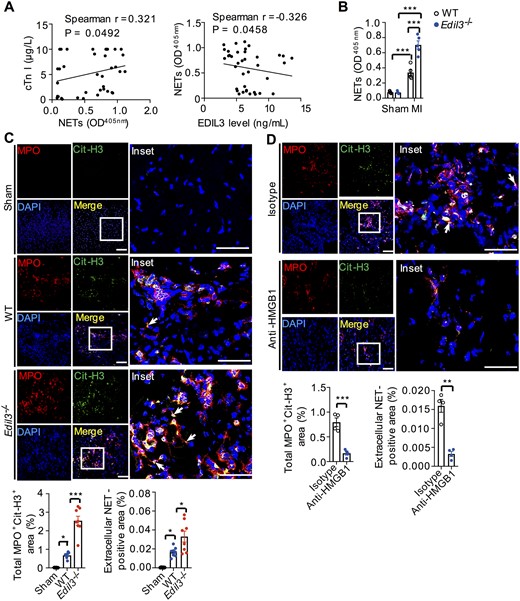
EDIL3 deficiency increased NET levels after MI. (A) Serum NET levels (MPO/DNA complex) in MI patients were detected with MPO/DNA complex ELISA and correlation with MI biomarker cTnI (left) and EDIL3 (right) levels was analysed. P < 0.05, n = 38 (Spearman’s correlation). (B) Serum NET levels were tested in WT and Edil3−/− mice on post-MI day 1. Control mice were treated by a sham operation. ***P < 0.001, n = 5 (one-way ANOVA). (C) NETs were identified by Cit-H3 (green), MPO (red), and DAPI (blue) in hearts from WT and Edil3−/− mice on post-MI day 3. Control mice were treated by a sham operation. The white arrows figure out extracellular NETs. NET-positive area was in the infarcted zone and the border zone, but not in the remote zone. NET-positive area (%) = (the co-localized area of MPO+ and CitH3+/the area of the tissue in each microscopic field using 20× objective) ×100%. Scale bar =50 μm (left column) or 100 μm (right column). *P < 0.05 and ***P < 0.001, n = 6–8 (one-way ANOVA). (D) Representative confocal immunofluorescence images of heart sections from WT mice treated with isotype or anti-HMGB1 on Day 3 after MI, scale bar= 50 μm (left column) or 100 μm (right column). ***P < 0.001, n = 4 (t-test).
9.7 NETs improved post-MI cardiac function in MI mouse model
To examine the role of NETs in cardiac remodelling, NEi, a widely used reagent to specifically inhibit NETosis,17,46 was used in LAD mouse model. Immunofluorescence images showed that NEi treatment resulted in a three-fold decrease of NET-positive area (Figure 5A). Moreover, NEi administration resulted in worse cardiac function (LVEF, 30.5% ± 2.5% vs. 43.4% ± 1.8%; P < 0.001), larger LV cavity, longer scar length, and more fibrosis than DMSO group on Day 14 after LAD ligation (Figure 5B–D). DNA is the main component of NETs. NET breakdown by DNase I is a common method to validate the function of NETs. After daily injection with DNase I for the initial 3 days after MI, NET-positive area was smaller than the vehicle group (Figure 5E). Consistent with NEi injection, on post-MI day 14, DNase I treatment resulted in higher mortality, worse cardiac function and more excessive fibrosis at infarcted region than vehicle treatment (Figure 5F–H). These results indicated that NETs contribute to the improvement of heart function in permanent ligation mouse model.
