





Abstract
Inherited arrhythmias are disorders caused by one or more genetic mutations that increase the risk of arrhythmia, which result in life-long risk of sudden death. These mutations either primarily perturb electrophysiological homeostasis (e.g. long QT syndrome and catecholaminergic polymorphic ventricular tachycardia), cause structural disease that is closely associated with severe arrhythmias (e.g. hypertrophic cardiomyopathy), or cause a high propensity for arrhythmia in combination with altered myocardial structure and function (e.g. arrhythmogenic cardiomyopathy). Currently available therapies offer incomplete protection from arrhythmia and fail to alter disease progression. Recent studies suggest that gene therapies may provide potent, molecularly targeted options for at least a subset of inherited arrhythmias. Here, we provide an overview of gene therapy strategies, and review recent studies on gene therapies for catecholaminergic polymorphic ventricular tachycardia and hypertrophic cardiomyopathy caused by MYBPC3 mutations.
This article is part of the Spotlight Issue on Inherited Conditions of Arrhythmia.
1. Introduction
Broadly defined, inherited arrhythmias are disorders caused by one or more genetic mutations that increase the risk of arrhythmia and result in life-long risk of sudden death.1 Inherited arrhythmia syndromes can be classified as disorders of electrophysiologic homeostasis [e.g. long QT syndrome (LQTS); catecholaminergic polymorphic ventricular tachycardia (CPVT)], structural diseases associated with arrhythmia [e.g. hypertrophic cardiomyopathy (HCM)], or a hybrid that involves both high propensity for arrhythmia in combination with structural manifestations (e.g. arrhythmogenic cardiomyopathy). Small molecules that modulate receptors (e.g. β-blockers) or ion channels (e.g. sodium channel blockers) have demonstrated efficacy in some types of inherited arrhythmias, but in many cases patients remain incompletely protected. Progressive myocardial disease is an intrinsic feature of many forms of inherited arrhythmia, and no currently available therapy successfully attenuates disease progression. Recent studies have hinted at the possibility of developing gene therapies for patients with inherited arrhythmias to both control dangerous arrhythmias and to limit disease progression by targeting underlying disease mechanisms. These next-generation therapies have the potential to greatly reduce side effects and improve clinical outcomes.
Herein, we review the current state of the art in this rapidly expanding and exciting field. Since the pathophysiology of these disorders is covered in depth by other reviews within this Spotlight Issue of Cardiovascular Research, we focus on the gene therapy approaches that might be used to treat inherited arrhythmias.
2. Currently available therapies for inherited arrhythmias
Arrhythmias caused by gene mutations represent up to 50% of unexplained deaths in young people.2 As many as 30–40% of inherited arrhythmia patients will first present with cardiac arrest or aborted cardiac arrest.3 This is especially true in patients with CPVT, where the ECG is normal at baseline and fully one-third will have cardiac arrest as the presenting symptom.4 Among the inherited arrhythmias, the most common cause of sudden death is HCM, which causes 36% of cases of sudden cardiac death in competitive athletes.5 Even with an accurate genetic diagnosis, determination of risk for inherited arrhythmia patients remains a challenge because arrhythmic episodes are probabilistic events driven by both internal and external factors. Large clinical studies with deep patient phenotyping have given rise to improved clinical guidelines for common syndromes such as LQTS6 and HCM,7 but ‘low-risk’ patients can still suffer from unpredictable life-threatening events. Therefore, choices for therapy, sports participation, frequency of follow-up, and life-style modifications for risk reduction remain ongoing clinical management challenges.
Choosing appropriate therapeutic options for patients can be difficult for many patients and must factor in the genetic disorder, known medical risk, patient preferences, age, lifestyle, and comorbidities. Beta-blockers remain the backbone for therapy in LQTS and CPVT but are not without therapy-related complications including depression8 and potential worsening of allergic and asthmatic conditions.9 Non-compliance, often secondary to side effects, is a major limitation of beta-blocker therapy in adolescents and young adults.10 Potentially life-threatening breakthrough arrhythmias on beta-blocker therapy are not uncommon in CPVT, affecting 25% of patients during an 8-year follow-up.11 This failure rate on beta-blockers prompted the search for additional therapies including the class I sodium channel blocker flecainide12,13 and left cardiac sympathetic denervation.14 While these advanced therapies have certainly improved outcomes for patients with inherited arrhythmias, failures and non-responders have been reported.
Implantable cardiac defibrillators (ICDs) can be the ultimate ‘safety net’ for patients with an ongoing risk of unstable ventricular arrhythmias and sudden cardiac death.15 Implanted either transvenously into the right ventricle or epicardially, these devices continuously monitor the heart rhythm. Upon detection of a potentially life-threatening arrhythmia, the ICD delivers electric current to the myocardium to interrupt the arrhythmia.16 In patients with documented cardiac arrest secondary to ventricular tachycardia (secondary prevention) or patients meeting high-risk criteria for arrhythmia (primary prevention), ICD implantation can be life-saving protection against future events. However, prediction of risk remains an imperfect science and ICD complications occur in up to 25–30% patients including inappropriate shocks, lead dislodgement/failure, infection, or need for extraction, which carries up to a 3% risk of major complications.17,18 Worse still, is ‘ICD storm’—repeated appropriate discharges, which can cause death in some patients despite appropriate therapy.19 ICD storm affected nearly 30% of CPVT patients with ICDs,20 leading to caution in the use of ICDs in this condition.
In addition to risk for arrhythmic episodes, many inherited arrhythmias, such as HCM21 or arrhythmogenic cardiomyopathy,22 involve progressive myocardial changes that exacerbate arrhythmic risk as well as impair pumping capacity, leading in some cases to the need for heart transplantation. No available therapy has been shown to attenuate these progressive myocardial changes. Often these diseases progress over many years to decades, affording a therapeutic window in which therapies that slow disease progression could have substantial clinical impact.
To summarize, the inherited arrhythmias are life-threatening disorders that are, in many cases, inadequately treated by currently available therapies, which currently are not targeted at the underlying genetic disorder. Addressing this unmet need requires novel therapies that directly target disease mechanisms.
3. Genetic mechanisms in inherited arrhythmias
Disease-causing gene mutations group into recessive and dominant categories. Generally, recessive mutations reduce the expression or activity of a gene product (loss-of-function) and manifest disease only when both alleles are affected, because in heterozygotes the remaining wild-type allele produces sufficient gene product to maintain cellular function. A well-studied albeit rare example of an arrhythmic recessive loss-of-function mutation is CASQ2-D307H, which causes CPVT.23 Calsequestrin 2 (CASQ2) is the major sarcoplasmic reticulum (SR) Ca2+ binding protein, and the D307H missense mutation abrogates Ca2+ binding by the mutant protein.24 Patients with a heterozygous CASQ2D307H/+ genotype do not manifest a clinical CPVT phenotype, because the remaining wild-type (WT) allele expresses sufficient CASQ2 protein, and the mutant protein does not interfere with the activity of the WT protein. However, patients with homozygous inactivating mutations in CASQ2 (e.g. CASQ2D307H/D307H) lack functional protein and manifest the disease.
