





Abstract
Pulmonary hypertension (PH) is a common complication of left heart disease (LHD, Group 2 PH) leading to right ventricular (RV) failure and death. Several loss-of-function (LOF) mutations in KCNK3 were identified in pulmonary arterial hypertension (PAH, Group 1 PH). Additionally, we found that KCNK3 dysfunction is a hallmark of PAH at pulmonary vascular and RV levels. However, the role of KCNK3 in the pathobiology of PH due to LHD is unknown.
We evaluated the role of KCNK3 on PH induced by ascending aortic constriction (AAC), in WT and Kcnk3-LOF-mutated rats, by echocardiography, RV catheterization, histology analyses, and molecular biology experiments. We found that Kcnk3-LOF-mutation had no consequence on the development of left ventricular (LV) compensated concentric hypertrophy in AAC, while left atrial emptying fraction was impaired in AAC-Kcnk3-mutated rats. AAC-animals (WT and Kcnk3-mutated rats) developed PH secondary to AAC and Kcnk3-mutated rats developed more severe PH than WT. AAC-Kcnk3-mutated rats developed RV and LV fibrosis in association with an increase of Col1a1 mRNA in right ventricle and left ventricle. AAC-Kcnk3-mutated rats developed severe pulmonary vascular (pulmonary artery as well as pulmonary veins) remodelling with intense peri-vascular and peri-bronchial inflammation, perivascular oedema, alveolar wall thickening, and exaggerated lung vascular cell proliferation compared to AAC-WT-rats. Finally, in lung, right ventricle, left ventricle, and left atrium of AAC-Kcnk3-mutated rats, we found a strong increased expression of Il-6 and periostin expression and a reduction of lung Ctnnd1 mRNA (coding for p120 catenin), contributing to the exaggerated pulmonary and heart remodelling and pulmonary vascular oedema in AAC-Kcnk3-mutated rats.
Our results indicate that Kcnk3-LOF is a key event in the pathobiology of PH due to AAC, suggesting that Kcnk3 channel dysfunction could play a potential key role in the development of PH due to LHD.

1. Introduction
Pulmonary hypertension (PH), which constitutes an abnormal increase in blood pressure in the pulmonary circulation, has seen its definition revised during the last PH world symposium, as mean pulmonary arterial pressure (mPAP) >20 mmHg.1 PH can be divided into five sub-groups, being Group 1, or pulmonary arterial hypertension (PAH). Other forms of PH, such as PH due to left heart disease (PH-LHD), have much higher incidence and prevalence, and although constitutes one of the most common forms of PH, roughly representing 50% of PH cases.2 However, molecular mechanisms involved in the development of PH-LHD are poorly documented.
The pathophysiology of PH-LHD is complex and is caused by several mechanisms: (i) an initial passive increase in left ventricular (LV) filling pressures more specifically left atrial (LA) pressure; (ii) pulmonary artery endothelial dysfunction; (iii) pulmonary vascular (artery and vein) remodelling; and (iv) right ventricular (RV) dysfunction and altered RV/pulmonary artery coupling.2
Pulmonary endothelial dysfunction seems to play a central role in PH-LHD in both preclinical models3–5 and patients.6 Endothelium-derived vasorelaxation is markedly impaired in pulmonary arteries from animals with PH-LHD induced by ischaemic7 or pressure overload conditions.3 Similarly, exacerbated pulmonary arterial and venous remodelling is observed in patients with both heart failure (HF) with reduced or preserved (HFpEF) ejection fraction,8,9 mostly explained by endothelial dysfunction and an imbalance between vasodilation/vasoconstriction and growth factor signalling.10 To date, most of the molecular pathways associated with PH-LHD are assumed to be common with PAH namely endothelial dysfunction, mitochondrial dysfunction, exacerbated inflammation.11 In PH-LHD, endothelial dysfunction is at least characterized by an increased endothelin-1 (ET-1) production in the pulmonary vasculature,12 as well as in ascending aortic constriction (AAC)-induced PH in rat.4 ET-1 inhibits KCNK3 function in pulmonary artery smooth muscle cell (PASMC) through Rho kinase-mediated phosphorylation,13 linking one of the main modulators of PH-LHD (ET-1) and KCNK3-dysfunction. KCNK3 protein, also known as TASK-1 (Twik-related-acid-sensitive-K+ channel) is an outward-K+ channel. KCNK3 regulates the resting membrane potential in several cell types, including PASMCs14 and RV cardiomyocytes.15 The role of KCNK3 dysfunction in the development of PAH was previously highlighted by the identification of several loss-of-function (LOF) mutations in the KCNK3 gene, in PAH patients.16,17 In PH Group 1, 16 different mutations were identified in PAH patients, 93.6% of them were missense mutations, most of them were heterozygous, and only one patient was identified with a homozygous mutation,16 and by the fact that KCNK3 mutation carriers’ are younger and have higher mPAP at diagnosis.14
We have recently developed the first Kcnk3-mutated rat line with a Kcnk3-LOF-mutation (Kcnk3Δ94ex1/Δ94ex1), which develops exaggerated PH when challenged with either monocrotaline or chronic hypoxia.18 Furthermore, Kcnk3-LOF-mutated rats are predisposed to pulmonary artery constriction, and have compromised endothelium-dependent and independent relaxation.18
Assuming the importance of KCNK3 in the modulation of pulmonary vascular tone, and the fact that endothelial dysfunction in PH-LHD should induce KCNK3 dysfunction, we hypothesize that KCNK3 dysfunction could be a key player in PH-LHD pathobiology and that Kcnk3-mutated rats are more susceptible to the development of PH secondary to LHD induced by LV pressure overload (AAC).
2. Methods
Due to space limitation, an expanded methods section is provided in the Supplementary material online.
2.1 Animals and surgical procedures
The animal facility is licensed by the French Ministry of Agriculture (agreement N° C92-019-01). This study was approved by the Committee on the Ethics of Animal Experiments (CEEA26 CAP Sud). The animal experiments were approved by the French Ministry of Higher Education, Research and Innovation (N°#7757). The animal experiments were performed conforming to the guidelines from Directive 2010/63/EU on 22 September 2010 of the European Parliament on the protection of animals used for scientific purposes and complied with the French institution’s guidelines for animal care and handling.
AAC was induced in rats under anaesthesia and analgesia, by placing a stainless steel hemoclip (0.6 mm internal diameter) on the ascending aorta via thoracic incision. The surgical procedure was carried out on 3-week-old rats (WT/Kcnk3Δ94ex1/Δ94ex1, Kcnk3Δ94ex1/+ rats), under anaesthesia with a ketamine-xylazine mix (respectively, 75–10 mg/kg and 0.3 mL/100 g, i.p.). Buprenorphine chlorhydrate (0.03 mg/kg, 0.2 mL/100g, s.c.) was administered twice daily for 3 days beginning at the end of the surgery. Age-matched Sham-operated animals (WT and Kcnk3-mutated) underwent the same procedure without placement of the clip. Echocardiographic measurement and closed-chest RV catheterization were performed in operated (AAC and Sham) animals 7 weeks after surgery. After RV catheterization, under profound anaesthesia (isoflurane 5%), the animals were euthanized by exsanguination (section of the descending abdominal aorta) and tissues harvest were performed in all animals.