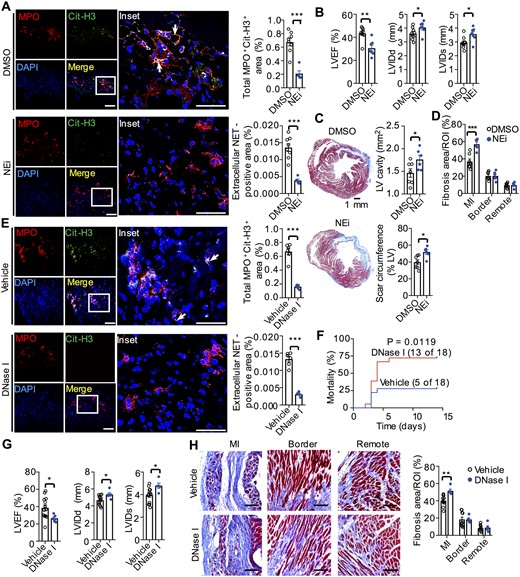
NETs improved cardiac healing after MI. (A) Representative confocal immunofluorescence images (left) and quantitation of NETs (right) of heart sections from WT mice treated with DMSO or NEi (50 μg per mouse) for 3 days after MI. Stained with Cit-H3 (green), MPO (red), and DAPI (blue). The white arrows figure out extracellular NETs. Total MPO+ CitH3+ (%) = (the co-localized area of MPO+ and CitH3+/the area of the tissue in each microscopic field using 20× objective) ×100%; Extracellular NET-positive area (%) = (Extracellular reticular structures in co-localized area of MPO+ and CitH3+/the area of the tissue in each microscopic field using 20× objective) ×100%. Scale bar = 50 μm (left column) or 100 μm (right column). ***P < 0.001, n = 5–7 (t-test). (B) Echocardiographic data for LVEF, LVIDd, and LVIDs on Day 14 after MI. *P < 0.05 and **P < 0.01, n = 7–9 (t-test). (C) Representative Masson’s trichrome staining of cardiac tissue in the DMSO and NEi groups on Day 14 after MI (left, scale bar = 1 mm). Quantitative analysis of the LV cavity size and scar length in both groups (right). *P < 0.05, n = 6–7 (t-test). (D) Quantitative analysis of the fibrosis content (blue) in the hearts of WT mice in the DMSO and NEi treated groups on Day 14 after MI. **P < 0.01 vs. DMSO group at the same area, n = 6–7 (t-test). (E) Representative confocal immunofluorescence images (left) and quantitation of NETs (right) of heart sections from WT mice treated with 200 U of DNase I or vehicle control for 3 days after MI. Stained with Cit-H3 (green), MPO (red), and DAPI (blue). Scale bar =50 μm (left column) or 100 μm (right column). ***P < 0.001, n = 5–6 (t-test). (F) Cumulative mortality within 14 days after MI in WT mice injected intravenously with 200 U of DNase I or vehicle. n = 18 (log-rank test). (G) The echocardiographic data for LVEF, LVIDd, and LVIDs on Day 14 after MI. *P < 0.05, n = 13 in vehicle group, n = 5 in DNase group (the deficiency of echocardiographic data for sixth mouse in (F) was due to unsuitable anaesthesia before operating echocardiography at the end of post-MI day 14) (t-test). (H) Representative collagen staining (left) and quantitative analysis of the fibrosis content (right) in the hearts of WT mice in the DNase I and vehicle groups on Day 14 after MI. We noted that the fibrosis area of control groups in (H) (40% on post-MI day 14) is smaller than that shown in Figure 1H (80% on post-MI day 28). This is because, during the development of MI, collagens will be accumulated with the time and the fibrosis area will be enlarged.74 Scale bar = 100 μm. **P < 0.01, n = 5–10 (t-test).
These observations seem to be inconsistent with a previous study on the ischaemia/reperfusion (I/R) model, where NETs were reported to increase ischaemia-related cardiac damage.47 NETs provide a DNA scaffold to stimulate coronary thrombosis,48,49 which can obstruct distal micro-circulation of the culprit artery in reperfusion injury, resulting in insufficient tissue perfusion and severe myocardial injury in I/R model. We therefore studied the possible detrimental involvement of thrombosis in NET-mediated cardiac healing in I/R model. We observed an abundant expression of fibrin, the marker of thrombosis,50 in the hearts of I/R model, but little fibrin in the LAD model (fibrin-positive area: 1.23% ± 0.3% vs. 0.09% ± 0.02%, Supplementary material online, Figure S16A). The injection of heparin, which blocked thrombus generation, significantly improved cardiac function in I/R model, but not in LAD model, confirmed more reperfusion injury affected by thrombosis in I/R model (Supplementary material online, Figure S16A and B). After blocking thrombosis by the heparin, DNase I treatment worsened cardiac function in I/R model (Supplementary material online, Figure S16B), which is consistent with DNase treatment results in LAD model. These results indicated that NETs benefit cardiac healing after ischaemia/reperfusion in the absence of thrombosis, which is consistent with our observation in LAD model.
