





Abstract
Macrophage-mediated inflammatory response represents a key pathophysiological process in a host of cardiovascular diseases including heart failure. Regardless of aetiology, heart failure is invariably preceded by cardiac hypertrophy. In the present study, we investigated the effect of macrophage-specific deletion of myocardin-related transcription factor A (MRTF-A) on cardiac hypertrophy and the underlying mechanism.
We report that when subjected to transverse aortic constriction (TAC), macrophage MRTF-A conditional knockout (CKO) mice developed a less severe phenotype of cardiac hypertrophy compared to wild-type (WT) littermates and were partially protected from the loss of heart function. In addition, there was less extensive cardiac fibrosis in the CKO mice than WT mice following the TAC procedure. Further analysis revealed that cardiac inflammation, as assessed by levels of pro-inflammatory cytokines and chemokines, was dampened in CKO mice paralleling reduced infiltration of macrophages in the heart. Mechanistically, MRTF-A deficiency attenuated the expression of integrin beta 2 (ITGB2/CD18) in macrophage thereby disrupting adhesion of macrophages to vascular endothelial cells. MRTF-A was recruited by Sp1 to the ITGB2 promoter and cooperated with Sp1 to activate ITGB2 transcription in macrophages. Administration of a CD18 blocking antibody attenuated TAC-induced cardiac hypertrophy in mice. Interaction between MRTF-A and the histone demethylase KDM3A likely contributed to IGTB2 transcription and consequently adhesion of macrophages to endothelial cells.
Our data suggest that MRTF-A may regulate macrophage trafficking and contribute to the pathogenesis of cardiac hypertrophy by activating ITGB2 transcription.

1. Introduction
Macrophage-mediated pro-inflammatory response serves as a double-edged sword in the regulation of internal homeostasis. On the one hand, macrophage-derived inflammatory cytokines and reactive oxygen species (ROS) exert bactericidal effects and facilitate wound closure thereby boosting host defense against pathogen invasion.1 On the other hand, uncontrolled or excessive inflammation can be blamed as a culprit for the architectural and functional disruption of the host in the pathogenesis of a host of cardiovascular diseases including atherosclerosis,2 aortic aneurysm,3 and heart failure.4 Typically, macrophages gain access to the cardiovascular system via interacting with vascular endothelial cells.5,6 Adhesion molecules expressed on the surface of endothelial cells interact with a specific receptor (e.g. integrins) expressed on the surface of macrophages allowing firm and sustained adhesion of macrophages to the vasculature initiating the pro-inflammatory response.7,8 In accordance, blockade of the macrophage-endothelial interaction is associated with attenuation of cardiac inflammation.9–11
Heart failure, defined as insufficiency of the heart to pump blood throughout the body to sustain normal life activities, has become one of the leading causes of death in both industrialized and developing nations.12 A wide range of causative factors, including hypertension, diabetes, structural heart disease, and dilative cardiomyopathy have been identified for heart failure.13 Regardless of the specific aetiology, heart failure is invariably preceded by a compensatory stage of cardiac hypertrophy. However, prolonged cardiac hypertrophy results in irreversible and permanent loss of heart function and predisposes the affected individuals to heart failure.14 Macrophage has been demonstrated to play a key role in the pathogenesis of cardiac hypertrophy. Increased macrophage infiltration in the heart is observed in patients with heart failure.15 Furthermore, macrophage depletion, either by antibody-mediated clearance, or clodronate liposome, or diphtheria toxin (DT)-mediated cytotoxicity, has been shown to attenuate cardiac hypertrophy and heart failure in mice.15–17
Myocardin-related transcription factor A (MRTF-A) belongs to a family of transcriptional modulators that also include myocardin, the founding member, and MRTF-B.18 Unlike myocardin and MRTF-B, both of which are required for heart development during embryogenesis, MRTF-A deficiency is well tolerated in mice with no overt cardiac phenotype under physiological conditions.19,20 Further studies have revealed that MRTF-A-null mice are protected from a host of cardiovascular diseases including atherosclerosis,21 septic shock,22 cardiac ischaemia–reperfusion injury,23 and cardiac hypertrophy.24 Specifically, it has been demonstrated by Kuwahara et al. that MRTF-A interacts with serum response factor (SRF), a sequence-specific transcription factor, to activate the transcription of natriuretic peptide A (NPPA) and natriuretic peptide A (NPPB). As a result, MRTF-A deficiency blunts the mechanostress-induced hypertrophic response of cardiomyocytes. MRTF-A has a ubiquitous expression pattern.18 Weng et al.25 have shown that endothelial MRTF-A can also contribute to cardiac hypertrophy by activating the transcription of endothelin (ET-1), which in turn acts as a pro-hypertrophic cue on cardiomyocyte. More recently, we have reported that MRTF-A plays a key role in regulating inflammation26 and production of ROS27 in macrophages. Here we report that macrophage-specific MRTF-A deficiency attenuates cardiac hypertrophy in mice. Mechanistically, MRTF-A epigenetically activates the transcription of integrin B2 (ITGB2) to mediate adhesion of macrophages to vascular endothelial cells thereby promoting cardiac inflammation. Therefore, targeting MRTF-A may present a promising solution in the prevention and treatment of heart failure.
2. Methods
2.1 Cell culture, plasmids, transient transfection, and reporter assay
RAW264 cells (ATCC) were maintained in DMEM supplemented with 10% FBS. Mouse peritoneal macrophages were isolated as previously described.23 MRTF-A expression constructs28 and ITGB2 promoter-luciferase constructs29 have been previously described. Small interfering RNAs were purchased from Dharmacon. Primary cardiac macrophages were isolated from the non-myocyte suspension by magnetic beads coated with anti-F4/80 antibody (Miltenyi Biotech). Transient transfection was performed with Lipofectamine 2000. Cells were harvested 48 h after transfection and reporter activity was measured using a luciferase reporter assay system (Promega).