Mutations can be dominant through two different mechanisms. In haploinsufficiency, a loss-of-function mutation in one allele causes disease because the second allele does not express sufficient gene product to maintain cellular function. For example, heterozygous frameshift mutations in MYBPC3 significantly lead to unstable nonsense mRNA and lower total MYBPC3 protein levels in human tissue,25 suggesting MYBPC3 haploinsufficiency as a disease mechanism. Alternatively, a mutation can be dominant through dominant-negative or gain-of-function mechanisms. A dominant-negative mechanism indicates that the mutation negatively impacts the activity of the remaining WT allele. For instance, the recently described dominant CASQ2-S173I mutation causes CPVT through a dominant-negative mechanism.26 CASQ2-S173I retains its ability to dimerize with the WT protein, but precludes higher order interactions between dimers, which is essential for the Ca2+ buffering activity of CASQ2. As a result, the mutant protein impairs the activity of the WT protein, resulting in the mutant allele being dominant. Gain-of-function mutations are also typically dominant. For example, dominant mutations in the type 2 ryanodine receptor (RYR2) cause CPVT most commonly through gain-of-function mechanisms that increase diastolic channel opening.27
The genetic mechanism underlying an inherited arrhythmia shapes the available therapeutic strategies. Gene therapies may be targeted at gene replacement for haploinsufficient MYBPC3 mutations or reduction of CASQ2 protein. Conversely, dominant-negative or gain of function mutations such as those in RYR2-mediated CPVT may be targeted through modulation of signalling pathways, direct inhibition of the aberrant protein, or alteration of the ratio of WT to mutant protein. Targeted gene therapies must be tailored to the specific pathogenic lesion and carefully designed to avoid off-target effects.
4. Gene therapy
Gene therapy, broadly defined as therapy based on the introduction of genetic material into cells, encompasses viral vectors, oligonucleotides, and modified mRNA (Figure 1). The first human gene therapy trial was performed almost 30 years ago.28 Initial excitement over the promise of gene therapy was tempered by the tragic death of Jessie Gelsinger in 1999 as a result of an overwhelming inflammatory response to an adenoviral gene therapy vector. Further technical refinement of gene therapy platforms, greater experience in the design and implementation of clinical gene therapy trials, and recent spectacular success of some gene therapy trials for immune, retinal, and neurological diseases has led to resurgent excitement in the prospects for gene therapy.29 In 2017, the United States’ Food and Drug Administration (FDA) approved voretigene neparvovec-rzyl (Luxturna), the first gene therapy treatment commercially available in USA. This adeno-associated virus (AAV) gene therapy is administered by subretinal injection to treat patients with retinal disease caused by RPE65 mutation. In the 2 years since approval of Luxturna, the number of ongoing, completed, and approved gene therapy trials increased from 230030 to over 4000 (search of clinicaltrials.org using the term ‘gene therapy’).
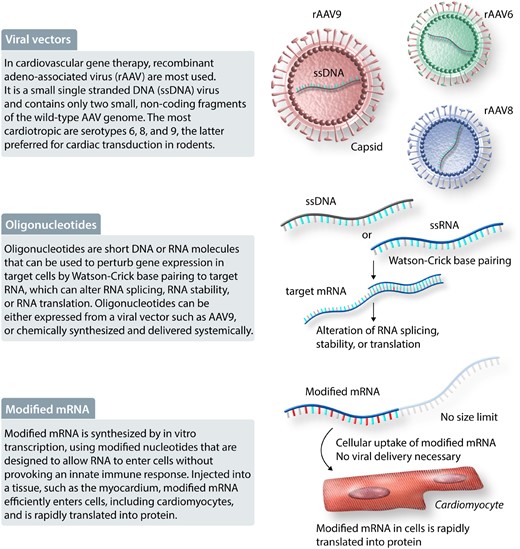
Gene therapy modalities. Viral vectors, oligonucleotides, and modified mRNA are some of the modalities available for gene therapy, defined as therapy based on the introduction of genetic material into cells. Abbreviations: rAAV, recombinant adeno-associated virus; ssDNA, small single-stranded DNA.
4.1 Gene therapy modalities
Several gene therapy modalities are being pursued (Figure 1), each with a unique range of potential activities, specificity, pharmacokinetic properties, and translational challenges (Table 1).
Summary of potential therapeutic modalities for inherited arrhythmias
| Parameter | Viral gene therapy | Oligonucleotides | Modified mRNA | Small molecules |
|---|---|---|---|---|
| Potential activities and benefits |
|
|
|
|
| Specificity |
|
|
|
|
| Dosing |
|
|
|
|
| Dosing frequency |
|
|
|
|
| Challenges |
|
|
|
|
| Parameter | Viral gene therapy | Oligonucleotides | Modified mRNA | Small molecules |
|---|---|---|---|---|
| Potential activities and benefits |
|
|
|
|
| Specificity |
|
|
|
|
| Dosing |
|
|
|
|
| Dosing frequency |
|
|
|
|
| Challenges |
|
|
|
|
Summary of potential therapeutic modalities for inherited arrhythmias
| Parameter | Viral gene therapy | Oligonucleotides | Modified mRNA | Small molecules |
|---|---|---|---|---|
| Potential activities and benefits |
|
|
|
|
| Specificity |
|
|
|
|
| Dosing |
|
|
|
|
| Dosing frequency |
|
|
|
|
| Challenges |
|
|
|
|
| Parameter | Viral gene therapy | Oligonucleotides | Modified mRNA | Small molecules |
|---|---|---|---|---|
| Potential activities and benefits |
|
|
|
|
| Specificity |
|
|
|
|
| Dosing |
|
|
|
|
| Dosing frequency |
|
|
|
|
| Challenges |
|
|
|
|
4.1.1 Viral vectors
Viral vectors have become gene therapy workhorses. Viruses have evolved to efficiently introduce and maintain genetic material in human cells. Recombinant viral vectors are engineered to take advantage of this ability, while minimizing potential toxic activities of WT viruses. For cardiovascular gene therapy, the most commonly used viral vector is based on AAV, a small single-stranded DNA virus that is not known to cause human disease.30 Recombinant AAV (rAAV) engineered for gene therapy contains only two small, non-coding fragments of the WT AAV genome that are required for rAAV packaging; the rest of the rAAV genome is dedicated to the genetic ‘cargo’.31 There are many rAAV serotypes, which differ by the viral proteins that comprise the virus’ outer shell, or capsid, which determines its cell tropism. Among the 12 naturally occurring AAV serotypes, the most cardiotropic in mice are serotypes 6, 8, and 9,32–34 with AAV9 generally being preferred for cardiac transduction via systemic, intravenous delivery. AAV6, AAV8, and AAV9 have typically been used for cardiac gene therapy in large animal and non-human primate models,35 but their relative tissue tropism in humans remains incompletely described. These naturally occurring serotypes have been further engineered to yield novel capsids with unique properties.34 Once an rAAV genome is introduced into a host cell, it enters the nucleus and is converted to double-stranded DNA. The genetic cargo is most often a transgene driven by a tissue-specific promoter. The rAAV genome cannot replicate with the host cell, but in non-dividing cells (like cardiomyocytes), it is episomally maintained for many years.