9.8 The DNA moiety of NETs primed Mertk−MHC-IIlo-int macrophage polarization via TRL9 pathway
Since NETs were reported to prime macrophages to release interleukin-1β, a pro-inflammatory cytokine,16 we next investigated the possible involvement of NETs in macrophage polarization. Incubated with NETs liberated from phorbol myristate acetate (PMA) treated neutrophils, monocytes showed two-fold increase in Mertk−MHC-IIlo-int (P3) macrophage percentage, whereas the inhibition of NETosis by NEi impaired Mertk−MHC-IIlo-int (P3) polarization (Figure 6A). As NETs are composite structures consisting of NE, DNA, and other neutrophil-derived granular proteins, we next investigated which component of NETs triggers Mertk−MHC-IIlo-int (P3) polarization. In contrast to NE treatment, which had no effect on Mertk−MHC-IIlo-int (P3) polarization, treatment with NETs or purified DNA from NETs increased Mertk−MHC-IIlo-int (P3) percentages by two-fold relative to the control group (Figure 6B). The “dNETs” group, which NETs were digested with DNase I, showed a similar Mertk−MHC-IIlo-int (P3) percentage with the control group without NETs treatment (Figure 6B). This indicated the lack of DNA in dNETs impaired the ability of NETs in Mertk−MHC-IIlo-int (P3) polarization, thereby reaffirming the crucial role for DNA in NET-induced Mertk−MHC-IIlo-int (P3) polarization.
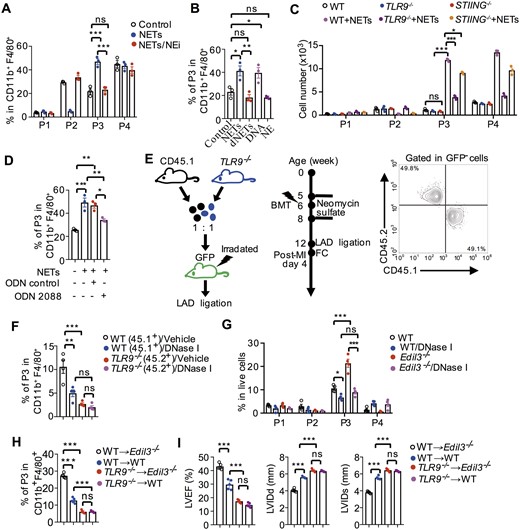
The DNA moiety of NETs primed pro-inflammatory macrophage polarization. (A) Flow cytometry analysis of macrophage polarization based on the expression of Mertk and MHC-II. Macrophage polarization in the presence of NETs, which were pre-treated with or without 5 μM NEi for 1 h. Cells in the control group were treated with 200 nM PMA without any NETs. ***P < 0.001, n = 3 (One-way ANOVA). (B) Macrophage polarization in the presence of NETs, dNETs, purified DNA from NETs, or NE. Digested NETs were formed by incubating NETs with DNase I (100 U/mL) for 30 min. *P < 0.05 and **P < 0.01, n = 3 (One-way ANOVA). (C) Quantitative analysis of macrophage subsets, which were polarized from monocytes of WT, TLR9−/− and STING−/− mice, in the presence or absence of NETs. *P < 0.05 and ***P < 0.001, n = 3 (One-way ANOVA). (D) Flow cytometry quantification of macrography subsets, which were polarized from WT monocytes incubated with TLR9 inhibitor (ODN 2088) or ODN negative control (tlrl-2088c) in the presence of NETs. Data in (A–D) are based on three independent experiments, *P < 0.05, **P < 0.01, and ***P < 0.001 (One-way ANOVA). (E) A diagram for the generation of bone marrow chimaera (left). GFP mice were used as recipient after irradiation and then reconstituted with CD45.1+ bone marrow cells in conjunction with equal number of TLR9−/− (CD45.2+) bone marrow cells. The success of reconstitution was confirmed by flow cytometry (right). (F) Macrophage polarization was analysed on post-MI day 4 in GFP+ recipient mice treated with vehicle or DNase I. **P < 0.01 and ***P < 0.001, n = 4 (One-way ANOVA). (G) On Day 4 after LAD ligation, flow cytometry quantification of macrophage polarization in the hearts of WT and Edil3−/− mice, treated with or without DNase I for 3 days. *P < 0.05 and ***P < 0.001, n = 4 (One-way ANOVA). (H) Flow cytometry analysis of macrophage polarization on post-MI day 4 in hearts of WT or Edil3−/− mice transplanted with bone marrow of WT or TLR9−/− mice. ***P < 0.001, n = 5 (One-way ANOVA). (I) Analysis of cardiac function in bone marrow chimaeras 14 days post- MI. ***P < 0.001, n = 5 (One-way ANOVA).