2.2 Animals
All the animal experiments were reviewed and approved by the intramural Ethics Committee on Humane Treatment of Experimental Animals. All animal procedures conform to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes or the NIH guidelines. Myeloid-specific deletion of MKL1 was achieved by crossing the Mkl1f/f strain27 with the LyzM-Cre strain.23 The breedings were conducted by Nanjing Biomedical Research Institute of Nanjing University. Pathological cardiac hypertrophy was induced in mice by the transverse aortic constriction (TAC) procedure as previously described.30 Cardiac functions were evaluated by echocardiography (GE Vivid 7 equipped with a 14-MHz phase array linear transducer, S12, allowing a 150 maximal sweep rate). Mice were anesthetized using 5 mg/mL ketamine and 2 mg/mL xylazine. The body temperature was maintained at 37°C using a heating pad. The animals were euthanized by pentobarbital sodium (100–120 mg/kg) to obtain their samples. In certain experiments, the mice were intraperitoneal injections of an anti-CD18 blocking antibody (Biolegend, Clone M18/2) or a corresponding isotype control IgG three times a week (100 μg per injection) following the TAC procedure.
2.3 RNA extraction and real-time PCR
RNA was extracted using an RNeasy RNA isolation kit (Qiagen). Reverse transcriptase reactions were performed using a SuperScript First-strand synthesis system (Invitrogen). Real-time PCR reactions were performed on an ABI STEPONE Plus (Life Tech) with primers and Taqman probes purchased from Applied Biosystems. Experiments were routinely performed in triplicate wells and repeated at least three times.
2.4 Protein extraction, co-immunoprecipitation, and western blotting
Whole-cell protein extraction and nuclear protein extraction were essentially performed as previously described.31,32 Specific antibodies or pre-immune IgGs (P.I.I.) were added to and incubated with cell lysates overnight before being absorbed by Protein A/G-plus Agarose beads (Santa Cruz). Precipitated immune complex was released by boiling with 1× SDS electrophoresis sample buffer. Western blot analyses were performed with commercially available antibodies: anti-MRTF-A (Santa Cruz, sc-32909), anti-Sp1 (Santa Cruz, sc-59), anti-KDM3A (Proteintech, 12835-1), anti-ITGB2 (Proteintech, 10554-1), anti-ANP (Thermo Fisher, 702539), anti-β-MHC (Thermo Fisher, MA1-83347), anti-collagen type I (Rockland, 600-401-103), anti-α-SMA (Sigma, A2547), and anti-β-actin (Sigma, A2228). All experiments were repeated at least three times. Full-scan un-cropped blots are presented in the Supplementary material online, Figure S11.
2.5 Chromatin immunoprecipitation (ChIP) and Re-ChIP
ChIP assays were performed essentially as described before.33–40 Briefly, chromatin was cross-linked with 1% formaldehyde. Cells were incubated in lysis buffer (150 mM NaCl, 25 mM Tris pH 7.5, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate) supplemented with protease inhibitor tablet. DNA was fragmented into 500 bp pieces using a Branson 250 sonicator. Aliquots of lysates containing 100 μg of protein were used for each immunoprecipitation reaction with the following antibodies: anti-MRTF-A (Santa Cruz, sc-32909), anti-Sp1 (Santa Cruz, sc-59), anti-KDM3A (Proteintech, 12835-1), anti-acetyl H3 (Millipore, 06-599), anti-dimethyl H3K9 (Millipore, 07-441), and anti-acetyl H3K9 (07-352). Precipitated genomic DNA was amplified by real-time PCR with primers spanning the human RhoJ gene promoters. A total of 10% of the starting material is also included as the input. Data are then normalized to the input and expressed as fold changes compared to the control group.
2.6 Macrophage adhesion assay
Macrophages were stained with a fluorescent die [2′,7′-Bis-(2-carboxyethyl)-5(6)-carboxyfluorescein tetrakis (acetoxymethyl) ester; Sigma] for 30 min at 37°C. After several washes with PBS, the cells were co-incubated for 30 min with endothelial cells. Unbound leucocytes were removed by washing and the number of adhered cells was visualized by fluorescence microscopy and analysed with Image-Pro Plus (Media Cybernetics).
2.7 Immunofluorescence staining
The paraffin-embedded sections were incubated with anti-F4/80 (BD Biosciences) followed by incubation with donkey secondary antibodies (Jackson ImmunoResearch). The nuclei were counterstained with DAPI (Sigma). Pictures were taken using an Olympus IX-70 microscope. For each primary antibody, we stained at least three slides from each individual mouse.
2.8 Flow cytometric analysis
The hearts were minced and digested with type II collagenase, elastase, DNase I (all from Sigma Aldrich) in a 37°C water bath with gentle agitation. Cell suspensions were incubated with anti-CD16/32 (2.4G2, BD Bioscience) at 4°C for 5 min to block nonspecific binding followed by incubation with anti-CD45, anti-Ly6G, anti-CD3, anti-CD11c, anti-MHC-II, and/or anti-NK1.1 (all from Biolegend) and sorting on a FACSAria III (BD Biosciences). The data were expressed as number of positive cells per heart.
2.9 Caspase activity assay
Caspase 3 activity in cardiac tissues was examined using a colorimetric kit (Ab39401, Abcam) according to vendor’s recommendations.
2.10 Statistical analysis
Two-sided t-test (for experiments involving two groups) or one-way ANOVA with post hoc Scheffe analyses (for experiments involving with at least three groups) were performed using an SPSS package. P values smaller than 0.05 were considered statistically significant (*).
