





This editorial refers to ‘Estrogen inhibits salt-dependent hypertension by suppressing GABAergic excitation in magnocellular AVP neurons’, by X. Jin et al., pp. 2263–2274.
Sex differences are evident in the severity of hypertension, the incidence of which is typically lower in pre-menopausal women than age-matched men, but the difference diminishes after menopause.1 In animal models of hypertension, including deoxycorticosterone acetate (DOCA)-salt,2 aldosterone,3 and angiotensin II4 models, females develop lower pressor responses than males. This sex-dependent difference in susceptibility to severe hypertension is lost following ovariectomy, consistent with an anti-hypertensive effect of endogenous estrogen in females. There are several mechanisms by which estrogen could confer protection against hypertension, including direct actions on the kidney, heart, and blood vessels, and indirectly via the central nervous system. There is evidence for a role of all three types of estrogen receptors (ER); the classical nuclear receptors, ERα5 and ERβ,5 and the novel G protein-coupled estrogen receptor 1,3 in mediating the anti-hypertensive effects of estrogen. Centrally, estrogen has been shown to regulate the activity of arginine vasopressin (AVP) neurons.6
AVP is a neurohormone produced by the hypothalamus, specifically in magnocellular neurons of the supraoptic nucleus and paraventricular nucleus, and is released to promote pressor responses to hypovolaemia or hyperosmolarity7 (see Figure 1). It binds to V1a receptors on vascular smooth muscle cells to cause vasoconstriction which increases total peripheral resistance.7 AVP also acts on V2 receptors in the late distal tubule or collecting ducts of the kidney to increase water retention and, hence, circulating blood volume. AVP production is regulated by a negative feedback system whereby changes in serum osmolarity and blood pressure are detected by baroreceptors that activate inhibitory gamma-aminobutyric acid (GABA) signalling in the hypothalamus. GABA is the main inhibitory neurotransmitter of the central nervous system, but under certain conditions such as hypertension or epilepsy, it can have excitatory effects on neurons.8 Neuronal chloride homeostasis is mainly regulated by the K+/Cl− co-transporter, KCC2, and Na+/K+/Cl− co-transporter, NKCC1. Chronically high salt intake disrupts the transmembrane chloride gradient required for inhibitory GABA signalling. This causes GABAergic signalling to switch from being inhibitory to excitatory, leading to excessive AVP production, water retention, and vasoconstriction and thus sustained increases in blood pressure8 (Figure 1).
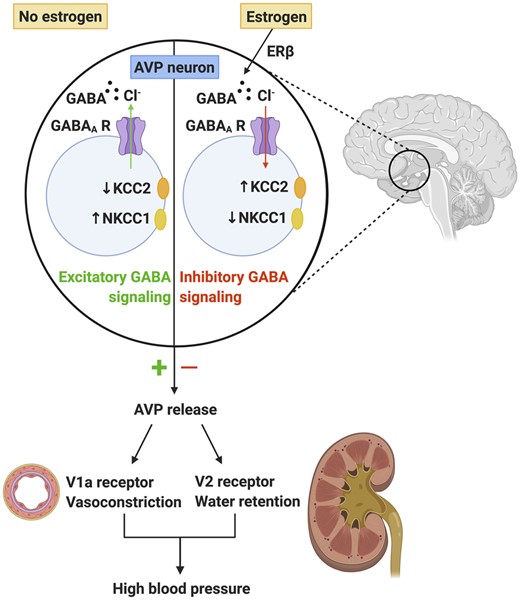
In the absence of estrogen, GABA signalling can become excitatory which leads to exacerbated arginine vasopressin (AVP) release and subsequent sustained increases in blood pressure. In the presence of estrogen via estrogen receptor β, GABA signalling remains inhibitory which prevents inappropriate release of AVP and blood pressure is normalized. Created with BioRender.com.