We further showed that DNA of NETs induces Mertk−MHC-IIlo-int (P3) polarization mainly through two major DNA sensors: TLR9 and stimulator of interferon genes.51 To verify this, WT, TLR9−/−, or STING−/− monocytes were incubated with or without NETs. Compared with WT monocytes, the number and the percentage of Mertk−MHC-IIlo-int (P3) was significantly decreased in TLR9−/− or STING−/− monocytes treated with NETs, while macrophage polarization was similar among these three monocytes in the absence of NETs (Figure 6C and Supplementary material online, Figure S17). Furthermore, monocytes pre-treated with TLR9 antagonist (ODN 2088) showed impaired NET-mediated Mertk−MHC-IIlo-int (P3) polarization (from 49.2% ± 3.9% to 34.1% ± 1.1%), whereas ODN negative control did not inhibit NET-mediated Mertk−MHC-IIlo-int (P3) polarization (Figure 6D). To confirm these findings in vivo, we adoptively transferred a 1:1 mixture of WT (CD45.1+) and TLR9−/− (CD45.2+) bone marrow cells into irradiated GFP mice (Figure 6E). Consistent with in vitro experiments, NET digestion by DNase I treatment led to lower CD45.1+ P3 percentages than the vehicle group (Figure 6F). Notably, at the same level of NETs in GFP bone marrow chimaera mice, TLR9−/− monocytes showed lower Mertk−MHC-IIlo-int (P3) percentages than WT monocytes (TLR9−/−, 2.68% ± 0.43% vs. WT, 10.57% ± 2.43%, Figure 6F). These data indicated that Mertk−MHC-IIlo-int (P3) macrophage polarization requires DNA sensors to transduce the signal from the DNA moiety of NETs, especially through the TLR9 pathways. Consistently, in WT and Edil3−/− mice after LAD ligation, DNase I treatment not only impaired Mertk−MHC-IIlo-int (P3) polarization, but also eliminated the difference of Mertk−MHC-IIlo-int (P3) polarization between WT and Edil3−/− mice (Figure 6G). A similar result was also observed in I/R mouse model with NEi treatment in the absence of thrombosis (Supplementary material online, Figure S16D). These results suggested that DNA moiety of NETs primed Mertk−MHC-IIlo-int macrophage polarization in Edil3−/− MI model. In order to validate the role of this macrophage subset in the clinical outcomes of MI, an experiment with Edil3−/− mice transplanted with TLR9−/− bone marrow cells were performed, the results showed that the protective effect of EDIL3 deficiency was abrogated in the absence of this subset (Figure 6H and I).
9.9 NET-mediated pro-inflammatory Mertk−MHC-IIlo-int macrophage polarization improved cardiac function of Edil3−/− mice
We suspected Mertk−MHC-IIlo-int (P3) macrophages are pro-inflammatory, because TLR9 was reported to respond to DNA to recruits MyD88 and the serine/threonine kinase IRAK to activate NF-κB-dependent pro-inflammatory pathway.13 Activated NF-κB in THP-1 macrophages down-regulates Mertk protein level,52 which partly explains the absence of Mertk in P3 macrophages. From the qPCR results of sorted Mertk−MHC-IIhi (P1), Mertk+MHC-IIhi (P2), Mertk−MHC-IIlo-int (P3), and Mertk+MHC-IIlo-int (P4) cardiac macrophages on Day 4 after MI, we found that pro-inflammatory markers (IL-1β and TNF-α) and anti-inflammatory markers (IL-10 and Mertk) were up- and down-regulated, respectively, in Mertk−MHC-IIlo-int (P3) macrophages relative to other macrophage subsets (Figure 7A), suggesting Mertk−MHC-IIlo-int macrophages (P3) were pro-inflammatory. Accordingly, compared with WT mice, higher levels of pro-inflammatory cytokines (IL-1β and TNF-α), and lower levels of anti-inflammatory cytokines (IL-10 and TGF-β) were observed in Edil3−/− mice on Day 4 after MI (Figure 7B), suggesting EDIL3 deficiency led to more pro-inflammatory Mertk−MHC-IIlo-int macrophages.