2.2 Statistical analyses
All statistical tests were performed using GraphPad Prism software (GraphPad, version 8.0 for Windows). All data were verified for normal distribution using Shapiro–Wilk normality. Positively skewed data was log-transformed for analysis, while for clarity reasons graphical representations are shown in their original scale. All values are reported as mean ± standard error of the mean. For experiments with more than five samples, the difference between groups was assessed by two-way ANOVA analysis completed by Sidak’s multiple comparisons test for post hoc analyses. For experiments with less than five samples, the difference between two groups was assessed by one-way ANOVA analysis completed by Dunn’s multiple comparisons test for post hoc analyses after non-parametric ANOVA (Kruskal–Wallis). Differences were considered statistically significant at P-values <0.05.
3. Results
3.1 AAC-induced LV pressure overload leads to concentric hypertrophy
To determine the role of Kcnk3 channel in the development PH-LHD, we attempted to induce PH secondary to LV pressure overload by performing ascending aorta constriction (AAC), in WT and Kcnk3-mutated rats (homozygous and heterozygous Kcnk3-mutated rat, Kcnk3Δ94ex1/Δ94ex1 and Kcnk3Δ94ex1/+, respectively) as compared to littermate Sham animals, as previously described.19 For clarity reasons, we present all results obtained in heterozygous Kcnk3-mutated rats in the Supplementary material online.
Three weeks old animals were submitted to AAC, and after 7 weeks, cardiac structure and function was evaluated by echocardiography. LV cavity sizes remained within the normal range, resulting in unaltered LV ejection fraction (LVEF) in our different genotypes (Figure 1A and B). AAC resulted in significant LV wall hypertrophy [as evaluated by LV wall thickness, as relative wall thickness] was also increased (Figure 1C). LV hypertrophy were confirmed at the whole organ and cardiomyocyte level (Figure 1D–F), and revealed a concentric hypertrophy pattern (Figure 1G), in both WT and Kcnk3-mutated animals submitted to pressure overload (AAC). Similar results were obtained in heterozygous Kcnk3-mutated rats (see Supplementary material online, Figure S1).
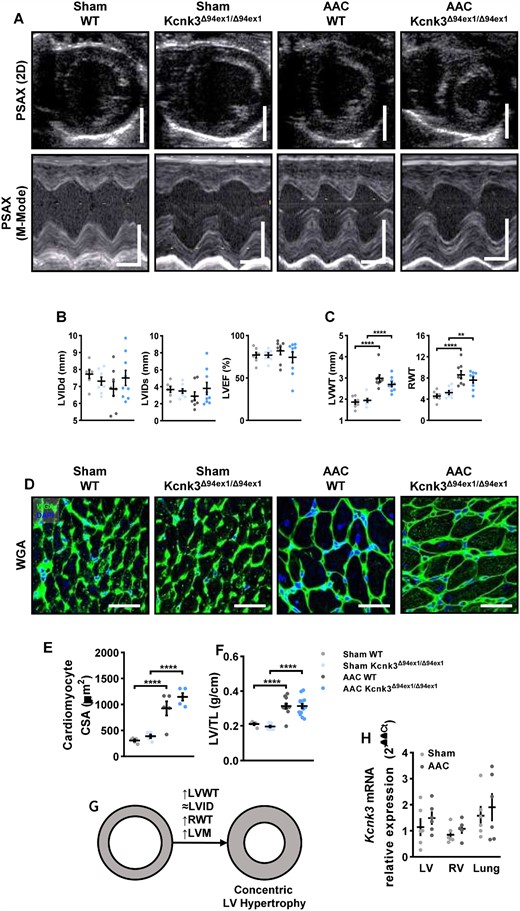
AAC-induced LV pressure overload leads to concentric hypertrophy. (A) Parasternal short axis (PSAX) projection with M-mode was used to quantify left ventricle structure and function. Scale bars, 5 mm (upper panels), horizontal bars represent 100 ms. (B) LViDd, LVIDs, LVEF in LV from Sham-WT (n = 7), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 7–8), AAC-WT (n = 8), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 9). (C) LVWT, RWT in LV from Sham-WT (n = 7), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 8), AAC-WT (n = 8), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 9). (D) Immunofluorescence images of LV sections stained with FITC-conjugated wheat germ agglutinin (WGA, 50 μg/mL, green) and 4′,6-diamidino-2-phenylindole (DAPI, blue). Scale bar, 50 μm. (E) Quantification of cardiomyocytes cross section area (CSA, five different rats for each group). (F) LV/TL in Sham-WT (n = 5), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 10), AAC-WT (n = 11), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 15). (G) Schematic representation of concentric LV hypertrophy. (H) Relative mRNA expression (2−ΔΔCt) of kcnk3 in LV, RV, lung from Sham-WT (n = 5–6), and AAC-WT rats (n = 5–6). Experiments were normalized to 18S mRNA and statistical analysis performed using ΔCt values. Data are represented as scatter dot plots, with mean ± SEM. Experiments presented in panels A–D and F–H were analysed using two-way ANOVA followed by Sidak’s post hoc test, and experiments presented in panel E were analysed using one-way ANOVA followed by Dunn’s post hoc test after Kruskal–Wallis test, **P < 0.01, ****P < 0.0001. LVEF, LVWT, cardiomyocyte CSA, and LV/TL were not normally distributed and were log-transformed prior to statistical analysis.
Of note, the aortic mean and peak gradient across the constriction were not different between WT and homozygous or heterozygous Kcnk3-mutated animals (see Supplementary material online, Figure S2). No changes in Kcnk3 mRNA expression were detected in either ventricles or lung after AAC-surgery (Figure 1H).
3.2 Kcnk3-LOF mutation leads to LV diastolic dysfunction
Despite similar LV hypertrophic responses and preserved LVEF, diastolic function was compromised in Kcnk3-mutated rats (Figure 2A). Doppler-derived trans-mitral flow showed an increase in peak early diastolic flow velocity (E-wave) in AAC-Kcnk3Δ94ex1/Δ94ex1 rats (Figure 2B). Furthermore, tissue Doppler imaging-derived mitral annular early diastolic peak myocardial velocity (e′) was significantly decreased in Kcnk3-mutated rats (Sham or AAC conditions), resulting in a quasi-significant increase in the E/e′ ratio in Kcnk3-mutated rats, suggesting increased LV filling pressures. Myocardial relaxation dynamics (IVRT, Figure 2C) were affected by surgery, but was uninfluenced by the genotype. A similar pattern was observed in Kcnk3Δ94ex1/+ rats, in which E/e′ was significantly increased, despite an apparently e′ velocity (see Supplementary material online, Figure S3A and B). The diastolic alterations present in Sham-Kcnk3-mutated rats suggest that Kcnk3-LOF mutation compromises myocardial compliance, predisposing Kcnk3-mutated rats to diastolic dysfunction.