Important strengths of rAAV are that they are safe, non-integrating, maintain expression for many years, and efficiently transduce many cell types, including cardiomyocytes. Another important strength is that expression can be precisely targeted to desired cell types, by selection of the appropriate capsid and transgene promoters,30,36 as well as inclusion of additional sequences that modulate gene product stability in on-target or off-target cells.37 One limitation of rAAV is that their capsids elicit both innate and adaptive immune responses.38 In large animals, the innate immune response has been reported to cause potentially dose-limiting toxicity.39 As a result of adaptive immunity and cross-reactivity with AAV present in the environment, as many as 40–60% of patients may have pre-existing neutralizing antibodies that exclude them from treatment with a therapeutic product.40 Furthermore, development of adaptive immune responses after initial treatment currently precludes rAAV re-administration. Being limited to single administration raises concerns about correct dosing, durability of therapy, and being able to transduce a sufficiently high percentage of target cells. Immunomodulatory regimens are under development to overcome pre-existing neutralizing antibodies or to permit repeated dosing.41,42 The rAAV cargo may itself elicit an immune response, if it is a gene product that is not normally expressed in the patient, for example, an exogenous, engineered protein, or a normally expressed protein that is absent from a patient due to homozygous gene mutation. A second important limitation of rAAV is the cargo capacity. The maximum size of rAAV’s genetic cargo is ∼4.5 kb, which is less than the size of some large sarcomere or ion channel genes. Methods to overcome this limitation are being developed, including recombination or trans-RNA splicing, or trans-protein splicing between dual AAVs,43–45 or trans-splicing to native pre-mRNA.46 A third consideration for rAAV, which differs from traditional small molecule therapy, is that rAAV will yield tissue that is a mosaic of transduced and non-transduced cells. The percentage of cells that must be transduced for therapeutic efficacy, vs. the feasibility of achieving this transduction efficiency, is likely to be a key determinant of whether or not a therapy will be successful. A fourth practical issue with rAAV is the high cost of large-scale GMP-grade manufacturing. It is anticipated that technical and process improvements as well as economies of scale will reduce rAAV manufacturing cost moving forward.
4.1.2 Oligonucleotides and modified mRNA
Oligonucleotides are short DNA or RNA molecules that can be used to perturb gene expression in target cells.47 Specificity of oligonucleotide therapy is achieved through Watson–Crick base-pairing to target RNA, which can alter RNA splicing, RNA stability, or RNA translation. This base-pairing specificity could potentially be used to selectively perturb a mutant gene product. Oligonucleotides can be either expressed from a viral vector such as AAV9, or chemically synthesized and delivered systemically. For synthetic oligonucleotides, chemical modifications have been developed to enhance stability, modulate specificity and target binding affinity, enhance cell uptake, and increase in vivo half-life. As a result of these modifications, synthetic oligonucleotide therapy can be achieved through subcutaneous dosing at weekly or even quarterly intervals. Currently, modifications to selectively transduce hepatocytes are available, but efficient or selective targeting of most other parenchymal cell types, including cardiomyocytes, is not yet possible.48
Modified mRNA is a relative newcomer to the gene therapy space.49 RNA is synthesized by in vitro transcription, using modified nucleotides that are designed to allow RNA to enter cells without triggering the innate immune response. Modified mRNA injected into a tissue, such as the myocardium, efficiently enters cells, including cardiomyocytes, and is rapidly translated into protein.50 Modified mRNA is attractive because it does not have a theoretical size limit, and it could potentially permit a gene to be administered like a drug, with predictable pharmacokinetics and dose response. Currently, a major barrier to most translational applications is the lack of a method to selectively and efficiently target cells of interest, such as cardiomyocytes, after systemic delivery. Combined with its relatively short half-life of ∼24–48 h, the potential applications are currently limited to those in which transient expression following myocardial injection could yield the desired response. However, we can anticipate broader application of this gene therapy technology as the delivery barrier is surmounted.
4.2 Gene therapy strategies
These gene therapy modalities can be deployed to treat inherited arrhythmias following a few therapeutic strategies, which are largely shaped by the underlying genetic mechanism.
4.2.1 Gene replacement
The most straightforward gene therapy strategy is gene replacement (Figure 2), in which a healthy gene copy is introduced into a cell, usually to replace a defective copy (recessive loss-of-function or haploinsufficiency). Indeed, the currently approved AAV therapies—Glybera (lipoprotein lipase), Luxturna (RPE65), Zolgensma (SMN)—use gene replacement strategies. Gene replacement would not directly address dominant-negative or gain-of-function mechanisms, but expression of a WT copy can result in down-regulation of the endogenous mutant allele or at least increase the fraction of WT transcript.51 For the most part, gene replacement requires nucleic acids that are too large to synthesize as oligonucleotides. Therefore, rAAV is most commonly used for gene replacement strategies, although one could envision exciting opportunities for modified mRNA if its delivery challenges could be solved.

Gene therapy strategies. Gene therapy strategies to treat inherited arrhythmias can act at the level of DNA or mRNA, depending on the genetic mechanism. Abbreviations: rAAVs, recombinant adeno-associated virus; CaMKII, Ca2+/calmodulin-dependent protein kinase II; CPVT, catecholaminergic polymorphic ventricular tachycardia; PTC, premature termination codon; NHEJ, non-homologous end-joining; HDR, homology-directed repair; ssODN, single-stranded oligonucleotide; GoF, gain-of-function, shRNA, short hairpin RNA; AON, antisense oligonucleotides; PTM, pre-trans-splicing molecule.
4.2.2 Allele-specific silencing
A second therapeutic strategy is to silence a mutant allele that causes disease through dominant-negative or gain-of-function mechanisms. Since these genetic mechanisms are dominant, the remaining allele is typically normal. This therapy is based on the hypothesis that the remaining allele would be sufficient for normal cell function, as long as the deleterious effects of the mutant allele could be attenuated or eliminated. Allele-specific silencing could be achieved through rAAV-mediated expression of a mutation-specific short hairpin RNA,52,53 or through an allele-specific antisense oligonucleotide,54 which ideally should suppress the mutant allele but not affect expression of the WT allele. This strategy would require a different therapeutic product for each mutation. This is feasible for cases where a single mutation accounts for a large number of patients. However, without a significant change in the regulatory landscape, this strategy would not be commercially feasible in disorders where any single mutation accounts for a small number of patients. In addition, this strategy becomes increasingly difficult as the nature of the mutation becomes more subtle; for example, it is not currently clear the extent to which allele-specific suppression can be reliably achieved for single-nucleotide variants.
4.2.3 Modulation of signalling pathways
A third therapeutic strategy is to modulate signalling pathways or biological processes essential for disease pathogenesis. This is conceptually similar to how most small molecule therapies work. Compared to small molecules, gene therapy approaches might increase the spectrum of targetable processes or permit cell type-specific inhibition that is difficult to achieve with small molecules. Most gene therapy strategies directed at treating acquired forms of heart failure are pursuing this general approach.55 One example of this strategy is the work by several groups to enhance cardiomyocyte Ca2+ handling in heart failure. In experimental heart failure models, and in human failing myocardium, SERCA2 is down-regulated. AAV-mediated expression of SERCA2 improved heart function in animal models.56,57 Unfortunately, a clinical trial of AAV-SERCA2 did not demonstrate efficacy.58 However, low-vector dosing in the trial prevented it from adequately testing the underlying strategy.