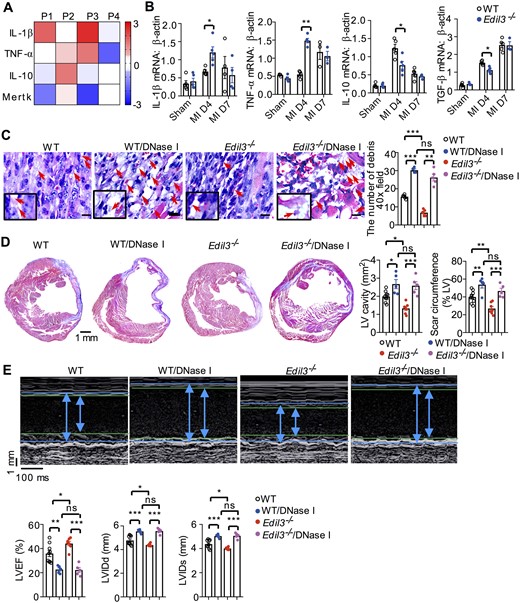
NET-mediated pro-inflammatory Mertk−MHC-IIlo-int macrophage polarization improved cardiac function of Edil3−/− mice after permanent LAD ligation. (A) mRNA levels of pro-inflammatory markers (IL-1β and TNF-α) and anti-inflammatory markers (IL-10 and Mertk), normalized to β-actin, for cardiac macrophage subsets sorted by flow cytometry 4 days after MI (n = 4). (B) mRNA levels of IL-1β, TNF-α, IL-10, and TGF-β, normalized to β-actin, in the hearts of WT and Edil3−/− mice at the indicated time points after MI. *P < 0.05 and **P < 0.01, n = 5 (t-test at the same time point). (C) On day 4 after MI, representative H&E staining for debris (red arrows) in WT and Edil3−/− mice, which treated with or without DNase I for 3 days (scale bar 10 μm). **P < 0.01 and ***P < 0.001, n = 4–5 (One-way ANOVA). (D) Representative Masson’s trichrome staining of post-MI day 28 cardiac tissue from WT and Edil3−/− mice, treated with or without DNase I for initial 4 days. Scale bar = 1 mm. Quantitative analysis of LV cavity size and scar length on Day 28 after MI in WT and Edil3−/− mice. *P < 0.05, n = 5–9 (One-way ANOVA). (E) Representative echocardiograms of WT and Edil3−/− mice on Day 28 after MI (left). Quantitative analysis of echocardiographic data for LVEF, LVIDd, and LVIDs. *P < 0.05, **P < 0.01, and ***P < 0.001, n = 5–9 (One-way ANOVA).
Pro-inflammatory macrophages were reported to prevent excessive collagen deposition by matrix degradation53 and remove necrotic cardiomyocyte debris by proteolysis and phagocytosis.54 In agreement with these reports, we found that, compared with WT mice, more pro-inflammatory Mertk−MHC-IIlo-int (P3) macrophages, less collagen and cellular debris, and better cardiac function were observed in Edil3−/− mice after LAD ligation. After DNase I treatment to digest NETs in vivo, the difference of macrophage polarization between WT and Edil3−/− mice were eliminated (Figure 6G), followed by the vanished differences in cellular debris clearance, LV cavity size, scar length, and cardiac function (Figure 7C–E). Same conclusion was found in I/R mouse model under heparin treatment (Supplementary material online, Figure S16). These results suggested that NET-polarized pro-inflammatory macrophages can improve cardiac function of Edil3−/− mice in the MI model.
10. Discussion
We demonstrated that EDIL3 levels were negatively correlated with MI severity in patients, which was consistent with decreased EDIL3 expression in infarcted hearts after LAD ligation. In mouse model, EDIL3 deficiency decreases post-MI mortality, improves cardiac function and ameliorates adverse remodelling. We further demonstrated that EDIL3 deficiency facilitates neutrophil recruitment and consequently NET-mediated pro-inflammatory Mertk−MHC-IIlo-int macrophage polarization, which in turn improves cardiac function by facilitating debris clearance in LAD model and I/R model. These findings not only revealed the role of EDIL3 in MI but also shed new mechanistic light on the ability of neutrophils to regulate the local inflammatory response.