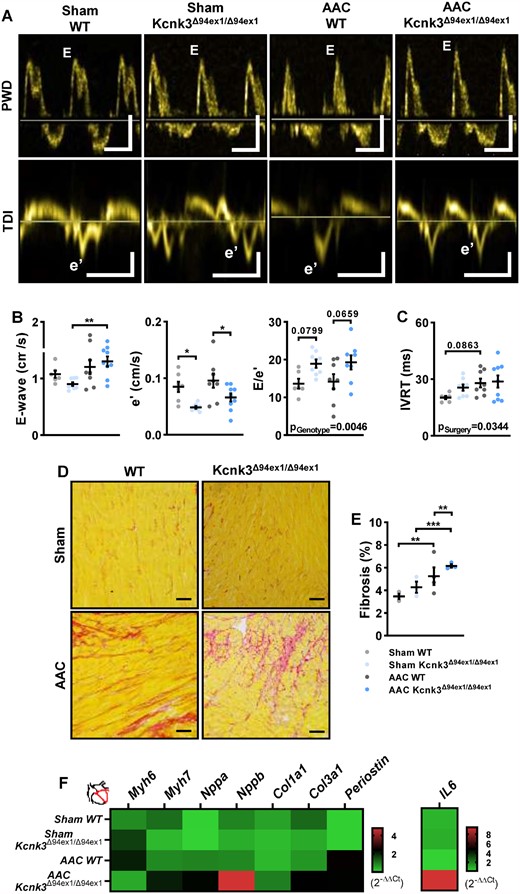
Kcnk3-LOF mutation leads to LV diastolic dysfunction. (A) Mitral inflow and mitral annulus velocity analysis, using pulse wave Doppler (PWD) and tissue-Doppler imaging (TDI). Vertical bars, 50 (upper) and 5 (lower panel) cm/s, horizontal bars represent 100 ms. (B) Left panel: E-wave, middle panel: e′, right panel: E/e′ from Sham-WT (n = 6–7), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 7–8), AAC-WT (n = 7–8), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 8–9). (C) IVRT from Sham-WT (n = 5), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 8), AAC-WT (n = 8), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 9). (D) Representative images of interstitial collagen deposition in LV tissue sections stained with sirius red from WT and Kcnk3-mutated sham or AAC rats. Scale bar, 100 µm. (E) Analysis of the percentage of LV interstitial collagen staining in Sham-WT (n = 3), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 3), AAC-WT (n = 4), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 3). (F) Relative mRNA expression (2−ΔΔCt) of Myh6, Myh7, Nppa, Nppb Col1a1, Col3a1, Periostin, and IL-6 (heatmap representation of the mean) in LV from Sham-WT (n = 6), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 5), AAC-WT (n = 6), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 6). Experiments were normalized by 18S mRNA and analysed using ΔCt values (for a full statistical analysis refer to the Supplementary material online). Data are represented as scatter dot plots, with mean ± SEM. Experiments presented in panels A–D and F were analysed using two-way ANOVA followed by Sidak’s post hoc test, and experiments presented in panel E were analysed using one-way ANOVA followed by Dunn’s post hoc test after Kruskal–Wallis test. *P < 0.05, **P < 0.01, and ***P < 0.001. Fibrosis was not normally distributed and was log-transformed prior to statistical analysis.
Underlying LV diastolic dysfunction, LV fibrosis was present in AAC rats, as shown by an increased proportion of interstitial fibrotic deposition (Figure 2D and E), even more pronounced in homozygous (Figure 2E) and heterozygous (see Supplementary material online, Figure S3C) AAC-Kcnk3-mutated rats. This increase was associated with an up-regulation of pro-fibrotic transcripts (Col3a1 and Periostin) in left ventricle from AAC-Kcnk3-mutated rats (Figure 2F, and see Supplementary material online, Table S1). Also associated with diastolic dysfunction, the left ventricle of AAC-Kcnk3Δ94ex1/Δ94ex1 rats (re)expressed the pathological hypertrophic markers atrial and brain natriuretic peptides (Nppa and Nppb), and shifted towards the HF prone myosin heavy chain beta gene (Myh7) (Figure 2F, and see Supplementary material online, Table S1). The pro-inflammatory cytokine, Il6, gene expression was up-regulated (six-fold).
3.3 Left atrial dysfunction underlines Kcnk3 LOF mutation-associated with LV diastolic dysfunction
In parallel to LV stiffness, we observed a significant dilation of the left atrium, associated with compromised LA function evaluated by LA emptying fraction (Figure 3A–C, and see Supplementary material online, Figure S3D and E), in homozygous and heterozygous Kcnk3-mutated rats submitted to AAC. As previously shown in human and rat heart,20 we observed an atrial predominance for Kcnk3 expression (Figure 3D). This indicates that KCNK3 plays an important role in modulating LA function, and its LOF could affect atrioventricular flow, impaired in Sham-Kcnk3-mutated rats (Figure 2B, see Supplementary material online, Figure S3A). Indeed, altered gene expression, with activation of pro-remodelling signalling in AAC-Kcnk3Δ94ex1/Δ94ex1 rats, could either trigger or result from LA structural and functional changes. Natriuretic peptides (Nppa and Nppb) and Myh7 up-regulation in LA tissue (Figure 3E, and see Supplementary material online, Table S1), as surrogates of hypertrophic and myocardial stretch signalling, and massive (>100-fold) increase in Periostin and Il6, markers of fibrosis and inflammation, fits with the enlarged and dysfunctional LA seen in AAC-Kcnk3Δ94ex1/Δ94ex1. These results demonstrate that Kcnk3-LOF mutation promotes LA remodelling and diastolic dysfunction in LV pressure overload condition, a common consequence of LV dysfunction playing a major role in patients with HFpEF.21
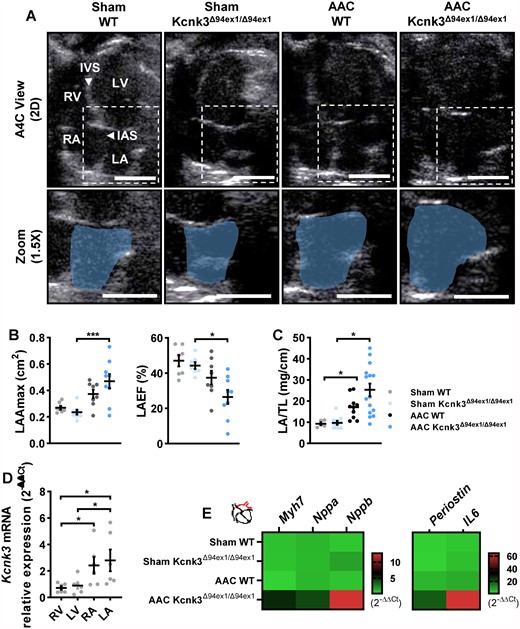
Left atrial (LA) dysfunction underlines Kcnk3-LOF mutation-associated LV diastolic dysfunction. (A) Using an apical four chamber projection (A4C) we quantified left atrial (LA) dimensions. IAS, interatrial septum; IVS, interventricular septum; LV, left ventricle; RA, right atria; RV, right ventricle. Dashed white boxes represent the zoomed sections in the lower panel, and the blue shaded area delineates the LA. Vertical bars, 0.5 cm, horizontal bars represent 100 ms. (B) Left panel: LAAmax, right panel: LA emptying fraction from Sham-WT (n = 7), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 7–8), AAC-WT (n = 8), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 9), and (C) LA/tibia length from Sham-WT (n = 5), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 10), AAC-WT (n = 10), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 15). (D) Relative mRNA expression (2−ΔΔCt) of Kcnk3 in LV, RV, LA, and RA from Sham-WT (n = 6). Experiments were normalized by 18S mRNA and analysed using ΔCt values. (E) Relative mRNA expression (2−ΔΔCt) of Myh7, Nppa, Nppb, Periostin, and IL-6 (heatmap representation of the mean) in LA from Sham-WT (n = 5), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 6), AAC-WT (n = 5), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 7). Experiments were normalized by 18S mRNA and analysed using ΔCt values (for a full statistical analysis refer to the Supplementary material online). Data are represented as scatter dot plots, with mean ± SEM. Experiments were analysed using two-way ANOVA followed by Sidak’s post hoc test. *P < 0.05 and ***P < 0.001. LAAmax, LA/TL, and Kcnk3 expression were not normally distributed and were log-transformed prior to statistical analysis.