3. Results
3.1 MRTF-A deletion in macrophages attenuates TAC-induced pathological cardiac hypertrophy in mice
We first determined the overall expression level and subcellular translocation of MRTF-A in cardiac macrophages in the course of pathological hypertrophy in mice. C57BL/6 mice were subjected to transverse aortic constriction (TAC) and sacrificed at 2 weeks and 4 weeks after the procedure. MRTF-A expression levels were progressively up-regulated in cardiac macrophages following the TAC procedure (Supplementary material online, Figure S1). There was also an appreciable nuclear migration of MRTF-A in cardiac macrophages at 2 weeks and 4 weeks following the surgery (Supplementary material online, Figure S1). In addition, it was determined by qPCR that MRTF-A activation in cardiac macrophages appeared to correlate with the induction of signature molecules associated with hypertrophy, inflammation, and fibrosis (Supplementary material online, Figure S2).
We then sought to answer a simple question: what is the effect of macrophage-specific MRTF-A deficiency on pathological cardiac hypertrophy in mice? To address this question, MRTF-A Flox mice (MRTF-Af/f) were crossed to Lyz2-Cre mice to generate macrophage conditional MRTF-A knockout mice (MRTF-ACKO/CKO); MRTF-A deletion in bone marrow-derived macrophages (BMDMs) was verified by Western blotting (Supplementary material online, Figure S3). Both WT and CKO mice were to subject to the TAC procedure to induce cardiac hypertrophy. The mice were sacrificed four weeks after the procedure for evaluation. As shown in Figure 1A and B, pressure overload significantly increased heart weight/body weight and heart weight/tibia bone length indicative of the development of cardiac hypertrophy; the CKO mice developed a less severe phenotype of cardiac hypertrophy compared to the WT mice. In addition, echocardiographic measurements of left ventricular systolic diameter (Figure 1C) and left ventricular posterior wall thickness ( Figure 1D) showed that TAC-induced cardiac hypertrophy was attenuated in CKO mice. These observations were corroborated by qPCR (Figure 1E–G) and Western blotting (Supplementary material online, Figure S4) examination of hypertrophic genes including ANP, BNP, and β-MHC. Finally, echocardiography indicated that MRTF-A deficiency in macrophages partially recovered heart function following the surgery (Figure 1H and I). Taken together, these data suggest that macrophage MRTF-A might play a role in the development and deterioration of pathological cardiac hypertrophy in mice.

MRTF-A deletion in macrophages attenuates TAC-induced pathological cardiac hypertrophy in mice. Macrophage MRTF-A conditional knockout (MRTF-ACKO/CKO) mice and wild-type (MRTF-Af/f) littermates were subjected to the TAC procedure or the sham procedure. (A) Heart weight vs. body weight. (B) Heart weight vs. tibia bone length. (C) LVSd values. (D) LVPWd values. (E–G) Hypertrophic genes were measured by qPCR. (H) EF values. (I) FS values. N = 5 mice for the sham groups and N = 6–7 mice for the TAC groups. Data represent mean ± SD. *P < 0.05, two-tailed t-test.
3.2 MRTF-A deletion in macrophages attenuates pressure overload-induced cardiac fibrosis in mice
Suppression of heart function during the de-compensatory stage of cardiac hypertrophy can be attributed to adverse cardiac remodelling. Therefore, we examined the effect of macrophage-specific MRTF-A deficiency on pressure overload-induced cardiac fibrosis. First, expression levels of pro-fibrogenic genes were determined by qPCR. Compared to the WT hearts, the CKO hearts displayed marked reduction in the levels of collagen type I (Col1a1, Figure 2A), collagen type III (Col3a1, Figure 2B), α-SMA (Acta2, Figure 2C), tissue inhibitor of metalloproteinase 1 (Timp1, Figure 2D), tissue inhibitor of metalloproteinase 3 (Timp3, Figure 2E), and lysyl oxidase (Lox, Figure 2F) suggesting that MRTF-A deficiency was associated with amelioration of cardiac fibrosis. This was further supported by protein expression levels of collagen type I and α-SMA (Supplementary material online, Figure S5). Picrosirius red staining (Figure 2G) and Masson’s trichrome staining (Figure 2H) confirmed that cardiac fibrosis was dampened as a result of MRTF-A deficiency in macrophages. Cardiac hydroxyproline quantification showed that collagenous tissue was less extensive in the CKO hearts than the WT hearts (Figure 2I). In addition, measurements of caspase 3 activity showed that cell death was attenuated in the CKO hearts compared to the WT hearts (Figure 2J). We thus conclude that MRTF-A deletion in macrophages may alleviate pressure overload-induced cardiac remodeling.
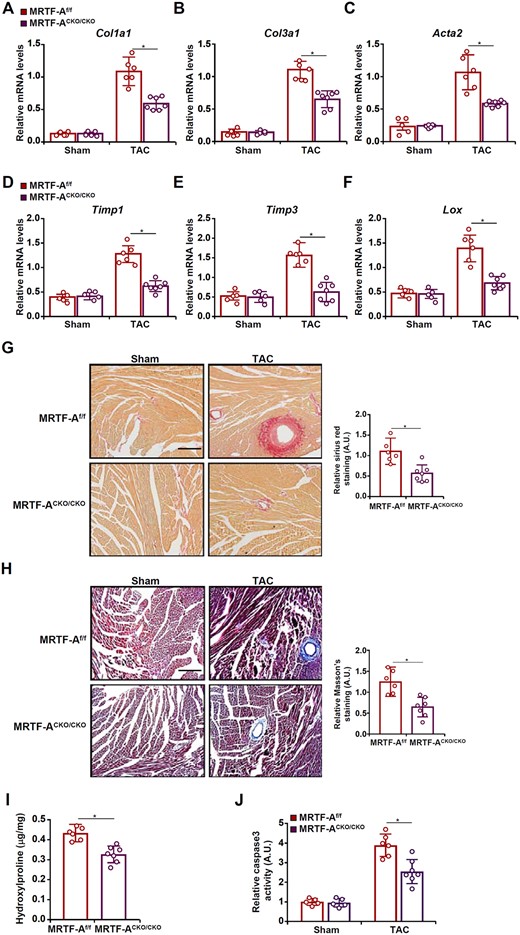
MRTF-A deletion in macrophages attenuates pressure overload-induced cardiac fibrosis in mice. Macrophage MRTF-A conditional knockout (MRTF-ACKO/CKO) mice and wild-type (MRTF-Af/f) littermates were subjected to the TAC procedure. (A–F) Fibrogenic genes were measured by qPCR. (G) Picrosirius red staining. (H) Masson’s trichrome staining. Scale bar, 50 µm. (I) Hydroxyproline levels. (J) Cell death was assessed with a colorimetric caspase 3 assay kit. N = 5 mice for the sham groups and N = 6–7 mice for the TAC groups. Data represent mean ± SD. *P < 0.05, two-tailed t-test.