EDIL3 displays versatility and participates in multiple pathogenic pathways based on its receptors. By binding to αVβ3 and αVβ5 integrins of endothelial cells or phagocytes and phosphatidylserine of platelet microparticles or apoptotic cells, EDIL3 contributes to the clearance of platelet microparticles or apoptotic cells.25,55 By blocking the activity of oxidized low-density lipoprotein, EDIL3 attenuates atherogenesis.56 Recently, EDIL3 was shown to interact with αLβ2 and αMβ2 integrins to inhibit neutrophil recruitment in acute peritonitis and inflammatory bone loss.12,57 In this study, we reported EDIL3 deficiency enhanced neutrophil recruitment and NETs mediated macrophage polarization after cardiac injury. In addition to neutrophil recruitment, other roles of EDIL3 in cardiac remodelling still need further investigation.
PTX-3, GDF-15, and EDIL3 are known endogenous regulators of leukocyte recruitment. However, the deficiency of PTX-3 and GDF-15 results in severe myocardial damage, while EDIL3 deficiency improves cardiac function. The differences in MI outcomes among these three molecules are probably due to their subtle differences in molecular mechanisms. Apart from binding to P-selectin to interfere with neutrophil rolling, PTX-3 also binds the complement component C1q, facilitating reperfusion-induced tissue damage.9 While both GDF-15 and EDIL3 inhibit neutrophil recruitment through β2 integrins, they may display different degrees of inhibition. First, GDF-15 counteracts the activation of β2 integrins. Its deficiency results in enhanced early-onset cardiac rupture due to excessive inflammation. Moreover, GDF-15 was shown to directly protect cardiomyocytes from ischaemic injury via the PI3K and Akt pathways.58 EDIL3, by contrast, inhibits neutrophil recruitment by competitively antagonizing the interaction between LFA-1 and ICAM-1. The degree of inhibition may be mild and thus does not lead to excessive inflammation. Therefore, the distinct mechanism of these three molecules results in different outcomes.
It seems like there is a discrepancy between our data for mouse model and clinical data: while low EDIL3 levels were associated with worse outcome in MI patients, EDIL3 deficiency in MI models leads to better outcome. Similar apparent contradictions were observed for the other two endogenous modulators of leukocyte recruitment: GDF-15 and PTX-3. While high levels of GDF-15 in MI patients were associated with worse outcome in cardiovascular diseases,59–61 studies using MI mouse model and I/R model both reported that GDF-15 deficiency resulted in greater infarct size and severer mortality.10,58 Similarly, high levels of PTX3 predicts a high risk of mortality in patients,62,63 whereas PTX3 null mice exhibit accentuated cardiac injury after I/R.9 The mechanism to explain the discrepancies of these three endogenous regulators remains unclear. Since EDIL3 expression is down-regulated by inflammatory stimuli, such as IL-17 and TNF-α,64,65 which were reported to be predictors of mortality and adverse outcomes in AMI patients,66 we speculate that the down-regulation of EDIL3 by inflammatory stimuli promotes the rapid recruitment of neutrophils to inflamed sites57 to clear debris and initiate reparative process, eventually improving cardiac function after MI. Indeed, we observe a 1.6- and 3.0-folds larger IL-17A and TNF-α levels, respectively, in low EDIL3 patients than in high EDIL3 patients, although the P-values are not significant due to the limited sample size (Supplementary material online, Table S1). In the future, discrepancies in all three leukocyte recruitment modulators require more in-depth analysis on larger patient cohort to reconcile.
Our discovery that NETs are beneficial for cardiac healing is somewhat surprising, given that NETs increase ischaemia-related cardiac damage.47 In our study, a higher level of fibrin, the marker of micro-thrombosis, was observed in I/R model than in LAD model. After blocking thrombosis by heparin, NETs removal worsened cardiac function in I/R model, which was consistent with LAD model. In a recent study using LAD model, Zhou et al.67 reported a seemingly contradictory phenomenon in LAD mouse model: different from ApoE−/− mice, cardiac healing of WT mice benefits from NETs, as shown by the increased infarct size after inhibiting NETosis by Cl-amidine (Figure 5a in Zhou’s paper), consisting with our results in LAD model.
NETs are composite structures consisting of NE, DNA, and other neutrophil-derived granular proteins. In this study, we reported DNA moiety of NETs primes Mertk−MHC-IIlo-int macrophage polarization in vitro and in vivo. The extracellular DNA of NETs are made up of mitochondrial and nuclear DNA (nDNA).68 Mitochondrial DNA (mtDNA) is rich of unmethylated CpG repeats and therefore shows stronger inflammatory characteristics than nDNA in the NETs of atherosclerosis.69 Further investigations are also needed to determine whether nDNA or mtDNA in NETs is the main cause for cardiac improvement.