3.4 Pulmonary hypertension is exaggerated in Kcnk3-mutated rats with AAC
At the 10-week endpoint, animals were submitted to right heart catheterization, using a closed-chest approach to evaluate severity of PH. Compared to Sham groups, AAC resulted in the development of mild PH in AAC-WT rats, which was more severe in heterozygous and homozygous Kcnk3-mutated rats (Figure 4A and B, and see Supplementary material online, Figure S4B). Right ventricular systolic pressure (RVSP) was strongly correlated with LA dilation (LA maximal area), when looking at AAC rats, irrespectively of genotype, suggesting that PH had a passive/post-capillary component (Figure 4C), as a consequence of LA enlargement/dysfunction by Kcnk3 LOF-mutation. Sham-Kcnk3-mutated rats showed no significant increase in RVSP as compared to Sham-WT (Figure 4A and B, and see Supplementary material online, Figure S4B), contrarily to what we have previously shown in older Kcnk3-mutated rats.18 This is likely due to the low number of sham animals.
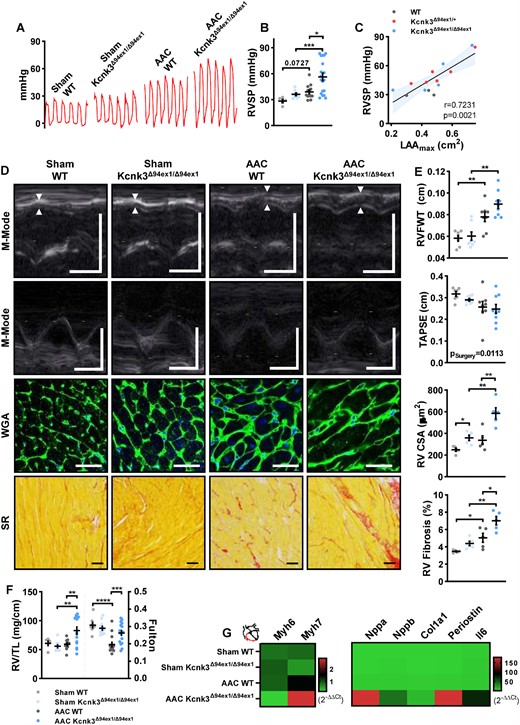
Pulmonary hypertension is exaggerated in Kcnk3-mutated rats with AAC. (A) Representation of the record of right ventricular pressure (RVP in mmHg) using right heart catheterization. (B) Quantification of RVSP (in mmHg) (SHAM; WT = 5, Kcnk3Δ94Ex1/Δ94Ex1 = 8, AAC; WT = 13, Kcnk3Δ94Ex1/Δ94Ex1 =19 rats). (C) Correlation (Spearman) between RVSP and LAAmax in AAC-WT (n = 3), AAC-Kcnk3Δ94Ex1/+ (n = 6), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 (n = 5) rats. (D) M-mode echocardiographic determination of RV free wall thickness (RVFW) and tricuspid annular plane systolic excursion (TAPSE, upper panels). Representative images of RV cardiomyocytes size (WGA staining) and RV interstitial collagen deposition (Sirius red staining, lower panels). Vertical bars, 0.5 cm (M-Mode) and horizontal bars represent 100 ms, 50 μm (WGA staining), and 100 μm (Sirius red). (E) First panel: RVFWT; and second panel: TAPSE from Sham-WT (n = 7), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 7–8), AAC-WT (n = 8), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 9). Third panel: RV cross-section area; and fourth panel: analysis of the percentage of RV interstitial collagen staining from Sham-WT (n = 5), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 5), AAC-WT (n = 5), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 5). (F) RV/tibia length ratio and Fulton index from Sham-WT (n = 5–8), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 9–11), AAC-WT (n = 9–14), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 14–17). (G) Relative mRNA expression (2−ΔΔCt) of Myh6, Myh7, Nppa, Nppb, Col1a1, Periostin, and IL-6 (heatmap representation of the mean) in RV from Sham-WT (n = 6), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 5), AAC-WT (n = 6), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 7). Experiments were normalized by 18S mRNA and analysed using ΔCt values (for a full statistical analysis refer to the supplemental material). Data are represented as scatter dot plots, with mean ± SEM. Experiments were analysed using two-way ANOVA followed by Sidak’s post hoc test after Kruskal–Wallis test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. RVSP, RV CSA, RV Fibrosis, RV/TL, and Fulton were not normally distributed and were log-transformed prior to statistical analysis.
As a consequence of the development of PH, AAC-Kcnk3-mutated rats developed more RV hypertrophy compared to AAC-WT rats. RV free wall thickness (Figure 4D and E, and see Supplementary material online, Figure S4C), cardiomyocyte size, and normalized RV weight were increased in AAC-Kcnk3-mutated rats (Figure 4D–F). Although the Fulton index decreased in AAC animals (Figure 4F), as a consequence of LV hypertrophy, it was higher in AAC-Kcnk3-mutated rats when compared with AAC-WT (Figure 4F and see Supplementary material online, Figure S4C), a sign of RV hypertrophy, fitting with the increased RVSP observed in these group. Despite increased PH severity and RV hypertrophy, no significant changes were observed in RV function, with only a mild non-significant decreased in tricuspid annular plane systolic excursion (TAPSE) (Figure 4D and E), when comparing all experimental groups. Interestingly, TAPSE was decreased in Sham and AAC-Kcnk3-mutated animals, when compared directly to Sham-WT rats (P < 0.05), suggesting that RV function was already compromised, albeit mildly, in animals with a Kcnk3-LOF mutation.
In AAC-Kcnk3Δ94ex1/Δ94ex1 rats, the development of PH and RV hypertrophy was paralleled with increased expression of overload/hypertrophic response-related genes (Nppa, Nppb and a shift in the Myh7/Myh6 relation, Figure 4G, and see Supplementary material online, Table S1), as well as overexpression of pro-inflammatory (Il6) and pro-fibrotic genes (Col3a1 and Periostin), which resulted in increased interstitial fibrosis deposition in AAC-Kcnk3Δ94ex1/Δ94ex1 rats (Figure 4D and E) compared to the AAC-WT rats. Overall, these results indicate that Kcnk3-dysfunction facilitates the development of PH-LHD.
Using frozen RV samples, we measured the myofilament properties of isolated skinned RV cardiomyocytes, where we found only a trend towards increased passive tension development in AAC-rats (WT and Kcnk3-mutated rats, see Supplementary material online, Figure S4D), without differences between genotypes. Active tension and calcium sensitivity were not changed (see Supplementary material online, Figure S4D).
Interestingly, and in agreement with the increase in RVSP, Sham-Kcnk3Δ94ex1/Δ94ex1 animals showed a significant increase in RV cardiomyocyte size (Figure 4E). Unexpectedly, and without changes in the analysed pro-remodelling transcripts, Sham-Kcnk3-mutated rats had a significantly higher percentage of fibrosis deposition in the right ventricle.
No differences were observed in mean carotid arterial pressure, while cardiac output was decreased in AAC-rats, irrespectively of the genotype (see Supplementary material online, Figure S4A and B), which could be a consequence of LV diastolic dysfunction leading to compromised LV filling.
The development of PH resulted in mild right atrial dilation together with non-significant increase in HF and pro-fibrotic gene expression, in Kcnk3-mutated rats (homozygous and heterozygous) (see Supplementary material online, Figure 5A–E).
