





This editorial refers to ‘Myocardin-related transcription factor A regulates integrin beta 2 transcription to promote macrophage infiltration and cardiac hypertrophy in mice’ by L. Liu et al., pp. 844–858.
Cardiac hypertrophy induced by haemodynamic overload is initially compensatory but eventually triggers maladaptive responses leading to heart failure.1 Inflammation is one of the most important mediators of the transition from adaptive to maladaptive cardiac hypertrophy during pressure overload.2 Pressure overload induces the recruitment of macrophages into the heart, which, in turn, triggers cytokine production, T-cell expansion, and adverse cardiac remodelling. Previous studies have shown that non-resident CCR2+ macrophages, rather than resident macrophages, mediate pathological hypertrophy during the late phase of pressure overload.3,4 Intervention to attenuate the recruitment and activation of CCR2+ macrophages is effective in preventing the development of heart failure in response to pressure overload. CCR2+ macrophages are up-regulated and recruited to the heart by CCL2, CCL7, and CCL12 chemokines.4 However, the detailed intracellular signalling mechanisms mediating the recruitment of non-resident macrophages have been unclear.
Liu et al. have shown that myocardin-related transcription factor A (MRTF-A)-mediated epigenetic regulation of integrin beta 2 transcription in LyzM-positive myeloid cells, including macrophages, plays an important role in both macrophage infiltration and pathological remodelling during pressure overload.5 The study provides important insights into the underlying mechanism of macrophage recruitment and its functional significance during pressure overload-induced cardiac hypertrophy. MRTF-A, a transcription factor co-factor, not only interacts with transcription factors, including Sp1, NF-κB, and STAT, but also recruits molecules involved in histone modification, including KDM3, a histone H3K9 demethylase, thereby allowing epigenetic regulation of genes involved in inflammation. Liu et al. demonstrated that macrophages utilize an epigenetic mechanism to facilitate their infiltration into the heart, thereby enabling prolonged activation of adverse cardiac remodelling (Figure 1). To my knowledge, this is the first study to identify a nuclear signalling mechanism involved in macrophage infiltration in the heart in vivo. The study provides an important hint to alleviating pathological remodelling during pressure overload.
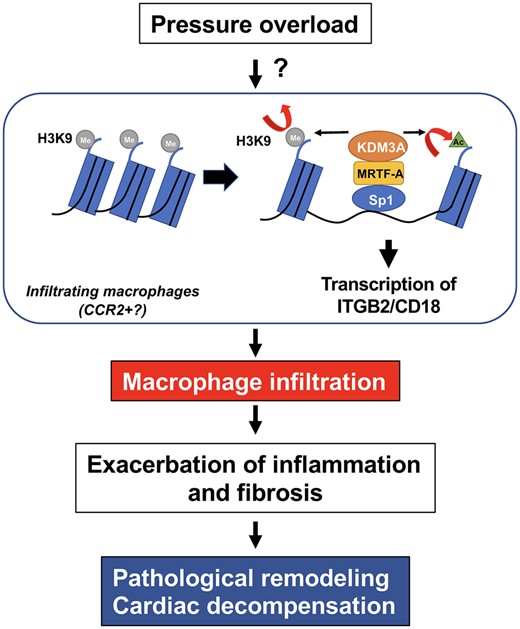
Pressure overload induces pathological remodelling in the heart through activation of an epigenetic mechanism, namely H3K9 demethylation and H3K9 acetylation, in macrophages where MRTF-A, a transcription factor co-factor, and KDM3A, a histone demethylase, are involved in this process. Up-regulation of ITGB2 promotes adhesion of macrophages to endothelial cells, which in turn promotes pathological remodelling in the heart. Detailed molecular mechanisms through which pressure overload activates the MRTF-A-KDM3A pathway remains to be elucidated. MRTF-A activates transcription of ITGB-2 through Sp1. The MRTF-A-KDM3A pathway may also activate other mechanisms of pathological hypertrophy through interaction of MRTF-A and other transcription factors, including NF-κB.
Although the study provides compelling evidence to support the involvement of macrophage MRTF-A during the development of pathological hypertrophy, important questions remain. Previous studies have shown that MRTF-A in other cell types is also involved in cardiac hypertrophy and cell growth responses.6 Thus, the relative importance of MRTF-A in macrophages vs. other cardiac cell types remains to be elucidated. If MRTF-A in macrophages, rather than in other cell types, is absolutely required, it would be interesting to clarify why macrophage MRTF-A is so important. In addition, since MRTF-A is involved in a wide variety of functions, including generation of cytokines and extracellular matrixes,7 it is possible that the effect of MRTF-A upon macrophage infiltration may also be mediated by other cellular mechanisms besides integrin beta 2.
It is becoming increasingly clear that non-resident macrophages comprise multiple distinct cell populations.2 Considering the importance of CCR2+ non-residential macrophages in mediating pathological hypertrophy, it is important to demonstrate the role of MRTF-A in CCR2+ macrophages. It would also be interesting to clarify whether MRTF-A is involved in all subpopulations of macrophages.
Liu et al. demonstrated that MRTF-A induces KDM-3A-induced H3K9-demethylation and that concurrent induction of H3K9-acetylation promotes Sp-1-mediated transcription of beta 2 integrin.5 Since MRTF-A also interacts with other transcription factors, including NF-kB, and epigenetic regulators, the molecular mechanism mediating the specificity of MRTF-A, namely the involvement of KDM-3A and Sp-1, should be clarified. It would also be important to show how the activity and the level of endogenous MRTF-A are regulated in the heart in response to pressure overload. This may involve chemokines such as CCL2 or other known mechanisms, including actin-associated proteins and miRNA. Knowledge obtained from such experiments could be useful to control endogenous MRTF-A and thereby alleviate the progression of heart failure.
In summary, Liu et al. demonstrated the involvement of the MRTF-A-KDM-3A pathway in mediating macrophage infiltration and pathological hypertrophy in response to pressure overload. The study suggests that MRTF-A and KDM-3A in macrophages could be promising targets to mitigate the progression of pressure overload-induced cardiac hypertrophy to heart failure. Given that MRTF-A and KDM-3A have multiple cellular functions in many cell types, it is essential to target the treatment to the right cell type, such as CCR2+ macrophages, with the right timing, namely during the transition to pathological remodelling.
The opinions expressed in this article are not necessarily those of the Editors of Cardiovascular Research or of the European Society of Cardiology.
Acknowledgement
The author thanks Daniela Zablocki for critical reading of the manuscript.
Conflict of interest: The authors declare no conflict of interest.
Funding
This work was supported in part by U.S. Public Health Service Grants HL67724, HL91469, HL102738, HL112330, HL138720, HL144626, HL150881, and AG23039, American Heart Association Merit Award 20 Merit 35120374, and the Fondation Leducq Transatlantic Network of Excellence 15CBD04.

{kind=link}