3.3 MRTF-A deletion in macrophages mitigates pressure overload-induced cardiac inflammation in mice
Macrophage-mediated cardiac inflammation is considered key to the development and deterioration of pathological cardiac hypertrophy. We asked whether MRTF-A deletion in macrophages may influence cardiac inflammation in the TAC model. QPCR examination indicated that pro-inflammatory cytokines, including interleukin 1 (Il-1b, Figure 3A), interleukin 6 (Il-6, Figure 3B), and tumour necrosis factor (Tnf-a, Figure 3C), were collectively down-regulated in the CKO hearts compared to the WT hearts. In addition, several chemokines known to promote macrophage infiltration, including chemokine C–C motif ligand 1 (Ccl1, Figure 3D), Ccl2 (Figure 3E), Ccl3 (Figure 3F), Ccl4 (Figure 3G), Ccl5 (Figure 3H), Ccl8 (Figure 3I), and chemokine C-X-C ligand 1 (Cxcl1, Figure 3J), were all reduced as a result of myeloid MRTF-A deficiency. Examination of protein levels of pro-inflammatory cytokines by ELISA confirmed that MRTF-A deficiency decreased cardiac inflammation (Supplementary material online, Figure S6). These observations were probably attributable to fewer F4/80+ macrophages infiltrated in the CKO hearts than the WT hearts (Figure 3K, Supplementary material online, Figure S7). In addition, FACS analysis showed that there was a decrease in neutrophil infiltration in the CKO hearts compared to the WT hearts. On the contrary, no differences in cardiac infiltration of T lymphocytes, NK cells, or dendritic cells were observed between the WT mice and the CKO mice (Supplementary material online, Figure S8).
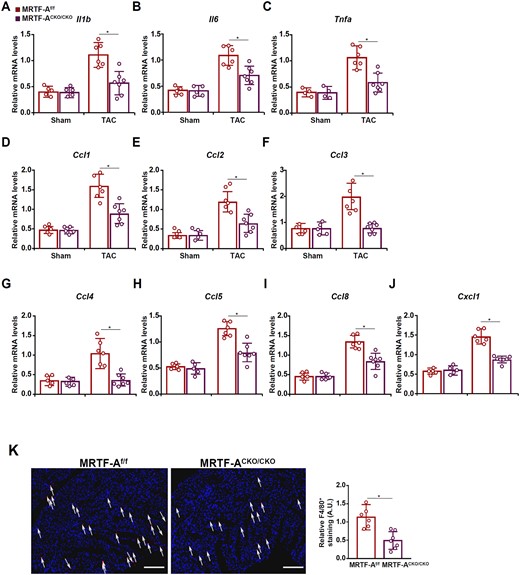
MRTF-A deletion in macrophages mitigates pressure overload-induced cardiac inflammation in mice. Macrophage MRTF-A conditional knockout (MRTF-ACKO/CKO) mice and wild-type (MRTF-Af/f) littermates were subjected to the TAC procedure. (A–J) Expression levels of pro-inflammatory cytokines and chemokines were measured by qPCR. (K) F4/80 staining and quantification. Scale bar, 50 µm. N = 5 mice for the sham groups and N = 6–7 mice for the TAC groups. Data represent mean ± SD. *P < 0.05, two-tailed t-test.
3.4 MRTF-A regulates adhesion of macrophages to endothelial cells
Circulating macrophages gain access to the heart via endothelium-dependent adhesion. Now that there was a defect in macrophage infiltration in the CKO hearts, we probed the possibility that MRTF-A deficiency might interfere with the macrophage–endothelial interaction. To test this hypothesis, we performed a series of leucocyte adhesion assays. MRTF-A knockdown by small interfering RNA (siRNA) significantly weakened the adhesion of cultured murine macrophages to endothelial cells (Figure 4A). Similarly, inhibition of MRTF-A activity with a small-molecule compound CCG-1423 crippled the adhesion of macrophages to endothelial cells (Figure 4B). We also isolated primary peritoneal macrophages from WT and MRTF-A KO mice. Compared to WT macrophages, MRTF-A KO macrophages exhibited a drastic reduction in adhesion to endothelial cells (Figure 4C).