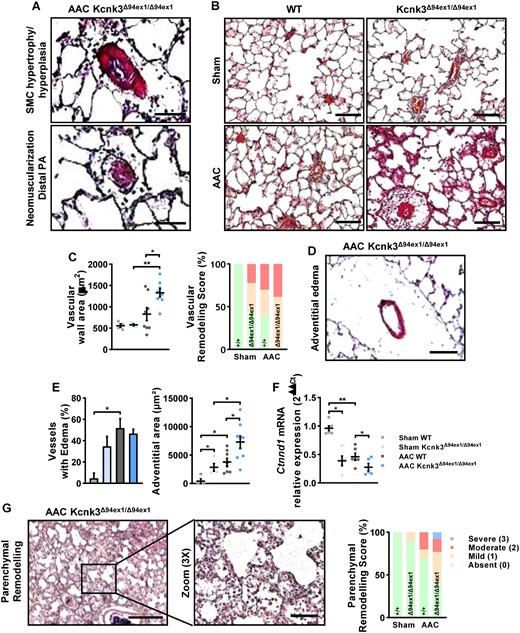
Analyses of pulmonary alteration in AAC-Kcnk3Δ94ex1/Δ94ex1 rats. (A) Representative photography of HES staining of lung from AAC-Kcnk3-mutated rats. Upper panel: Smooth muscle cell hypertrophy/hyperplasia in pulmonary arteries, Lower panel: neomuscularization of the small distal and normally not muscularized pulmonary vessels. Scale bars, 50 (upper) and 15 μm (lower). (B) HES staining on lung slice from Sham and AAC WT or Kcnk3Δ94ex1/Δ94ex1 rats. Scale bar, 100 μm. (C) Left panel: quantification of vessel wall area (µm2). Right panel: analysis of the vascular remodelling score in Sham and AAC WT or Kcnk3Δ94ex1/Δ94ex1 rats. Sham-WT (n = 4), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 4), AAC-WT (n = 8), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 8). (D) Illustrative of lung perivascular oedema in AAC-Kcnk3-mutated rats. Scale bar, 50 μm. (E) Left panel, percentage of vessels with large adventitia (which could be measured). Right panel, quantification of adventitia surface area (µm2) in Sham-WT (n = 4), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 4), AAC-WT (n = 8), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 9). (F) Relative mRNA expression (2−ΔΔCt) of Ctnnd1 (encoding p120Catenin) in lung from Sham-WT (n = 6), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 5), AAC-WT (n = 6), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 5). Experiments were normalized by 18S mRNA and analysed using ΔCt values. (G) Left panel, illustrative images of severe lung injury with prominent enlargement of alveolar septa and inflammatory exsudates observed in AAC-Kcnk3-mutated rats. Scale bar, 500 and 100 μm. Right panel, analysis of the parenchym alteration score in Sham-WT (n = 4), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 4), AAC-WT (n = 4), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 4). Data are represented as scatter dot plots, with mean ± SEM. Experiments presented in panels A–B and D–F were analysed using two-way ANOVA followed by Sidak’s post hoc test, and experiments presented in panels C and G were analysed using one-way ANOVA followed by Dunn’s post hoc test after Kruskal–Wallis test. *P < 0.05, **P < 0.01. Vascular wall area, oedema, and adventitial area were not normally distributed and were log-transformed prior to statistical analysis.
3.5 Extensive pulmonary vascular remodelling supports the development of PH in AAC- Kcnk3Δ94ex1/Δ94ex1 rats
Supporting the development of PH in AAC-rats and in comparison with Sham-group, we observed profound pulmonary structural remodelling in AAC-Kcnk3-mutated rats as media hypertrophy/hyperplasia in pulmonary arteries and neomuscularization of the small distal and normally not muscularized pulmonary vessels (Figure 5A and B). Semi-quantitative morphometric analysis of pulmonary vessels remodelling including arterioles and veinules indicated a greater proportion of pulmonary vessel remodelling in Sham-Kcnk3-mutated rats vs. Sham-WT, and that all pulmonary vessels were remodelled in AAC-Kcnk3-mutated rats compared to AAC-WT rats (Figure 5C). The pulmonary vessels wall area was increased in AAC-Kcnk3-mutated rats (Figure 5C and see Supplementary material online, Figure S6A).
We observed the presence of adventitial/perivascular oedema in AAC-induced PH animals (Figure 5D and E, and see Supplementary material online, Figure S6B), as well as a decrease in Ctnnd1 expression (Figure 5F), which codes for p120-catenin, an important regulator of endothelial permeability. Despite that, adventitial area was significantly higher in Kcnk3Δ94ex1/Δ94ex1 rats (Figure 5E, and see Supplementary material online, Figure S6B).
Interestingly, both adventitial area was increased and Ctnnd1 expression was decreased in Sham-Kcnk3-mutated rats, suggesting that the Kcnk3-LOF compromises endothelial integrity, leading to perivascular leakage, and ultimately supporting increased pulmonary pressures,22 and sensitizing the pulmonary circulation to an exaggerated response to passive pressure increases.
Not only the vasculature, but also the lung parenchyma was altered in AAC-induced PH, and aggravated with Kcnk3-LOF. In most of AAC-Kcnk3-mutated rats we observed severe lung injury with prominent enlargement of alveolar septa (Figure 5G), and inflammatory exudates (Figure 6D). Due to the lack of validated markers capable of differentiating arteries and veins, the assessment of vein remodelling in lung vascular diseases is an ambitious task, requiring accurate identification of venous vessels. In rodents, a reliable histologic item to identify pulmonary veins is based on the presence of a cardiomyocyte coat around pulmonary veins. Unfortunately, we did not find enough cardiomyocytes coat pulmonary veins to quantify their remodelling. However, when we succeeded to identify some cardiomyocytes-coated pulmonary veins in AAC-rats and we observed a severe interstitial oedema in the adventitial layer (see Supplementary material online, Figure S7A). In AAC-rats, we also observed that pulmonary veins wall thickness was increased with severe adventitial oedema and lymphangiectasis (see Supplementary material online, Figure S7B), indicating that large pulmonary veins were remodelled in AAC-rats.