This editorial is focused on the study by Jin et al.9 in the current issue who report that estrogen protects against DOCA-salt-induced hypertension by blocking GABAergic excitation of AVP neurons to inhibit AVP release. AVP can contribute to salt-dependent hypertension and the authors had previously reported that GABAergic excitation of AVP neurons contributes to DOCA-salt-induced hypertension in rats.10 In the current study, male and female rats were uninephrectomized and administered DOCA and 1% NaCl for drinking to induce hypertension. A cohort of females were ovariectomized, and some of them were treated with estrogen. The authors confirmed previous findings that males and ovariectomized females develop greater pressor responses to DOCA-salt than do intact females. Similarly, estrogen supplementation blunted the pressor response to DOCA-salt in ovariectomized females.
Reflecting the blood pressure responses, plasma AVP levels were higher in ovariectomized females than in intact or estrogen-treated ovariectomized females. Acute administration of a V1a receptor antagonist reduced blood pressure to a greater extent in the ovariectomized females, whereas intracerebroventricular (ICV) infusion of estrogen reduced blood pressure and the proportion of AVP neurons with GABAergic excitatory postsynaptic potential in the supraoptic nucleus. Furthermore, ICV infusion of ICI182780, a non-selective ERα/β antagonist, prevented these effects of estrogen, suggesting that central ERs are responsible for mediating these estrogenic effects. The authors delved further and found that selective antagonism of ERβ but not ERα blocked the effect of estrogen on GABAergic excitation, thus pointing to ERβ as the key target. Central administration of estrogen also reduced expression of NKCC1 and increased KCC2 in the supraoptic nucleus, and prevented the inhibitory-to-excitatory transition of GABAergic signalling. Interestingly, treatment with CLP290, a KCC2 activator, also reduced blood pressure and plasma AVP levels in DOCA-salt-treated ovariectomized females through the same protective pathway as estrogen. In males, estrogen had similar protective effects on blood pressure and plasma AVP levels via suppression of GABAergic excitation in AVP neurons.
Overall, the study by Jin et al. is a comprehensive one that ties together known concepts: the modulatory effect of estrogen on blood pressure and the role of AVP in salt-dependent hypertension (Figure 1). Furthermore, it provides evidence of a novel central mechanism by which estrogen may attenuate hypertension by central modulation of AVP release and activity. Strengths of the study are the inclusion of males and females and the demonstration that the relevant anti-hypertensive pathways identified are applicable in both sexes, which is important knowledge for potential translational outcomes.
While the authors speculate that drugs which target GABAergic excitation in AVP neurons could be used therapeutically for salt-dependent hypertension, it would be difficult to selectively target AVP neurons. It may be more feasible to target direct effects of AVP, such as via V1a receptors to prevent vasoconstriction or V2 receptors to reduce water reabsorption, since AVP receptor antagonists are already used to treat hyponatraemia.11 Furthermore, the study only tested the acute effects of a V1a receptor antagonist, and so it will be important that future work carefully examines the effects of chronic administration of AVP antagonists in salt-dependent hypertension. Moreover, as inhibition of AVP activity by endogenous estrogen is likely to occur normally in pre-menopausal women, it is likely that targeting AVP receptors to treat hypertension would only be effective in men or post-menopausal women.
As mentioned earlier, there are several mechanisms by which estrogen can modulate hypertension, and these also include anti-inflammatory actions. As hypertension is now established to be a chronic inflammatory condition,12 interactions between the immune system and AVP-induced increases in blood pressure seem worthy of future exploration. Indeed, AVP is reported to exert pro-inflammatory effects. For example, mouse and human CD4+ T cells express AVP receptors, and AVP infusion in mice throughout gestation elevates interferon-γ and interleukin-17 levels in the maternal plasma, and increases T helper 1 and T helper 17 cells and decreases T regulatory cells in the maternal spleen.13 Not only are T cells necessary for the full development of hypertension in several animal models, including DOCA-salt,3,14 but estrogen can reduce DOCA-salt hypertension through an action on T regulatory cells.2 Therefore, future studies could also examine whether AVP can modulate T cell responses to elevated blood pressure in an estrogen-sensitive manner.
Conflict of interest: none declared.
The opinions expressed in this article are not necessarily those of the Editors of Cardiovascular Research or of the European Society of Cardiology.

{kind=link}