4.2.4 Modulation of splicing
Altered splicing is a fourth therapeutic strategy, applicable to situations in which the effects of a mutation can be attenuated by inducing exon skipping or inclusion. The best example of this strategy is Duchenne’s muscular dystrophy (DMD). The most common DMD mutations alter the translational reading frame, with most mutations occurring within exons that encode the central rod domain, which contains 24 spectrin repeats. In the majority of cases, exon skipping could ‘reframe’ these frameshift mutations to yield an internally deleted protein that retains partial activity.59 The first antisense oligonucleotide-based drug, eteplirsen, was approved by the FDA for use in patients with DMD caused by frameshift mutations in exons 48–50, which are ‘reframed’ by eteplirsen-induced skipping of exon 51.60 Efficiency of oligonucleotide-induced exon-skipping in vivo is low, in part due to inefficient oligonucleotide entry into muscle cells, and clinical efficacy of eteplirsen remains unclear.59
4.2.5 Genome editing
Genome editing is a recently developed therapeutic strategy61 that may be deployed for treatment of some inherited arrhythmias. CRISPR/Cas9 is an endonuclease that is targeted to specific sequences by an engineered guide RNA (gRNA). The targeted sequence is cleaved by Cas9 with remarkable specificity and efficiency in vivo, including in post-mitotic cardiomyocytes.62 Repair by non-homologous end-joining results in generation of insertion or deletion mutations at the cleavage site, which can induce frameshift mutations or exon skipping. Indeed, Cas9 and gRNA delivered by rAAV were shown to efficiently induce exon skipping in mouse and large animal models of DMD.63–65 The ability of rAAV-delivered Cas9 to achieve therapeutic allele-specific ablation of a gain-of-function mutation was recently demonstrated in a mouse model of Wolf–Parkinson–White syndrome associated with cardiac hypertrophy caused by a PRKAG2H530R/+ mutation.66 rAAV-mediated Cas9 and gRNA delivery selectively ablated the mutant allele and restored cardiac morphology and function. Cas9 has been engineered to permit targeted epigenetic modification, including gene activation and gene repression,67 and selective base editing.68 Most recently, Cas9 was fused to reverse transcriptase to create ‘prime editing’, which promises to enable highly efficient and precise somatic genome modification.69 However, this large editing enzyme exceeds the cargo capacity of rAAV, so that its deployment will require transduction by two rAAVs, or further engineering to reduce its size.70
5. Experimental evidence for gene therapy in inherited arrhythmias
In this section, we outline the current experimental evidence supporting gene therapy to treat specific inherited arrhythmias in model systems, most commonly mouse models and induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs).
5.1 Catecholaminergic polymorphic ventricular tachycardia
CPVT is a highly malignant inherited arrhythmia disorder characterized by recurrent arrhythmias induced by emotional stress or exercise.71 While the estimated prevalence is 1:10 000, cases of CPVT are frequently the most common diagnosis in studies of sudden cardiac death in the young, suggesting under-diagnosis.72 Mutations in RYR2 cause the majority (∼60%) of CPVT cases. Much less frequently, CPVT is caused by CASQ2 mutations, which are classically recessive,23,73 although recently rare dominant CASQ2 mutations have been described.26,74 Both RYR2 and CASQ2 mutations result in abnormal diastolic Ca2+ release from SR during adrenergic stimulation (Figure 3).75 Gene therapy approaches for CPVT caused by dominant mutations in RYR2 or recessive mutations in CASQ2 have shown promising efficacy in experimental model systems, as summarized in the following sections.
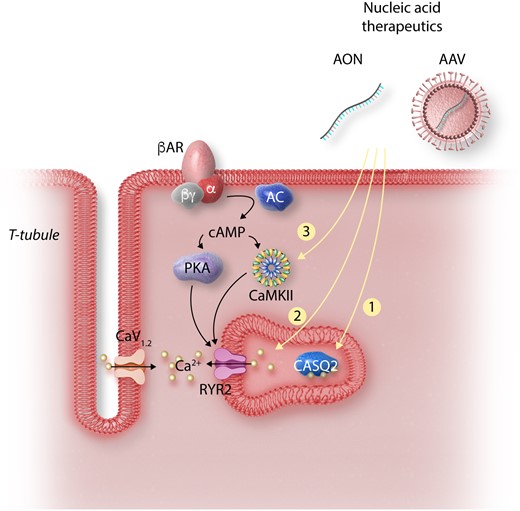
Strategies for gene therapy in CPVT. Cardiomyocyte depolarization drives Ca2+ through the L-type Ca2+ channel (CaV1.2) stimulating Ca2+ release from RYR2. Adrenergic stimulation activates PKA and CaMKII, which alters Ca2+ handling by phosphorylating multiple targets. In CPVT, CaMKII phosphorylation of RYR2 unmasks the abnormalities caused by RYR2 or CASQ2 mutations, resulting in increased Ca2+ leak and arrhythmia. Strategies for CPVT gene therapy target key signalling events in the pathogenesis of the disorder: (1) AAV-mediated expression of wild-type CASQ2 restores levels in SR to correct the autosomal recessive form of CPVT. (2) Ablation or suppression of mutant RYR2 allele by CRISPR or RNA interference results in more wild-type RYR2 tetramers. (3) Inhibition of CaMKII using an AAV-delivered selective inhibitory peptide suppresses abnormal Ca2+ release. Abbreviations: AON, antisense oligonucleotides; siRNA, small-interfering RNA; RYR2, ryanodine receptor 2; CASQ2, calsequestrin 2; CaMKII, Ca2+/calmodulin-dependent protein kinase II; CaV1.2, L-type Ca2+ channel, PKA, protein kinase A; AC, adenylate cyclase; βAR, beta-adrenergic receptor.

Strategies for gene therapy in HCM. HCM is mainly caused by dominant mutations in sarcomere genes, with the majority of mutations found in MHY7 or MYBPC3. The mechanisms by which HCM mutations in sarcomere genes cause arrhythmias are incompletely understood. Gene therapy strategies that have been tested to prevent arrhythmias and other HCM phenotypes have included: (1) gene replacement, (2) allele-specific silencing, and (3) modulation of Ca2+ signalling pathways. Abbreviations: AON, antisense oligonucleotides; siRNA, small-interfering RNA; GoF, gain-of-function; CaMKII, Ca2+/calmodulin-dependent protein kinase II.