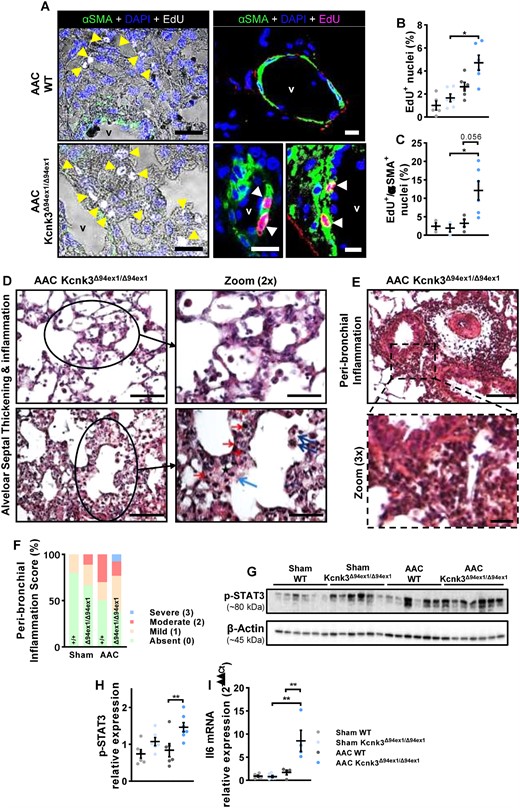
Characterization of pulmonary remodelling in AAC-rats. (A) Immunofluorescent staining of frozen rat lung sections and confocal imaging with Click-iT 5-ethynyl-2′-deoxyuridine (EdU; white or pink nuclei = EdU-positive nuclei = proliferating cells) in combination with α-smooth-muscle actin (α-SMA; in green) in AAC-WT and AAC-Kcnk3-mutated rats. Counterstain was DAPI (blue). EdU positive nuclei are indicated by yellow/white arrows, and vessel lumen is indicated (v). Scale bar, 50 μm (left panels) and 20 μm (right panels). (B) Quantification of the percentage of lung proliferating cells (% of EdU-positive nuclei) in Sham-WT (n = 5), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 6), AAC-WT (n = 6), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 6). (C) Quantification of the proportion of EdU positive + αSMA positive cells in in Sham-WT (n = 3), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 3), AAC-WT (n = 4), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 6). (D) Upper panel, in a group of AAC-kcnk3-mutated rats alveolar wall appeared thickened. The alveolar septa enlargement is characterized by the affluence of inflammatory cells (mostly mononucleated), and at some degree to alveolar oedema. Lower panel, in severe cases the alveolar septa enlargement is hypercellular, with many inflammatory cells (red arrow), macrophages (blue arrow) and interstitial oedema (black star). Endoalveolar macrophages are also visible (dark blue arrows). No fibrin deposits nor alveolar necrosis are observed. Scale bars, 100 and 50 (zoom) μm. (E) Severe peri-bronchial/perivascular inflammation, extending in peri-bronchiolar alveoli. Scale bars, 100 and 25 (zoom) μm. (F) Analysis of the peri-bronchiolar inflammation score in Sham-WT (n = 4), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 4), AAC-WT (n = 4), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 4). (G) Western blot images of the phosphorylation of STAT3 protein in lung lysates. (H) Quantification of the phosphorylation of STAT3 protein in lung from Sham-WT (n = 6), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 6), AAC-WT (n = 6), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 (n = 7) rats (see the complete unedited gel in see Supplementary material online, Figure S8). (I) Relative mRNA expression (2−ΔΔCt) of Il-6 in lung from Sham-WT (n = 6), Sham-Kcnk3Δ94Ex1/Δ94Ex1 (n = 6), AAC-WT (n = 4), and AAC-Kcnk3Δ94Ex1/Δ94Ex1 rats (n = 4). Experiments were normalized by 18S mRNA and analysed using ΔCt values. Data are represented as scatter dot plots, with mean ± SEM. Experiments presented in panels A–B, D–E, G–H, and I were analysed using two-way ANOVA followed by Sidak’s post hoc test, and experiments presented in panels C and F were analysed using one-way ANOVA followed by Dunn’s post hoc test after Kruskal–Wallis test,***P < 0.001. *P < 0.05, **P < 0.01, and ***P < 0.001. Edu+ and Edu+/αSMA+ and Stat3 phosphorylation were not normally distributed, and were log-transformed prior to statistical analysis.
Similarly to Kcnk3Δ94ex1/Δ94ex1, we also found a significant increase in pulmonary vessel wall thickening, adventitia area in Kcnk3Δ94ex1/+ (see Supplementary material online, Figure S6A and B) and the presence of foci alveolar wall thickening in AAC-Kcnk3Δ94ex1/+ rats (data not shown), with significant parenchymal remodelling (see Supplementary material online, Figure S6C).
3.6 Characterization of pulmonary remodelling
To evaluate the consequence of Kcnk3-LOF in AAC context on in situ lung cell proliferation, we visualized cells undergoing DNA replication by imaging the incorporation of 5-ethynyl-2′-deoxyuridine (EdU; white or pink nucleus in Figure 6). We observed very few EdU-positive cells in the lung parenchyma of Sham-WT or Sham-Kcnk3Δ94ex1/Δ94ex1 rats (data not shown). Underlying the pulmonary structural changes, we found an increase in lung proliferative cells (Figure 6A–C), including PASMCs, as the amount of α-smooth-muscle actin and EdU positive cells was augmented in AAC-Kcnk3-mutated rats (P-value = 0.056) (Figure 6C). A larger accumulation of proliferating CD34+ cells was observed in lung from AAC-Kcnk3Δ94ex1/Δ94ex1 rats compared with AAC-WT rats (see Supplementary material online, Figure S7C).
The analysis of lung histology (HES) revealed that moderate alveolar septa enlargement was mostly characterized by the affluence of inflammatory cells (mostly mononuclear: monocytes, lymphocytes). In more severe cases, we observed that the alveolar septa enlargement was hypercellular, with many inflammatory cells (monocytes, lymphocytes), macrophages, and interstitial oedema. Endo-alveolar macrophages were also visible (Figure 6D). Peri-bronchial inflammation was obvious in ACC-Kcnk3-mutated rats (Figure 6E), whose histological score was aggravated compared with WT animals (Figure 6F, and see Supplementary material online, Figure S6E). Elevated levels of Il-6 are described to hyper-activate STAT3 signalling mediating the cell proliferation and cell survival.23 In association with elevated lung Il-6 mRNA and pulmonary cell proliferation in AAC-Kcnk3Δ94ex1/Δ94ex1 rats, we found an increase of STAT3 phosphorylation (Figure 6G and H).
4. Discussion
Our findings, summarized in Figure 7, suggest that KCNK3 dysfunction plays a role in the development PH-LHD. In the context of LV pressure overload, we propose that Kcnk3-LOF mutation promotes (i) disruption of pulmonary endothelium integrity which leads to lung perivascular oedema and PA adventitial remodelling, (ii) inflammatory signalling (IL-6) which leads to increase PASMC proliferation promoting pulmonary vascular and parenchymal remodelling. (i) and (ii) contribute to the increase in pulmonary vascular resistances. (iii) Cardiac fibrosis links LV diastolic dysfunction to decreased LA compliance and abnormal pulmonary venous remodelling. All together these events act in favour of the aggravation of PH and the consequent RV hypertrophy and dysfunction in Kcnk3-mutated rats. Our results are of major interest since understanding of molecular mechanisms involved in PH-LHD pathobiology are yet to be defined.
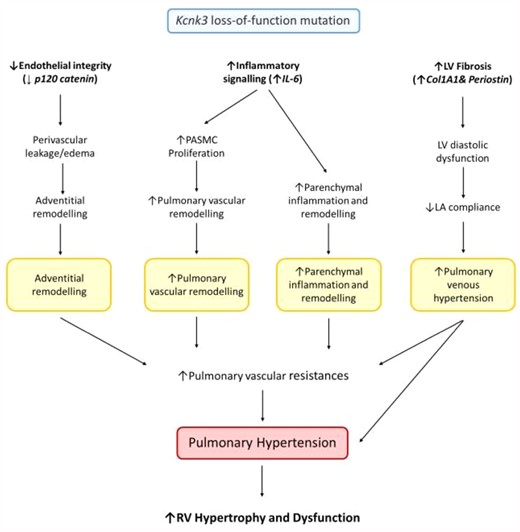
Proposed sequence of events arising from Kcnk3-LOF mutation in context of LV pressure overload. In context of LV pressure overload, we propose that Kcnk3-LOF mutation promotes (i) the disruption of pulmonary endothelium integrity (with a decreased expression of p120 catenin in lung from Sham-Kcnk3-mutated vs Sham-WT rats) which leads to lung perivascular oedema and PA adventitial remodelling, (ii) the inflammatory signalling (Increased expression of IL-6 in lung, LV, RV and LA from AAC-Kcnk3-mutated rats) which leads to increase PASMC proliferation and promoting pulmonary vascular remodelling and parenchymal remodelling. (i) and (ii) contribute to the increase of pulmonary vascular resistances, and (iii) the cardiac fibrosis (with an increased expression of Col1A1 and periostin in LV and RV from AAC-Kcnk3-mutated rats) link to LV diastolic dysfunction, decreased of LA compliance and abnormal pulmonary venous remodelling. All together these deregulations act in favour of the aggravation of pulmonary hypertension and the consequent RV hypertrophy and dysfunction. All these events are responsible for the development of more severe PH-induced by LV pressure overload in Kcnk3-mutated rats.