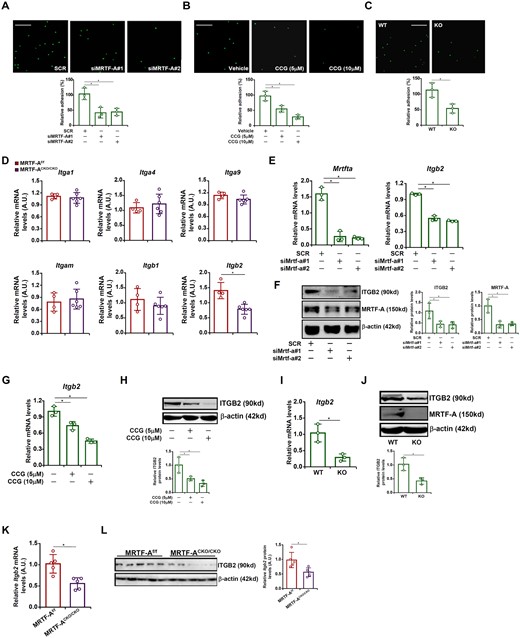
MRTF-A regulates adhesion of macrophages to endothelial cells by activating integrin β2 expression. (A) RAW cells were transfected with siRNA targeting MRTF-A or scrambled siRNA (SCR). Leucocyte adhesion assay was performed and quantified as described in Methods. Scale bar, 50 µm. N = 3. (B) Primary peritoneal macrophages were isolated from MFCKO mice and WT mice. Leucocyte adhesion assay was performed and quantified as described in Methods. Scale bar, 50 µm. N = 3. (C) RAW cells were treated with or without CCG-1423 (10 µM) for 24 h. Leucocyte adhesion assay was performed and quantified as described in Methods. Scale bar, 50 µm. N = 3. (D) Macrophage MRTF-A conditional knockout (MRTF-ACKO/CKO) mice and wild type (MRTF-Af/f) littermates were subjected to the TAC procedure. Expression levels of integrins were examined by qPCR. N = 4 mice for the MRTF-Af/f group and N = 6 mice for the MRTF-ACKO/CKO group. (E, F) RAW cells were transfected with siRNA targeting MRTF-A or scrambled siRNA (SCR). ITGB2 expression was examined by qPCR and Western. N = 3. (G, H) RAW cells were treated with or without CCG-1423 (10 µM) for 24 h. ITGB2 expression was examined by qPCR and Western. N = 3. (I, J) Primary peritoneal macrophages were isolated from MRTF-ACKO/CKO mice and WT mice. ITGB2 expression was examined by qPCR and Western. N = 3. (K, L) Primary cardiac macrophages were isolated from MRTF-ACKO/CKO mice and WT mice subjected to the TAC procedure. ITGB2 expression was examined by qPCR and Western. N = 5 mice for each group. Data represent mean ± SD. *P < 0.05, two-tailed t-test.
3.5 MRTF-A regulates integrin β2 transcription in macrophages
Previous studies have demonstrated that multiple members of the integrin (ITG) family may serve as receptors for endothelial adhesion molecules to mediate adhesion of macrophages to endothelial cells.41 QPCR profiling of ITG expression in the WT hearts and CKO hearts revealed that integrin β2 (ITGB2), but not ITGA1, or ITGA4, or ITGA9, or ITGM, or ITGB1, was down-regulated in the CKO hearts (Figure 4D). In cultured macrophages, depletion of MRTF-A by siRNAs significantly reduced ITGB2 expression at both mRNA (Figure 4E) and protein (Figure 4F) levels. Inhibition of MRTF-A by CCG-1423 also markedly repressed ITGB2 expression (Figure 4G and H). Peritoneal macrophages isolated from MRTF-A KO mice expressed less ITGB2 than those isolated from WT mice (Figure 4I and J). More importantly, ITGB2 expression was lower in primary cardiac macrophages isolated from the MRTF-A CKO hearts than from the WT hearts following the TAC procedure (Figure 4K and L). To confirm that MRTF-A relied on ITGB2 to promote macrophage adhesion, a rescue experiment was performed. MRTF-A knockdown macrophages were infected with lentivirus carrying an ITGB2 expression vector, which largely normalized the ability of macrophages to adhere the endothelial cells (Supplementary material online, Figure S9A). Similarly, ectopic ITGB2 over-expression rescued the adhesion of macrophages to endothelial cells despite MRTF-A inhibition by CCG-1423 (Supplementary material online, Figure S9B).
To determine whether regulation of ITGB2 by MRTF-A occurred at the transcriptional level, an ITGB2 promoter-luciferase construct was transfected into HEK293 cells. Over-expression of MRTF-A dose-dependently activated the ITGB2 promoter (Figure 5A). On the contrary, interference with MRTF-A activity by a dominant-negative (DN) MRTF-A (Figure 5B) or the small-molecule compound CCG-1423 (Figure 5C) suppressed the ITGB2 promoter. A close examination of the ITGB2 promoter sequence revealed a conserved binding site for the transcription factor Sp1. Sp1 levels were elevated in cardiac macrophages following the TAC procedure (Supplementary material online, Figure S1) and appeared to correlate with the induction of hypertrophic/fibrogenic/inflammatory marker genes in the heart (Supplementary material online, Figure S2). Therefore we hypothesized MRTF-A might interact with Sp1 to activate ITGB2 transcription. Indeed, co-expression of MRTF-A and Sp1 synergistically activated the ITGB2 promoter (Figure 5D). Enrichment of both Sp1 and MRTF-A was detected on the ITGB2 promoter in primary cardiac macrophages isolated from the murine hearts following the TAC procedure (Figure 5E). In accordance, mutagenesis of the conserved Sp1 site abrogated activation of the ITGB2 promoter by MRTF-A (Figure 5F), suggesting that Sp1 might be responsible for MRTF-A recruitment. In support of this notion, ChIP assay showed that Sp1 depletion significantly dampened the binding of MRTF-A to the ITGB2 promoter (Figure 5G). In addition, Re-ChIP assay confirmed that a Sp1-MRTF-A complex was clearly detectable on the ITGB2 promoter (Figure 5H). Collectively, these data demonstrate that MRTF-A likely contributes to macrophage adhesion by activating ITGB2 transcription.