5.1.1 CPVT caused by recessive mutations in CASQ2
With a coding region of 1.2 kb, full-length CASQ2 is easily packaged into rAAV for delivery to the heart. As proof of the concept that AAV-mediated CASQ2 gene replacement could treat CPVT caused by CASQ2 deficiency, researchers in the Priori laboratory used AAV9 to express full-length WT CASQ2 in the hearts of Casq2 null mice. Mice with complete absence of CASQ2 protein75 were treated with AAV9-CASQ2 particles (2.6×1011 vg per new-born mouse) and studied at 20 weeks of age. The exogenously expressed CASQ2 protein localized appropriately with RYR2 in isolated adult CMs, suppressed triggered activity in isolated cells, and prevented catecholamine-induced bi-directional VT, a hallmark of CPVT.76 In a follow-up study, the authors showed that AAV9-CASQ2 was similarly effective in Casq2R33Q/R33Q mice, which harbour a human missense mutation that reduces protein level and alters CASQ2 interaction with Ca2+. The protective effects remained at 1 year of age. Furthermore, AAV9-CASQ2 treatment was also effective when given to adult Casq2 mutant mice at a dose of 5.2×1011 vg/mouse.77 Importantly, these studies showed that transduction of ∼40% of ventricular cardiomyocytes was sufficient to protect mice from catecholamine-induced arrhythmia,77 potentially due to the transduced cardiomyocytes protecting untransduced cardiomyocytes through a source-sink relationship, as predicted by computational simulation.78 Kurtzwald-Josefson et al.79 further demonstrated the effectiveness of AAV9-CASQ2 in mice engineered to express CASQ2-D307H, a non-functional mutant protein, instead of WT CASQ2, and found a dose–response relationship between the level of CASQ2 protein expression and protection from ventricular arrhythmias.
5.1.2 CPVT caused by dominant mutations in RYR2—allele-specific silencing
The majority of CPVT patients have dominant heterozygous mutations in RYR2.71 While segregating RYR2 mutations in kindreds have been described,4 the majority of patients with CPVT have de novo mutations in RYR2.80 Currently, over 150 different RYR2 mutations associated with clinical CPVT have been identified, with most clustering in four ‘hot spot’ regions.81 The functional RYR2 channel is a homotetramer. Conceptually, a gene replacement strategy could potentially be effective against most RYR2 mutations by inducing down-regulation of the endogenous mutant allele and by reducing the fraction of protein tetramers containing a mutant RYR2 polypeptide. Unfortunately, RYR2’s gargantuan size (cDNA length 14 901 nucleotides) precludes packaging into AAV particles. Therefore, alternative gene therapy strategies have been tested.
Researchers in the Priori lab pursued RNA interference targeting a mutant Ryr2 allele.53 This strategy is supported by the lack of observable arrhythmogenic or functional phenotype in mice lacking one Ryr2 allele from targeted heterozygous knockout.82 In the well-studied Ryr2R4496C/+ CPVT mouse, a dominant missense mutation causes CPVT.83 Bongianino et al. screened over a dozen siRNA duplexes for their ability to selectively silence the mutant allele. One specific siRNA (siRYR2-U10) induced an 80% reduction in the R4496C mutant allele while reducing the WT allele by <20%. The authors used a microRNA template to engineer an expression cassette that would express the two strands of the siRNA under the cytomegalovirus (CMV) promoter. This expression cassette was packaged into rAAV9 (AAV9-miRNA-U10) and delivered to mice. AAV9-miRNA-U10 [4.2×1012 vg per 30-day-old mouse (P30)] reduced the ratio of mutant to WT transcript from 1:1 to 1:2, without reducing the overall transcript level. This was calculated to increase the fraction of WT tetramers from 6.25% to 19.75%. AAV9-miRNA-U10 administration at either P8 or P30 reduced delayed after-depolarizations and triggered activity in isolated CPVT cardiomyocytes, and protected CPVT mice from catecholamine-induced ventricular tachycardia. Importantly, AAV9-miRNA-U10 did not adversely affect ventricular systolic function.53 This study provided proof-of-concept that AAV-mediated suppression of a dominant RYR2 mutant allele could potentially be effective therapy.
Genome editing offers an unprecedented method for permanent suppression of mutant or aberrant proteins.61 To determine if somatic, allele-specific genome editing is a viable treatment for RYR2-related CPVT, the Wehrens laboratory tested whether AAV9-delivered CRISPR/Cas9 and gRNA could protect Ryr2R176Q/+ CPVT mice from ventricular arrhythmia.84 In this study, the authors first designed a gRNA that targeted a silent restriction site (3 nucleotide difference compared to the WT sequence) that had been engineered into the genome 15 bp upstream of the R176Q missense mutation when the knockin allele was created.85 The therapeutic rAAV was administered to P10 mice at 1×1012 vg per mouse. At 5–6 weeks of age, programmed ventricular stimulation demonstrated significant reduction in induced arrhythmias in Ryr2R176Q/+ mice treated with the therapeutic rAAV. Deep sequencing demonstrated indel mutations at the Cas9 cleavage site in 21% of the mutant cDNA, compared to 0.2% of the WT cDNA, confirming strong allele specificity. This result also suggests that therapeutic efficacy can be achieved with as little as 20% reduction of mutant transcripts. While there was no evidence for indels and top predicted off-target sites, there was evidence for insertion of AAV internal tandem repeats as well as whole viral genomes within the cut site of Ryr2, which likely inactivate the affected alleles. Total Ryr2 was reduced by 25–30% on both mRNA and protein levels, but a potential deleterious effect on cardiac function was not evaluated.84 The significance of viral DNA insertion in this study requires further evaluate and suggests that similar events should be examined when using AAV-mediated CRISPR genome editing. Overall, this study suggested that Cas9-mediated allele-specific cleavage may be an effective therapeutic approach for CPVT caused by dominant RYR2 mutations.
The demonstration of specific allele targeting for RYR2 mutations as a method to treat RYR2-related CPVT is certainly an exciting development. However, several important questions remain. The genome-editing strategy presented by Pan et al.84 was an idealized case using a previously inserted three base polymorphisms with an associated PAM near the target mutation, which greatly facilitated the design of the allele-specific gRNA. The aforementioned work on Cas9 genome editing of a point mutation in PRKAG2,66 or of siRNA targeting of RYR2R4496C,53 indicates that single-nucleotide variants can be selectively targeted, although the frequency with which this can be achieved using Cas9 or siRNA remains to be determined. As an alternative, corrective sequence replacement by homology-directed repair (HDR) would overcome the challenge of allele-specific targeting, but the efficiency of HDR in non-dividing cells and the competing occurrence of frameshift-inducing non-homologous end-joining may be limiting.86 More recent advances in using base-editors or ‘prime-editing’ may circumvent the limited efficiency of traditional sequence correction, but there are currently no studies using these technologies in cardiomyocytes.68,69
Technical considerations aside, at a strategic level allele-specific therapies will be challenging to translate in disorders like CPVT, where there are over 150 different mutations,71 and each individual mutation affects a low number of individuals. As a result, an allele-specific strategy would require a different therapeutic vector for each mutation, and each vector would need to be designed and tested as a separate product. This will complicate pre-clinical development, as each specific mutation will need a different pre-clinical model. More importantly, this fragmentation of the patient population into patients with specific mutations will complicate performance of clinical trials to establish efficacy and safety of each product, and make it difficult to make each product therapeutically viable. Therapeutic strategies which are mutation-independent or are applicable to a more broad range of pathogenic genotypes are likely to be more translationally viable.