KCNK3 is expressed in the rat and human heart,20 with a clear atrial predominance, which is maintained in end-stage HF.24 We have previously shown that Kcnk3 expression is decreased in the right ventricle of PH animals and PAH patients, as well as in the pressure overloaded right ventricle.15 To understand the potential role of KCNK3 in LV pressure overload-induced PH, we measured its expression in heart and lungs. Kcnk3 levels were unaltered by AAC surgery, in both left ventricle and right ventricle, and lungs. However, we observed enhanced expression in both right and left atria, in agreement with previously published literature, confirming that KCNK3 might play an important role in the function of atrial chambers. In human, both KCNK3 up-regulation and KCNK3-LOF mutations are associated with detrimental atrial electrophysiological alterations.25 The important role of KCNK3 in left atrium could explain the LA enlargement in AAC-Kcnk3-LOF-mutated rats. Directly or indirectly, LV diastolic dysfunction was associated with LA enlargement and functional compromise in AAC-Kcnk3-mutated rats. Inactivation of KCNK3 can lead to atrial enlargement when induced in the experimental context.26 Signs of adverse LA remodelling in HF include increased expression of myosin heavy chain-β (Myh7),27 natriuretic peptides,28 and Periostin,29 all of which were up-regulated in the left atrium of Kcnk3-mutated rats with LV pressure overload, and underlie the molecular mechanisms that resulted in compromised LA structure and function in these animals. Increased pulmonary pressures were highly correlated with LA size, which reveals the link between LA structural remodelling and the severity of PH.
In the current work, a Kcnk3-LOF-mutation resulted in LV dysfunction, characterized by a pro-fibrotic remodelling and diastolic impairment, despite a similar degree of pressure-overload (gradient) and LV hypertrophic response. Bertero et al.,30 recently demonstrated that KCNK3 knockdown up-regulated the expression of Col3a1 and Lox in human pulmonary fibroblasts, which could explain the increased expression of pro-fibrotic genes and fibrosis in all cardiac chambers. In association with increased RV and LV fibrosis in AAC-Kcnk3-mutated rats, we found an increase in Periostin expression. Periostin is known to modulate the transition of fibroblasts to myofibroblasts, collagen fibrillogenesis, extracellular matrix synthesis, as well as the inflammatory response.31 Moreover, Bruns et al.,32 recently demonstrated that Periostin-secreted from cardiac fibroblast drives cardiomyocyte dedifferentiation, contributing to RV dysfunction in experimental PH. In our study increased Periostin in the four cardiac chambers could partly explain exaggerated peri-vascular inflammation and cardiac fibrosis.
By contrast to our findings, a recent work using global kcnk3-deficient mice, submitted to transverse aortic constriction (TAC), develop reduced cardiac hypertrophy and preserved cardiac function compared to WT-TAC.33 These opposite effects could be explained either by species/strain-specific differences, as in Wistar rats Kcnk3 expression is two-fold higher in the right ventricle as compared to left ventricle,15 while in the current manuscript, using Sprague-Dawley rats, we found no differences between the two ventricles. Moreover, in mice pulmonary vascular KCNK3 is not functional in PASMCs (Manoury et al., 2011),18 while KCNK3 plays a key role in the homeostasis of the pulmonary vasculature. Recently, detailed species-related differences in the response to pressure overload34 have been described, fitting with the compromised ejection fraction in WT mice,33 opposed to our WT rats.
Either as a consequence of increased pulmonary resistances, direct RV effects of Kcnk3 decreased expression,15 or Kcnk3-LOF-mutation in AAC animals, worsened RV hypertrophy and function in response to LV pressure overload, is an important observation, since a worse RV function predicts a deteriorated functional capacity in HF patients,35 and affects treatment response in patients with HFpEF,36 making its evaluation in PH-LHD37 a requirement. RV cardiomyocyte hypertrophy and fibrosis in Sham-Kcnk3-mutated rats, suggests that the mutation itself promotes maladaptive remodelling, potentially having a direct consequence on RV adaptation in the context of LV pressure overload. The moderate deterioration RV function in our model could be explained by the improvement in RV perfusion secondary to AAC, recently shown to increase right coronary artery perfusion, leading to attenuation of RV functional and structural decline after MCT-exposure.38
Generalized remodelling, involving most lung structures are associated with retrograde pressure build up-induced capillary injury.11 Pulmonary arterial (pre-capillary component) and venous (post-capillary component) remodelling has been recently characterized in detail in patients with HF, irrespectively of EF, and correlated with disease severity.8 Accordingly, AAC-Kcnk3-mutated animals, showed significant alterations in both pulmonary arteries and veins, with both lumen-compromising medial hypertrophy and neomuscularization of pulmonary vessels.
Adventitial restructuring, characterized by perivascular cuffing (fluid accumulation in the interstitial surrounding the vessels), which was previously described as a consequence of elevated pulmonary venous pressure,22 was present in both AAC-groups (similar percentage of vessels with adventitial oedema), but amplified in homozygous Kcnk3-mutated rats (larger adventitial area), which could be explain by decreased Ctnnd1 expression. Kcnk3-LOF-mutation was associated with lower Ctnnd1 (p120-catenin) expression, a gatekeeper of endothelial barrier function and integrity,39 even without LV pressure overload, predisposing these animals to endothelial injury, and asserting a causative role. We have previously shown that Kcnk3-mutated rats undergo endothelial-to-mesenchymal transition, with loss of CD31 expression, further contributing to endothelial barrier impairment. Unfortunately, we did not measure lung weight as a measure of congestion.
In parallel, extensive parenchymal remodelling, characterized by alveolar septal thickening was patent in overloaded Kcnk3-mutated lungs. Similar alterations contribute to limited respiratory efficiency and effort capacity in HF patients with PH.35 As indicated by our present results, alveolar septal enlargement observed in AAC-Kcnk3-mutated rats is at least the consequence of the increased CD34+ cells proliferation. CD34 is a marker of endothelial cells which is also used as a marker of haematopoietic stem cells, haematopoietic progenitor cells as well other non-haematopoietic cell types, including vascular endothelial progenitors 1 and embryonic fibroblasts.40 Since Dierick et al.,41 found that a population of pulmonary progenitor cells PW1+/CD34+ are recruited in mice and rat experimental PH models, we could hypothesized that alveolar septal enlargement could also due to a larger accumulation of PW1+/CD34+ cells.