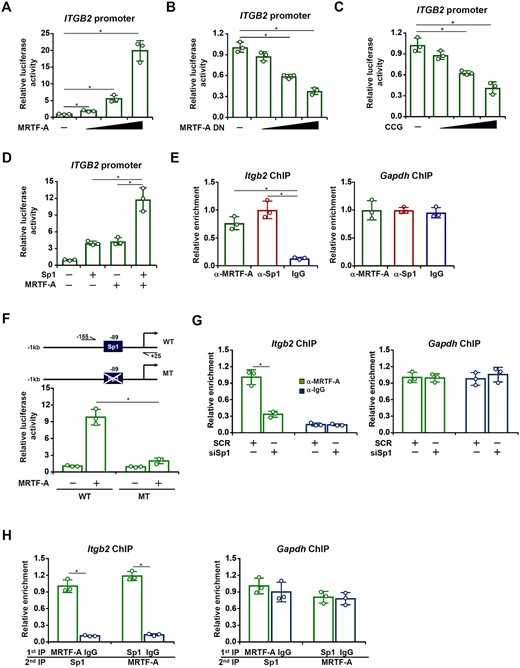
MRTF-A interacts with Sp1 to activate ITGB2 transcription. (A) An ITGB2 promoter–luciferase construct was transfected into HEK293 cells with or without MRTF-A. Luciferase activities were normalized by both protein concentration and GFP fluorescence. (B) An ITGB2 promoter–luciferase construct was transfected into HEK293 cells with or without MRTF-A DN. (C) An ITGB2 promoter–luciferase construct was transfected into HEK293 cells with or without CCG-1423. (D) An ITGB2 promoter–luciferase construct was transfected into HEK293 cells MRTF-A and/or Sp1. (E) Primary cardiac macrophages were isolated from C57BL/6 mice subjected to the TAC procedure. ChIP assays were performed with indicated antibodies. (F) Wild-type or mutant ITGB2 promoter-luciferase construct was transfected into HEK293 cells with or without MRTF-A. Luciferase activities were normalized by both protein concentration and GFP fluorescence and expressed as relative luciferase unit normalized to the control group. (G) RAW cells were transfected with siRNA targeting MRTF-A or scrambled siRNA (SCR). ChIP assays were performed with anti-MRTF-A or IgG. (H) Re-ChIP assays were performed with RAW cells lysates using indicated antibodies. N = 3 for all the experiments. Data represent mean ± SD. *P < 0.05, two-tailed t-test.
3.6 ITGB2 blockade attenuates cardiac hypertrophy in mice
In order to authenticate the functional relevance of ITGB2 (CD18) in macrophage adhesion and cardiac hypertrophy in vivo, a CD18 blocking antibody or an isotype control IgG was administered in mice following the TAC procedure. Compared to the control IgG group, mice receiving the blocking antibody displayed an attenuated phenotype of cardiac hypertrophy as evidenced by smaller heart weight/body weight ratios (Figure 6A), heart weight/tibia length ratios (Figure 6B), LSVd values (Figure 6C), and LVPWd values (Figure 6D). In addition, ITGB2 blockade partially normalized post-TAC heart functions measured by ejection fraction (Figure 6E) and fractional shortening (Figure 6F). Consistently, these changes were associated with a decline in cardiac macrophage infiltration (Figure 6G). Picrosirius red/Masson’s trichrome stainings (Figure 6H) and hydroxyproline quantifications (Figure 6I) indicated that ITGB2 blockade likely contributed to the reduction in cardiac fibrosis. QPCR analysis confirmed that ITGB2 blockade led to down-regulation of genes involved in hypertrophy, fibrogenesis, and inflammation (Figure 6J).
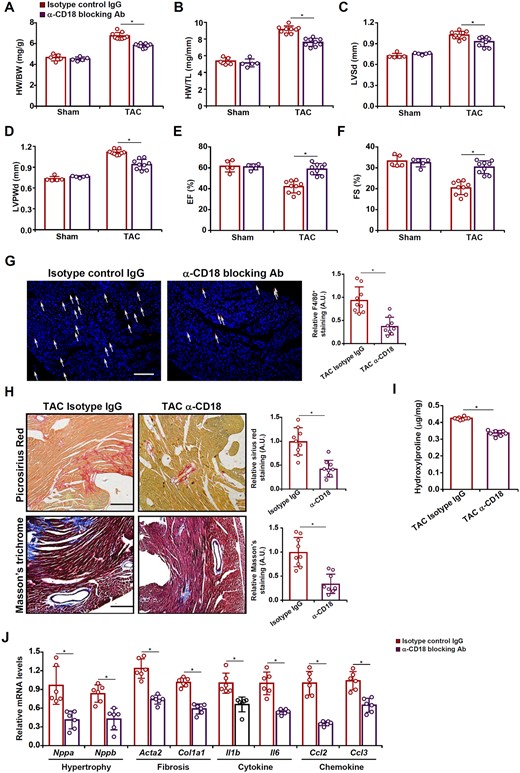
ITGB2 blockade attenuates cardiac hypertrophy in mice. C57BL/6 mice were injected with a CD18 blocking antibody or an isotype IgG following the TAC procedure. (A) Heart weight vs. body weight. (B) Heart weight vs. tibia bone length. (C) LVSd values. (D) LVPWd values. (E) EF values. (F) FS values. (G) F4/80 staining. (H) Picrosirius red staining and Masson’s trichrome staining. (I) Hydroxyproline levels. (J) Gene expression levels were examined by qPCR. N = 5 mice for the sham groups and N = 9 mice for the TAC groups. Data represent mean ± SD. *P < 0.05, two-tailed t-test.