We found that increased neutrophils and inflammatory macrophages in Edil3−/− mice contribute to improvement of cardiac healing, which seems contradictory to conventional anti-inflammation strategy. In fact, more and more evidences indicate that proper healing after MI requires a well-balanced initiation of neutrophils and the following macrophage polarization.30 The depletion of neutrophil in mice led to impaired cardiac function, increased fibrosis and progressive heart failure after MI.30 Despite the effectiveness of anti-CD11/CD18 integrin approaches to reduce infarct size via inhibiting neutrophil migration in animal models, little beneficial effects were observed in three clinical trials targeting β2 integrins in MI patients.42 Similarly, depletion of inflammatory monocytes resulted in larger areas of debris and necrotic tissue due to the requirement of debris clearance by acute inflammation and proteolytic influx from Ly-6Chi monocytes.1 MerTK is critical for the phagocytosis of apoptotic cells by macrophages.70,71 Previously, MerTK-positive macrophages have been reported to be reparative via increasing clearance of apoptotic cells and play protective role in myocardial injury.72,73 In our research, we discovered that Edil3−/− mice exhibited increased polarization of MerTK−MHC-IIlo-int macrophages, but no difference in the percentage of MerTK-positive macrophages. We further characterized MerTK−MHC-IIlo-int macrophages as pro-inflammatory and discovered that their polarization is TLR-9 dependent both in vitro and in vivo. These finding are consistent with the protective role of pro-inflammatory macrophages in MI. Thus, anti-inflammatory treatments in the setting of MI should be careful to design.
Overall, we demonstrated that EDIL3 deficiency prevents adverse cardiac remodelling through NET-mediated polarization of pro-inflammatory macrophage. Thus, EDIL3 may be a candidate target for the treatment of MI. Moreover, the new neutrophil-macrophage crosstalk identified herein may become an important consideration for the development and application of anti-inflammatory treatments for MI.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
J.L. and K.C. conceived and designed this project. X.W. and J.L. drafted the manuscript. X.W., Z.X., and S.Z. conducted majority of experiments and analysed the data. Z.C. collected clinical data. C.Z. revised the initial draft. Y.H. and Z.C. helped with establishment of LAD mouse model. N.H., Z.H., X.T., and Z.W. helped with flow cytometry analysis. X.L., H.Z., H.Z., S.C., and J.C. helped with in vitro experiments. F.Z., W.W., and Y.Z. helped with in vivo experiments, such as NEi injection and DNase injection. All authors provided critical discussions, proof-read the draft, and gave final approval of the manuscript.
Acknowledgements
We thank Dr Thomas Quertermous (Stanford University) for kindly providing Edil3−/− mice, Dr Yang Zhang (University of Michigan) for protein structure and function predictions, and Dr Ling Qi (University of Michigan) for insightful discussions. We also thank Zhen Yang and HuiFang Li (Core Facility, West China Hospital, Sichuan University) for their support.
Funding
This work was supported by the National Natural Science Foundation of China (31872739 to K.C., 81602763 to X.T., 81703132 to Z.W.), the National Science and Technology Major Project (2018ZX09303006-001-006 to J.L. and 2019ZX09201004-003 to J.L.) and the China Postdoctoral Science Foundation funded project (2016M592673 to X.T., 2017T100700 to X.T. and 2018M631087 to Z.W.), the Applied Basic Research Program of Sichuan Province (2021YJ0420 to Z.W.), the Postdoctoral Fund for West China Hospital (2019HXBH075 to Z.W.), the Fundamental Research Funds for the Central Universities (2019SCU12041 to Z.W., the Postdoctoral Foundation of Sichuan University).
Conflict of interest: There is no conflict of interest to declare.
Data availability
The data underlying this article are available in the article and in its Supplementary material online.
Translational perspective
We established EDIL3 as a critical regulator of neutrophil recruitment and macrophage polarization during post-MI cardiac remodelling. EDIL3 may be a candidate prognostic biomarker and drug target for cardiovascular diseases. The novel pathways and mechanisms revealed in this study have renewed our understanding of the role of leukocyte adhesion inhibitors in cardiovascular disease. Meanwhile, our study reaffirmed the indispensable role of inflammation in the healing process, thereby prompting the re-evaluation of post-MI anti-inflammatory treatments.
References
Author notes
Xiaoqiong Wei, Song Zou, Zhonghui Xie, Zhen Wang and Nongyu Huang contributed equally as co-first authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}