5.1.3 CPVT gene therapy targeting Ca2+/calmodulin-dependent protein kinase II
A hallmark of CPVT is that the baseline electrocardiogram is normal, and ventricular tachycardia is typically precipitated by catecholaminergic stimulation induced by exercise or emotions. This suggests that signalling pathways activated by catecholamines are required to ‘unmask’ the arrhythmic potential of CPVT mutations. In turn, this suggests that a therapeutic strategy designed to block these signalling pathways may be effective, allele-independent therapy. Catecholamines activate cardiac β-adrenergic receptors, which stimulate multiple downstream signalling cascades. Among these are cAMP-dependent protein kinase-A (PKA)87 and Ca2+/calmodulin-dependent protein kinase II (CaMKII),88 which phosphorylate multiple targets to increase heart rate, ventricular contractility, and myocardial performance. Among these targets is RYR2, which is phosphorylated by PKA and CaMKII, resulting in enhanced Ca2+ release from the SR following β-adrenergic receptor stimulation. This increased Ca2+ release augments myocardial performance as part of the ‘fight-or-flight’ response but can be deleterious in the context of disease.89–91 Most CPVT-associated RYR2 mutations are gain-of-function and increase diastolic Ca2+ release in the context of sympathetic activation.27 In both murine and iPSC-CM CPVT models, CaMKII inhibition by the small molecule KN-93 normalized Ca2+ handling and reduced arrhythmia,92,93 suggesting that CaMKII is an essential link between catecholamine stimulation and arrhythmia in CPVT. While KN-93 is a potent CaMKII inhibitor, it has other electrophysiological effects including potassium channel blockade, limiting the conclusion that CaMKII activity is required to unmask the arrhythmic phenotype in CPVT.
To establish the critical importance of CaMKII activation in the pathogenesis of CPVT and uncover arrhythmia mechanisms, Park et al. created the first engineered tissue model of CPVT by plating iPSC-CMs on micro-modelled gelatin substrates94 and then using optogenetics95 to enable optical point stimulation.96 Propagation of the resulting Ca2+ wave across the tissue was measured using a fluorescent Ca2+-sensitive dye. To simulate exercise, the ‘opto-chips’ were subjected to rapid pacing and isoproterenol stimulation. Whereas opto-chips assembled from WT iPSC-CMs exhibited uniform Ca2+ wave propagation under all simulated exercise conditions, CPVT (RYR2R4651I/+) opto-chips were vulnerable to re-entry, a tissue level equivalent of ventricular tachycardia, under conditions of simulated exercise. Re-entry of CPVT opto-chips was inhibited by the potent and highly specific CaMKII inhibitor autocamtide-2-related inhibitory peptide (AIP)97 but not by a PKA inhibitor. Furthermore, mutation of serine 2814 of RYR2, the site phosphorylated by CaMKII, to alanine in the context of the RYR2R4651I mutation fully prevented re-entry induced by simulated exercise. In contrast, ablation of the PKA site at RYR2-2808 did not alter the vulnerability of CPVT opto-chips to re-entry induced by simulated exercise. These data established that CaMKII phosphorylation of RYR2 at S2814 is necessary to unmask the arrhythmia phenotype in CPVT and identified CaMKII as a CPVT therapeutic target.96
CaMKII has been a therapeutic target for cardiovascular disease for over a decade, as CaMKII knockout is well-tolerated in the heart and is protective against heart failure and arrhythmia.98,99 However, a longstanding barrier to the development of clinically viable CaMKII small molecule inhibitors is the essential role of CaMKII outside of the heart. In contrast to the heart, where CaMKII activation is dispensable and even exacerbates heart disease induced by multiple different insults,100 CaMKII function is necessary in the central nervous system, where it is fundamental for learning and memory.101 To achieve cardiac-selective CaMKII inhibition, Bezzerides et al.102 used cardiotropic AAV9 and the cardiac-selective troponin T promoter to drive expression of AIP selectively in the heart. Administration to neonatal animals at P3 resulted in robust and specific expression in cardiomyocytes but not in other organs, including the brain. Treatment of RYR2R176Q/+ CPVT mice at either P3 or 6 weeks of age blocked arrhythmias induced by catecholamine stimulation or by programmed ventricular stimulation. AIP also significantly reduced triggered activity in isolated adult CPVT cardiomyocytes, consistent with reversal of the arrhythmia phenotype at a cellular level. Testing in iPSC-CMs derived from multiple different CPVT patients with diverse pathogenic genotypes suggests that this therapeutic approach is mutation-independent,102 although more mutations need to be studied. Further optimization of the inhibitory peptide may enhance its specificity and potency.98,103 This proof-of-concept study demonstrated the potential utility of AAV-delivered peptide inhibitors to the heart for therapeutic benefit and opens up exciting possibilities for a possible durable, single-dose treatment for patients with CPVT.
5.2 Hypertrophic cardiomyopathy
With an estimated prevalence of 1:500, HCM is the most common cardiac genetic disease.21 HCM is characterized by left ventricular hypertrophy, unexplained by secondary aetiologies, and a non-dilated left ventricle with preserved ejection fraction. The ventricular hypertrophy is often asymmetric, most commonly resulting in the greatest hypertrophy of the basal interventricular septum. The hypertrophied myocardium can cause left ventricular outflow tract obstruction, and the thickened muscle has poor diastolic function. There is often left atrial enlargement. At the cellular level, cardiomyocytes have increased size and disorganized arrangement, and there is increased interstitial fibrosis. Patients are at risk for ventricular tachycardia, ventricular fibrillation, and sudden death. Approximately a quarter of patients also develop atrial fibrillation, which is poorly tolerated due to diastolic dysfunction. In general, non-sustained ventricular tachycardia is associated with severity of hypertrophy and supraventricular arrhythmias.
HCM is caused by dominant mutations in sarcomere genes.104 Mutations in at least 10 different sarcomere genes have been shown to cause HCM, with mutations in nearly twice as many additional genes likely to be causative.21,105,106 The majority of patients (70–80%) carry mutations in MYH7 or MYBPC3, which encode β-myosin heavy chain and cardiac myosin binding protein-C (cMyBP-C), respectively (Figure 4).21,105,106 The mechanisms by which HCM mutations in sarcomere genes cause arrhythmias is unknown, but likely involves abnormalities of myofilament Ca2+ sensitivity that lead to perturbations in cardiomyocyte Ca2+ handling,107 coupled with interstitial fibrosis, myocardial ischaemia, and myocyte disarray.
Currently available treatment options for HCM are primarily prescribed to alleviate symptoms by reducing heart rate and improving cardiac relaxation and energetics. Surgical myectomy is used to relieve outflow tract obstruction, and ICDs and antiarrhythmic drugs are used to prevent sudden death.7,21,105 These therapies do not target the underlying disease pathophysiology or modify the evolution of the primary myocardial disease.
Identified risk factors for ventricular arrhythmia include several parameters indicative of the extent of cardiac remodelling and/or ischaemia: severe cardiac hypertrophy, extensive late gadolinium enhancement on cardiac MRI (indicative of cardiac fibrosis), and severe left ventricular outflow tract obstruction.108 These risk factors point out the important role of progressive myocardial disease in creating the arrhythmia-vulnerable substrate in HCM. Therefore, gene therapies targeting HCM have so far mainly focused on preventing these structural alterations and disease progression by addressing the underlying genetic component rather than targeting arrhythmogenesis per se. One way to achieve this is to correct the mutation in the germline, as has been shown recently for a pathogenic MYBPC3 variant.109 This study demonstrated that co-injection of human sperm that carried a 4-bp pathogenic MYBPC3 deletion with Cas9 genome editing components into oocytes with a normal MYBPC3 allele resulted in 72% of embryos with two normal MYBPC3 alleles. Whereas post-zygotic Cas9 genome editing resulted in mosaic embryos, this pre-zygotic modification was free of mosaicism. Possible side effects for the resulting embryo are unclear since whole-genome sequencing identified multiple new insertions and deletions that occurred within poly-A or poly-GT repeats.109 Ultimately, this study showed that CRISPR/Cas9 is a powerful tool to combat inherited diseases, albeit with a myriad of unresolved ethical and safety issues.