3.7 MRTF-A recruits KDM3A to activate ITGB2 transcription
Since MRTF-A is known to regulate transcription by engaging the epigenetic machinery, a series of ChIP assays were performed to determine whether MRTF-A deficiency would alter the chromatin structure surrounding the ITGB2 promoter. As shown in Supplementary material online, Figure S10A and B, acetylation levels of histone H3 and H3K9, usually associated with actively transcribed chromatin, were down-regulated on the ITGB2 promoter, but not the GAPDH promoter, in cells depleted of MRTF-A. In contrast, dimethyl H3K9, an antagonistic marker of acetyl H3K9 and usually associated with repressed chromatin, was increased on the ITGB2 promoter following MRTF-A knockdown (Supplementary material online, Figure S10C). Congruently, the binding of KDM3A, a dedicated H3K9 demethylase, was weakened on the ITGB2 promoter in MRTF-A deficient macrophages (Supplementary material online, Figure S10D). Treatment of the MRTF-A inhibitor CCG-1423 also down-regulated H3K9 acetylation (Supplementary material online, Figure S10E) but up-regulated H3K9 dimethylation (Supplementary material online, Figure S10F) on the ITGB2 promoter, which might be accounted for by a reduction in KDM3A binding (Supplementary material online, Figure S10G). Similar observations were made in primary peritoneal macrophages isolated from the MRTF-A KO mice and the WT mice: levels of acetyl H3K9 (Supplementary material online, Figure S10H) were lower whereas levels of dimethyl H3K9 (Supplementary material online, Figure S10I) were higher on the ITGB2 promoter in MRTF-A KO macrophages compared to WT macrophages. KDM3A binding to the ITGB2 promoter was significantly weaker in the KO macrophages than in the WT macrophages (Supplementary material online, Figure S10J). More importantly, ChIP assays performed with primary cardiac macrophages showed diminished acetyl H3K9 levels (Supplementary material online, Figure S10K) and elevated dimethyl H3K9 levels (Supplementary material online, Figure S10L) accompanying reduced KDM3A recruitment (Supplementary material online, Figure S10M) on the ITGB2 promoter in the CKO hearts than in the WT hearts. Co-immunoprecipitation assay (Supplementary material online, Figure S10N) and Re-ChIP assay (Supplementary material online, Figure S10O) confirmed that MRTF-A and KDM3A formed a complex.
3.8 KDM3A regulates ITGB2 transcription and macrophage adhesion
Finally, the functional relevance of the MRTF-A-KDM3A interplay was evaluated. Reporter assay showed that whereas KDM3A alone was unable to influence the ITGB2 promoter activity, it enhanced the activation of the ITGB2 promoter by MRTF-A (Figure 7A). Silencing KDM3A by siRNA down-regulated ITGB2 expression at both mRNA (Figure 7B) and protein (Figure 7C) levels. Consistently, KDM3A knockdown was accompanied by an increase in H3K9 dimethylation (Figure 7D) and a simultaneous decrease in H3K9 acetylation (Figure 7E) on the ITGB2 promoter. Finally, KDM3A depletion in macrophages disrupted their adhesion to the endothelial cells (Figure 7F). Combined, these data suggest that MRTF-A recruits KDM3A to activate ITGB2 transcription and promote macrophage adhesion.
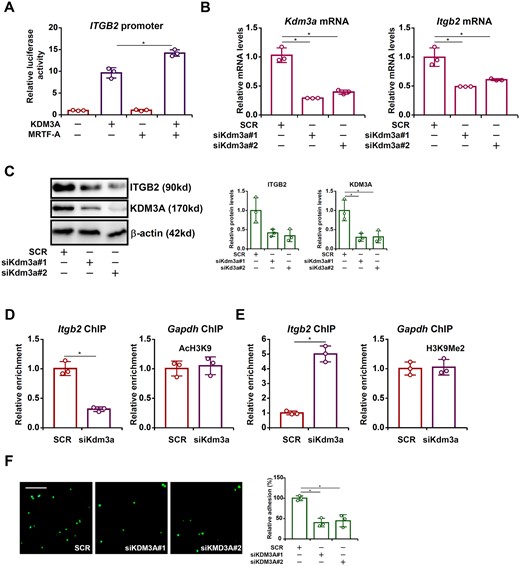
KDM3A regulates ITGB2 transcription and macrophage adhesion. (A) An ITGB2 promoter-luciferase construct was transfected into HEK293 cells MRTF-A and/or KDM3A. (B–F) RAW cells were transfected with siRNA targeting KDM3A or scrambled siRNA (SCR). ITGB2 expression was examined by qPCR and Western. ChIP assays were performed with anti-acetyl H3K9 and anti-dimethyl H3K9. Leucocyte adhesion assay was performed and quantified as described in Methods. Scale bar, 50 µm. N = 3 for all the experiments. Data represent mean ± SD. *P < 0.05, two-tailed t-test.
4. Discussion
Cardiac hypertrophy is defined as an outgrowth in size of cardiomyocytes.This process, critical to the development of heart failure, is hardly an autonomous one solely reliant on the events taking place in the muscle cells. Instead, different types of non-cardiac cells, including endothelial cells, myeloid cells, adipose cells, and fibroblast cells, have been proposed to play a role in the regulation of cardiac hypertrophy.42 Macrophages can potentially contribute to the pathogenesis of cardiac hypertrophy by producing and releasing a host of pro-inflammatory mediators, including TNF-α,43,44 IL-1β,45 and IL-6,46 which act on cardiomyocytes to stimulate the hypertrophic response. Though pointed out by Kuwahara et al.24 that MRTF-A can contribute to transcriptional activation of both ANP and BNP in cardiomyocytes, the exclusive reliance on a systemic MRTF-A knockout (KO) animal model in that investigation did not definitively answer the question as to whether the ability of MRTF-A to promote cardiac hypertrophy in vivo can be fully accounted for by its regulation of pro-hypertrophic response in the myocardium. In fact, our previous investigation has shown that endothelial-specific MRTF-A depletion, with a Tie2-derived shRNA system, attenuated pressure overload-induced cardiac hypertrophy in mice.25 We show here that myeloid-derived MRTF-A mediates the interaction between circulating macrophages and the vasculature thereby promoting cardiac inflammation and pathological hypertrophy (Figure 7G). This model, of course, does not foreclose the possibility that MRTF-A could directly regulate the synthesis of pro-inflammatory mediators per se in macrophages. Indeed, our previously published studies have provided unequivocal evidence that MRTF-A, presumably via interacting with the master regulator of pro-inflammatory transcription NF-κB, can bind to the promoters of pro-inflammatory mediator genes (e.g., Il1b, Tnfa, and Il6) and activate transcription by recruiting an H3K4 methyltransferase complex in macrophages.22,26,47 Thus, it is likely that MRTF-A could contribute to cardiac inflammation and hypertrophy by simultaneously modulating the abundance of macrophage infiltration and the magnitude of pro-inflammatory mediator production.