In vivo, KCNK3 inhibition induced proliferation of CD4+ and CD8+ lymphocytes,42 suggesting an activation of inflammatory processes. Moreover, kcnk3-deficient mice and fibroblast-derived induced pluripotent stem cells from patients with a Kcnk3-LOF mutation, showed an increased sensitivity to inflammatory stimulus,43 with increased inflammatory mediators expression and an exaggerated infiltration of inflammatory cells in the lung. In harmony, increased IL6/STAT3 activation, which was recently demonstrated to play an important role in experimental and human PH-LHD,44 was evident in AAC-Kcnk3-mutated rats, leading to extensive peri-bronchial inflammatory infiltrates, and overall increased pulmonary cellular proliferation, including PASMC, sustaining pulmonary vascular wall remodelling.
In AAC-Kcnk3-mutated rats, the inadequate pulmonary vascular remodelling and PH could shed some light on the development of unexplained disproportionate PH in LHD.45 In some cases of PH-LHD, severe pulmonary vascular alterations persisted despite resolution of the underlying LV disease.11 Our observation that Kcnk3-LOF exaggerates pulmonary vascular remodelling in the context of LV pressure overload, suggests that KCNK3-dysfunction might be associated with a lack of reverse remodelling, and therefore non-response to treatment. Recently, in AAC-induced LV pressure overload with pulmonary vascular remodelling, surgically removing the aortic constriction, normalized pulmonary vascular structure,46 therefore using this approach would dissect if structural changes remain in the presence of a Kcnk3-mutation.
Importantly, vascular changes were already present in Sham-Kcnk3-mutated rats, which could explain the exaggerated pulmonary remodelling in response to LV pressure overload in a Kcnk3-LOF-mutated rats. Underlying remodelling in unstressed conditions describes a pre-capillary component in AAC-Kcnk3-mutated animals, which could explain the different response to LV pressure overload in the absence and presence of a Kcnk3-LOF mutation, and suggests that Kcnk3-dysfunction or reduced expression could not only worsen PH, but also compromise its reversibility/treatment in the context of HF.
Several works have used either TAC or AAC to induce LHD or LV pressure overload and to study pulmonary vascular changes.19,44 Although they might not replicate all the phenotypical characteristics of group 2 PH, it is clear that pulmonary vascular changes develop in response to a primary insult to the left ventricle, and have granted experimental evidence for PH-LHD treatment, including sildenafil.3 Inhaled iloprost showed a pulmonary-specific vasodilator effect in rats with AAC-induced PH,5 which was confirmed in patients with HFpEF.47 In animal model of PH-HFpEF, treprostinil attenuated the development of PH.48 Both of these prostanoids stimulate KCNK3 currents in human PASMC,13 providing that stimulation of KCNK3 activity might play a beneficial role in PH-LHD.
4.1 Limitations
Other works have shown that PH-LHD development of experimental PH-LHD, with measurement of LV filling pressures in both AAC with19 or without metabolic syndrome associated LV disease.44 Unfortunately we were unable to measure LV pressures, using our right heart catheterization, where LA or pulmonary capillary wedge pressure measurement is not possible.49 Despite that, we show a clear concentric LV hypertrophy phenotype, characteristic of LV pressure overload in the rat.34
Our echocardiographic analysis lacks E/A wave analysis. Due to the elevated heart rate of small animals, and equipment capabilities, E and A wave separation was not observed in all animals. Indeed, we were only able to measure the A wave in around 60% of animals, and without differences between groups, due to the small number of measurements (data not shown). Despite that, in the animals where peak separation occurs, E > A, allowing us to measure E peak velocity with a significant degree of confidence in all animals, we showed that homozygous AAC-Kcnk3-mutated rats had an increased E/e′ ratio.
The access to lung from patients with PH-LHD is extremely difficult, since most of these do not undergo to surgical procedures, we are not able to measure KCNK3 function and expression in human PH-LHD patients. Furthermore, the genetic variations of PH-LHD patients have been barely studied,50 consequently no information is available regarding the presence of mutation in KCNK3.
We acknowledge that our model does not represent a pure phenotype of passive PH-LHD. Despite that, we undoubtedly demonstrate that the presence of a Kcnk3-LOF-mutation aggravates pulmonary vascular remodelling and PH, generating new hypothesis for the understanding of the pathophysiology.
5. Conclusion
PH secondary to LHD is a common disease, but without specific therapies and a poorly understood pathophysiology. With this work, we established KCNK3 as an important mediator of PH in the context of LV pressure overload. Using Kcnk3-mutated rats and a model of AAC-induced LV concentric hypertrophy, we revealed a compromised left heart compliance, and disproportionate pulmonary remodelling, associated with pro-inflammatory and proliferative processes, ultimately leading to exaggerated PH and RV dysfunction. Either by sensitizing the pulmonary circulation directly, or by worsening LV/LA function, we established the importance of KCNK3 in this pathology, and have opened the way for the development of new therapeutic options for this condition.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
M.L., P.M.-F., M.-R.G., and F.A. participated in the research design. M.L., P.M.-F., M.-R.G., H.L.R., R.A., A.B., V.C., C.R.-M., V.D., and F.A. conducted the experiments and performed the data analysis. All authors draft the manuscript for important intellectual content the manuscript. All authors reviewed and revised the final version and approved manuscript submission.
Acknowledgements
This work benefited from the facilities and expertise of TEFOR—Investissement d’avenir—ANR-II-INSBS-0014. We thank Dr Ignacio Anegon, Laurent Tesson, and Séverine Ménoret from Transgenic Rats and Immunophenomics Core Facility (TRIP), platform TRIP–(Immunology–Nantes) for generating Kcnk3-mutant rats. The authors thank Dr J. Sabourin from INSERM UMR S1180, Faculté de Pharmacie, Université Paris-Saclay for critical reading of the manuscript. We wish to thank the staff at the ANIMEX platform for caring for the rat lines.
Conflict of interest
M.H. has relationships with drug companies, including Actelion, Bayer, GSK, Novartis, and Pfizer. In addition to being investigators in trials involving these companies, other relationships include consultancy services and memberships to scientific advisory boards. The other authors have no conflicts of interest.
Funding
This study was supported by grants from the French National Institute for Health and Medical Research (INSERM), the Université Paris-Saclay, the Marie Lannelongue Hospital, the French National Agency for Research (ANR) [grant no. ANR-18-CE14-0023 (KAPAH)]. F.P. received funding from Fondation maladies rares in the frame of the ‘Small animal models and rare diseases’ programme to generate the Kcnk3-mutaed rats. M.L. is supported by Therapeutic Innovation Doctoral School (ED569). P.M.-F., R.A., and C.B.-S. were funded by Portuguese Foundation for Science and Technology through Grant UID/IC/00051/2013 (COMPETE_2020, POCI) and projects IMPAcT (Ref. PTDC/MED-FSL/31719/2017; POCI-01-0145-FEDER-031719) and NETDIAMOND (Ref. POCI-01-0145-FEDER-016385).
Data availability
The authors declare that all supporting data are available within the article and its Online Data Supplement.
References
Pulmonary hypertension (PH) due to left heart disease (PH-LHD) has higher incidence and prevalence than other forms of PH. However, no pulmonary-targeted drugs are available in clinical practice. The difficulties to have access to PH-LHD patient tissues, and the small number of preclinical models available compromise our understanding of the molecular mechanisms underlying PH-LHD. Using unique Kcnk3-loss-of-function-mutated rats, the present results demonstrate for the first time that KCNK3 loss-of-function is a key event in the pathobiology of experimental PH due to AAC, suggesting that the Kcnk3 channel could play a potential key role in the development of PH due to LHD.
Author notes
Mélanie Lambert and Pedro Mendes-Ferreira authors contributed equally to the work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}