5.2.1 Gene replacement in HCM
Gene replacement therapy is of specific interest for pathogenic truncating MYBPC3 mutations, which mostly result in cMyBP-C haploinsufficiency. Importantly, infants carrying bi-allelic truncating mutations in MYBPC3 develop a HCM or DCM phenotype after birth that quickly evolves to heart failure and death within 1 year.110 Initial studies were done in mouse models harbouring homozygous Mybpc3 variants that lead to cMyBP-C haploinsufficiency, thereby mimicking the genetic situation of infants.111 These two studies used lentivirus to deliver full-length murine Mypbc3 to the myocardium of homozygous Mybpc3-null mice, resulting in improved contractile function of skinned cardiomyocytes in vitro and ventricular function in vivo. These studies provided proof-of-concept of the benefit of Mybpc3 gene replacement, but lentivirus is unlikely to be a translatable myocardial gene therapy modality because it requires direct myocardial injection, does not efficiently transduce cardiomyocytes, and randomly integrates into the genome, resulting in potentially genotoxic effects. Mearini et al.51 used AAV9 at 4 different doses to deliver full-length WT Mybpc3 cDNA to mice homozygous for a prevalent human mutation that mutates the splice donor site of exon 6 (Mybpc3KI/KI mice), yielding a hypomorphic allele that expresses 80% less protein.112 A single systemic delivery of AAV9-Mybpc3 to P1 mice dose-dependently normalized left ventricular mass at 34 weeks of age. The level of full-length cMyBP-C protein was increased, albeit not to normal levels, and accumulation of mutant mRNAs and consequently the production of potentially dominant-negative truncated gene products was suppressed.112 Furthermore, transduction of engineered heart tissue derived from cardiac cells of neonatal Mybpc3-targeted knock-out or knock-in mice with a similar rAAV9 construct encoding either WT or a phosphomimetic cMyBP-C restored force of contraction to control levels.113,114
To test MYBPC3 gene replacement in a human background, Prondzynski et al.115 took advantage of iPSC technology and generated patient-specific iPSC-CMs from an individual carrying a pathogenic truncating MYBPC3 variant. The rAAV-mediated delivery of the human full-length MYBPC3 cDNA in patient-specific iPSC-CMs restored MYBPC3 mRNA and cMyBP-C protein levels 7 days after transduction, and reduced iPSC-CM hypertrophy. Although MYBPC3 gene transfer to unrelated control iPSC-CMs resulted in higher levels of mRNA, stoichiometry of the sarcomere was preserved and cMyBP-C protein level was unchanged.115 Similar results were obtained by adenovirus-mediated expression of full-length MYBPC3 in human embryonic stem cell-derived cardiomyocytes carrying a truncating MYBPC3 mutation.116 Whereas gene replacement by expression of full-length MYBPC3 has been successful in mouse and human HCM iPSC-CM models, replacement via an RNA trans-splicing approach was less successful.115,117,Trans-splicing repaired mRNA was detected but the efficiency (0.14% in mice and 1% in iPSC-CMs) was not enough to yield functional benefit. Similarly, RNA trans-splicing approach worked in mice, but to a low efficiency and did not rescue the cardiac dysfunction.117 These studies show that full-length MYBPC3 gene replacement corrects pathogenic functional abnormalities in cardiomyocytes and heart tissue.
5.2.2 Antisense oligonucleotides in HCM
As an alternative strategy to replace deficient gene transcripts, oligonucleotide-mediated exon skipping or suppression of the mutant allele has been evaluated in HCM. Antisense-based exon skipping was tested in the aforementioned Mybpc3KI/KI mice,118 in which a G > A mutation of the exon 6 splice donor site causes aberrant mRNA splicing.112 Antisense oligonucleotides that induced skipping of Mybpc3 exons 5 and 6, expressed using rAAV9 and U7 RNA polymerase III promoters, suppressed the effects of the endogenous mutation, abolished cardiac dysfunction, and prevented the development of left ventricular hypertrophy when administered to new born, but not 4-week-old, mice.118
Antisense-based allele-specific suppression was also tested for the dominant HCM mutation MYH7R403Q. The encoded mutant protein is stable, incorporates into the sarcomere, and causes dominant-negative effects; meanwhile, hemizygosity of the analogous Myh6 was associated with normal heart function in mice,119 and MYH7 mutations are unlikely to be pathogenic via haploinsufficiency.120 Therefore, treatment goals are not gene replacement but elimination of the damaging missense transcript. One study utilized RNA interference by AAV9-mediated delivery of small-interfering RNAs (siRNAs) targeting the dominant Myh6R403Q allele in mice, a mouse model of human HCM caused by MYH7R403Q mutation. AAV9 delivery of a specific short hairpin RNA selectively reduced mutant allele expression by 29% without affecting expression of the WT allele. This moderate reduction of the mutant allele was sufficient to delayed progression of hypertrophy and fibrosis.52 These studies suggest that antisense-mediated exon skipping or allele-specific suppression is potentially an effective therapeutic strategy.
5.2.3 Modulation of signalling pathways in HCM
Intracellular Ca2+ handling is altered in HCM cardiomyocytes and likely contributes to arrhythmia risk.107 In myocardium from patients with sarcomere mutations, CaMKII was upregulated and activated, although phosphorylation of RYR2 was not significantly increased.121 This was also found in a murine HCM model.122 At the same time, SERCA2 abundance and SR Ca2+ uptake were reduced.121 The impact of correcting these Ca2+ handling-related abnormalities in HCM has been investigated. Transgenic expression of a CaMKII inhibitory peptide ameliorated HCM phenotypes caused by transgenic expression of a dominant mutant form of cardiac troponin T, TNNT2R92W.122 Interestingly, this CaMKII inhibitory strategy exacerbated rather than ameliorated HCM phenotypes caused by transgenic expression of the closely related TNNT2R92L. This difference was thought to reflect a role of CaMKII in the early progression of HCM caused by the R92W but not the R92L mutation.
SERCA2 overexpression has also been tested in HCM caused by transgenic overexpression of TPM1-E180G.123 Adenoviral expression of SERCA2 in neonatal mice reduced hypertrophy and fibrosis and normalized haemodynamics when compared with untreated mice at 3 months of age. Alternatively, SERCA2 activity can be increased by suppression of its endogenous inhibitor, PLN (phospholamban). TPM1-E180G transgenic mice that lacked PLN showed improved ventricular function, reduced collagen deposition, and normalized cardiac size.124 Unfortunately, it is unlikely that this approach is applicable to humans, since heterozygous PLN loss-of-function in humans causes dilated cardiomyopathy.125 Nevertheless, these studies provide proof-of-concept that the progression of at least some forms of HCM can be attenuated by normalization of Ca2+ handling.