There still exist possibilities that fibroblast-derived or adipose-derived MRTF-A may also contribute to the pathogenesis of cardiac hypertrophy. For instance, MRTF-A is a known activator of chemokines.22 It has been previously suggested that cardiac fibroblast cells can produce abundant chemokines in response to mechanic stretch.48 Therefore, it is tempting to speculate that MRTF-A may promote cardiac inflammation and consequently hypertrophy by modulating the production of chemokines in fibroblasts to stimulate immune cell homing. Alternatively, Nobusue et al.49 have observed that MRTF-A may regulate adipose cell differentiation and adipokine production in vitro. Since a wide range of adipokines have been demonstrated to exert modulatory effects on cardiac hypertrophy,50 it is reasonable to conjecture that attenuation of cardiac hypertrophy in MRTF-A-null mice may be reflected by an altered adipokine secretome. These alternative scenarios, yet to be tested in new animal models, combined with our data summarized here reinforce the notion that MRTF-A possesses cell-specific roles in regulating cardiac hypertrophy and that targeting MRTF-A may lead to multiple, instead of a singular, beneficial effects.
One of the highlights regarding the current report is that the histone H3K9 demethylase KDM3A, in cooperation with MRTF-A, regulates ITGB2 transcription to promote macrophage adhesion. Few investigations in the past have examined the role of KDM3A in regulating macrophage function. Instead, there is evidence to suggest that KDM3A may indirectly regulate macrophage trafficking. For instance, Osawa et al.51 have observed that hypoxia induces KDM3A expression in epithelial cells, which leads to up-regulation of chemokines and macrophage infiltration. Zhang et al.52 have also observed that endothelial KDM3A promotes macrophage homing by activating the transcription of colony-stimulating factor 1 (CSF1). Of interest, it has been reported that hypoxia can directly stimulate the expression of KDM3A in macrophages, which presumably is responsible for the up-regulation of pro-inflammatory cytokines.53 Recently, Liu and colleagues have presented evidence to show that systemic KDM3A over-expression promotes whereas KDM3A knockout attenuates cardiac hypertrophy in mice.54 Mechanistically, these authors focused exclusively on a cardiomyocyte-specific role of KDM3A to account for the observed phenotypes without addressing the cell-specific roles. Similar to MRTF-A, KDM3A is ubiquitously expressed.55 Thus, whether non-cardiac cell KDM3A may contribute to pathological cardiac hypertrophy remains unknown and certainly deserves further studies.
Despite the advances proffered by the present study with regard to the regulation of cardiac hypertrophy by myeloid MRTF-A, a few limitations dampen its significance. First, the LyzM-Cre driver used in this study is not specific for macrophages; Cre activity driven by the LyzM promoter can be detected in all granulocytes and some dendritic cells.56 Because MRTF-A has a role in the migration and/or adhesion of neutrophils57 and dendritic cells,58 the observed phenotype of MRTF-A CKO mice cannot be simply be accounted for by the deficiency of macrophage adhesion. Indeed, our data showed that MRTF-A deficiency led to decreased neutrophil infiltration in the heart (Supplementary material online, Figure S8). Therefore, it is likely that MRTF-A may regulate cardiac inflammation and hypertrophy by modulating the behaviours of multiple immune cell lineages. Second, dynamic changes in macrophage composition/phenotype are observed in and contribute to the development and progression of cardiac hypertrophy. It remains undetermined whether MRTF-A may play a role in this process. Previously An et al.59 have reported that MRTF-A over-expression in macrophages, achieved through the CD11b-Cre driver, promotes colitis in mice likely by skewing classic macrophage polarization although no mechanistic insights were provided. The only published study investigating the genome-wide binding pattern of MRTF-A reveals that MRTF-A displays preferences for several DNA motifs of sequence-specific transcription factors including STAT.60 Because STAT proteins play a prominent role determining macrophage phenotype, it is plausible to speculate that MRTF-A could influence macrophage composition/phenotype in the context of cardiac hypertrophy (and possibly other macrophage-associated pathophysiological processes) by interacting with STATs. This lingering issue certainly warrants further attention.
In summary, our data allude to a novel mechanism whereby the MRTF-A-KDM3A axis in macrophages may contribute to cardiac hypertrophy. Since neither MRTF-A nor KDM3A is indispensable for normal heart functions, small-molecule inhibitors against MRTF-A and/or KDM3A can be considered as potentially safe and effective therapeutic solutions to prevent heart failure.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
Y.X. conceived the project; L.L., M.K., Q.W.Z., L.M., and Y.Y. designed experiments, performed experiments, collected data, and analysed data; Y.X. wrote the manuscript with input from the others; Y.Y. and Y.X. secured funding and provided supervision.
Conflict of interest: none declared.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81725001 and 81870326), Priority Academic Program Development of Jiangsu Higher Education Institutions, Six Talent Peaks Project of Jiangsu Province, Key Laboratory of Emergency and Trauma (Hainan Medical University), Ministry of Education (KLET-201915).
Data availability
The data and analytic methods will be made upon reasonable request. The study materials will only be made available if sufficient material can be provided to other researchers for purposes of reproducing the results or replicating the procedure.
References
We report a novel mechanism whereby myocardin-related transcription factor A (MRTF-A) regulates macrophage trafficking in pathological cardiac hypertrophy. Given that MRTF-A has a well-established role in regulating hypertrophic response in cardiomyocytes, our data provide a renewed rationale for targeting MRTF-A in the prevention and treatment of heart failure.
Author notes
The first four authors contributed equally to the study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}