5.3 Other arrhythmia disorders
Arrhythmia is a common phenotype in many dilated cardiomyopathies.126 However, with some cardiomyopathies, such as deletion of arginine 14 of PLN (PLN-R14del),127,128 arrhythmias are a central feature of the disease. This mutation causes 10–15% of dilated cardiomyopathy and arrhythmogenic cardiomyopathy in the Netherlands.129 This dominant mutation causes irreversible super-inhibition of SERCA2.128 The iPSC-CMs harbouring the PLN-R14del mutation exhibited frequent Ca2+ transient irregularities. An AAV vector that expressed antisense RNA against endogenous PLN and a PLN cDNA resistant to this antisense RNA restored normal Ca2+ handling in the mutant PLN-R14del iPSC-CMs.130
Congenital long QT syndrome is characterized by prolongation of the QT interval on the surface ECG leading to repolarization abnormalities and predisposing patients to life-threatening ventricular arrhythmias including torsades de pointes.131 With an estimated prevalence of 1:2000,132 congenital long QT syndrome is the most prevalent channelopathy. While there are >13 genes associated with LQTS, the large majority of patients have dominant mutations in just three genes, KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3).80 These mutations reduce repolarization reserve by impairing potassium flux or prolonging inward depolarizing currents, increasing cardiac action potential duration. RNA interference has also been shown to restore normal potassium currents in a model of LQTS type 2 (LQT2).133 To model LQT2, the authors introduced both WT KCNH2 and a construct containing the KCNH2 mutation E637K134 into HEK293 cells. Patch-clamp electrophysiology demonstrated impaired potassium channel function consistent with a typical LQT phenotype. After transfection with an optimized siRNA targeted against the mutant channel, there was nearly complete restoration of potassium currents to WT levels. While this study further demonstrates the potential feasibility of allele-specific targeting for treating channelopathies, it is limited by the need for allele-specific therapy. Given the sheer number of pathogenic variants associated with LQTS and the low number of individuals treatable by any given allele-specific reagent, commercializing allele-specific knockdown will be challenging. Incomplete expression or correction of repolarization within the myocardium may further exacerbate LQT syndrome by contributing to electrical inhomogeneity and worsening the risk for developing arrhythmia.
6. Conclusions and future perspectives
6.1 Current challenges to clinical translation
As summarized in the preceding sections, studies in small animals have provided proof-of-concept for several gene therapies for inherited arrhythmias that would be desirable to move towards clinical trials. However, moving from proof-of-concept to clinical translation requires addressing several important challenges.
Robust expression in the heart and minimal extra-cardiac expression are key factors for selection of promoters to direct cardiac gene therapy. The CMV promoter and its derivatives have been used in multiple preclinical studies and even in the first-in-human cardiac gene therapy trial because of its robust expression.135–137 However, the CMV promoter has widespread activity and cardiac targeting by AAV isotype selection is insufficient to restrict expression to the heart.138 Promoters of cardiac-specific genes including cardiac troponin T (cTnT) and alpha myosin heavy chain (αMHC) have improved specificity for cardiac tissue with comparable expression levels to CMV.139 While the minimal cTnT promoter is particularly attractive given its small size (∼420 bp), adding additional cis-regulatory elements may be beneficial in some scenarios to augment the level and specificity of expression.140 However, to date, there have not been large animal studies to test the effectiveness of rationally designed cardiac-specific promoters for gene therapy.
While preclinical studies in mice have largely used AAV9 as the preferred serotype for effective cardiac transgene expression, human trials may use other AAV serotypes because of high liver transduction and some CNS penetration.138,141 AAV6 and AAV8 have also shown robust cardiac expression in both rodent and large animal models137,142 and do not efficiently penetrate the blood–brain barrier. However, these serotypes also robustly transduce the liver.142 Capsid engineering promises to overcome these limitations and several reports of chimeric AAVs have already demonstrated reduced transduction of the liver and brain, but further research will be necessary to establish the ideal rAAV for cardiac gene therapy.31,143 Capsid engineering may also reduce the impact of pre-existing neutralizing antibodies.31
Therapies that modulate cardiac rhythm carry the conceptual risk of pro-arrhythmia. This risk bears special consideration in the case of AAV gene therapy, which will create inhomogeneities within the myocardium as a result of cardiomyocytes that have and have not been transduced by AAV. Thorough evaluation of pro-arrhythmia risk requires testing in larger animals that have heart rates, size, and cardiac physiology comparable to humans.
The immune response to gene therapy is another area of active investigation. Activation of the innate immune response, resulting in potentially lethal systemic inflammation, has been reported in large animals that have received large AAV doses. Repeated vector dosing, which could facilitate high-level transduction or improve durability of gene therapy, is currently precluded by development of neutralizing antibody after single AAV administration. Although AAV transduction of cardiomyocytes itself results in minimal inflammatory response, the potential for deleterious immune responses should be considered when the therapeutic cargo encodes a foreign protein.
6.2 Concluding thoughts
The last decade has seen exciting advances in gene therapy and their applications to inherited arrhythmias. Gene replacement therapy with AAV vectors has yielded miraculous results in the clinic for non-cardiac diseases. Challenges for cardiac gene therapy using viral vectors include demonstration of efficacy at transduction efficiencies achievable in humans, and demonstration of safety at these doses. Unforeseen off-target effects, development of proarrhythmia, durability of therapy, and lack of reversibility after treatment remain important concerns for translation. Developing methods that overcome pre-existing neutralizing antibodies and that permit repeated dosing would greatly enhance the clinical utility of viral vectors, improve safety, and address concerns over therapeutic durability. Reducing the enormous cost of clinical scale AAV production will be essential to make viral gene therapy economically feasible. For oligonucleotides and modified mRNAs, development of methods that enable efficient and selective cardiomyocyte delivery would be game changing.
Authors’ contributions
V.J.B. and M.B. drafted the original manuscript. L.C. and W.T.P. edited the manuscript. V.J.B. and M.P. designed the figures.
Acknowledgements
We would like to thank Suellen Lopes Oliveira for her help in designing the figures.
Conflict of interest: V.J.B. and W.T.P. hold a provisional patent on gene therapy for inherited arrhythmias by AAV-mediated CaMKII inhibition. L.C. holds a patent on gene therapy vectors for treating cardiomyopathy that was licensed to DiNAQOR, and is member of the DiNAQOR Scientific Advisory Board. W.T.P. is a consultant for AveXis.
Funding
W.T.P. and V.J.B. were supported by funding from the U.S. Office of the Assistant Secretary of Defense for Health Affairs through a Technology/Therapeutic Development Award of the Peer-Reviewed Medical Research Program (PR181262/W81XWH-19-1-0473) and by NIH (HL140197 to V.J.B.; UH3 HL141798 to W.T.P.). L.C. was supported by funding from the DZHK (German Centre for Cardiovascular Research) and the University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
References
null null,
European Heart Rhythm Association, Heart Rhythm Society

{kind=link}
{kind=link}
{kind=link}
{kind=link}