





Abstract
The aryl hydrocarbon receptor (AHR) mediates 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced toxicity that can lead to chloracne in humans. A characteristic of chloracne, in contrast to acne vulgaris, is shrinkage or loss of sebaceous glands. Acne vulgaris, on the other hand, is often accompanied by excessive sebum production. Here, we examined the role of AHR in lipid synthesis in human sebocytes using distinct classes of AHR ligands. Modulation of AHR activity attenuated the expression of lipogenic genes and key proinflammatory markers in the absence of canonical DRE-driven transcription of the AHR target gene CYP1A1. Furthermore, topical treatment with TCDD, which mediates DRE-dependent activity, and SGA360, which fails to induce DRE-mediated responses, both exhibited a decrease in the size of sebaceous glands and the number of sebocytes within each gland in the skin. To elucidate the mechanism of AHR-mediated repression of lipid synthesis, we demonstrated that selective AHR modulators, SGA360 and SGA315 increased the protein turnover of the mature sterol regulatory element-binding protein (mSREBP-1), the principal transcriptional regulator of the fatty acid synthesis pathway. Interestingly, selective AHR ligand treatment significantly activated the AMPK-dependent kinase (AMPK) in sebocytes. Moreover, we demonstrated an inverse correlation between the active AMPK and the mSREBP-1 protein, which is consistent with the previously reported role of AMPK in inhibiting cleavage of SREBP-1. Overall, our findings indicate a DRE-independent function of selective AHR ligands in modulating lipid synthesis in human sebocytes, which might raise the possibility of using AHR as a therapeutic target for treatment of acne.
Acne is a chronic inflammatory disease of the skin that affects many people during adolescence and can persist into adulthood. Although the pathophysiology of acne is poorly identified, several factors in addition to excessive sebum production contribute to the development of acne lesions, including modified keratinization process, alterations in sebum composition, colonization of hair follicles by the commensal bacterium Cutibacterium acnes, and induction of inflammatory signaling in the skin (Thiboutot et al., 2009). Bacteria utilize sebum as a growth substrate in the skin. Sebum is a complex mixture of lipids secreted by sebaceous glands and is considered a key factor in acne pathogenesis. Triglycerides and fatty acids comprise the major portion of human sebum. Additional sebum components include wax esters, squalene, and to a lesser extent cholesterol esters, cholesterol and free fatty acids (Greene et al., 1970). Specialized cells of the sebaceous gland, sebocytes, accumulate lipid droplets in the cytosol as they differentiate, and mature sebocytes subsequently release their content into the follicular duct as a result of complete cellular disintegration (Niemann and Horsley, 2012). Previous studies demonstrated lipids in human sebum to have antioxidant, antimicrobial, proinflammatory and anti-inflammatory activity; thus, sebum is an essential component of the skin (Smith and Thiboutot, 2008). Elevated secretion of sebum, also known as seborrhea, however, is a common feature of acne patients, and the exact mechanism by which excess sebum leads to the formation of acne is not clear.
A potential mediator of increased sebum production is a class of transcription factors called the sterol response element binding proteins (SREBPs) that modulate the expression of multiple genes involved in lipid and cholesterol synthesis and uptake. The transcriptional activity of SREBPs is regulated by sterol levels in cells. The precursor form of SREBP (pSREBP) resides in the membrane of the endoplasmic reticulum (ER), where low intracellular sterol levels lead to mobilization of pSREBP to the Golgi. Upon sequential proteolytic cleavage in the Golgi, the transcriptionally active N-terminal fragment of SREBP (mSREBP) translocates into the nucleus, where it modulates the transcription of numerous genes involved in the lipogenic pathway. However, high intracellular sterol levels restrict the precursor SREBP to the ER membrane, and de novo cholesterol synthesis is diminished (Brown and Goldstein, 2009). SREBPs also actively function in the transcriptional regulation of lipid metabolism. There are three forms of SREBPs; SREBP-1a, SREBP-1c, and SREBP-2 encoded by two genes SREBP-1 and SREBP-2 in humans (Yokoyama et al., 1993). SREBP-2 mainly functions in cholesterol metabolism, whereas SREBP-1a and SREBP-1c have been implicated in both cholesterol and fatty acid synthesis pathways. Despite its lower transcriptional activity compared with SREBP-1a, SREBP-1c is recognized as a key transcription factor involved in fatty acid biosynthesis.
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that belongs to the basic helix-loop-helix/PER-ARNT-SIM family of proteins. In the presence of an agonist, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), AHR translocates to the nucleus, where it forms a heterodimer with aryl hydrocarbon receptor nuclear translocator (ARNT). The AHR/ARNT complex then binds to dioxin response elements (DREs) in the promoter region of a wide variety of target genes and modulates their transcription in a cell-, tissue-, and context-specific manner. Three different classes of AHR ligands are agonists, antagonists, and selective AHR modulators (SAhRMs). Agonists are capable of both inducing DRE-driven transcriptional activity and mediating attenuation of acute phase gene expression (Murray et al., 2010). In contrast, SAhRMs exhibit the ability to mediate repression of acute phase gene expression with weak (eg, SGA315) or no DRE-mediated activity (eg, SGA360). An antagonist fails to induce any known AHR activity and suppresses both agonist and SAhRM from mediated activity. Prolonged exposure to the potent AHR agonist TCDD leads to toxicity due to dysregulation of gene expression by the AHR. The characteristic phenotype of dioxin-mediated toxicity in humans is chloracne, a hyperkeratotic skin disorder with sebaceous gland hypotrophy (Kurokawa et al., 2009; Ramot et al., 2009; Suskind, 1985). Whether chloracne is a DRE-driven response to TCDD exposure or a non-DRE response is not known.
We have previously demonstrated that AHR activation attenuates the expression of genes involved in fatty acid and cholesterol synthesis in a DRE-independent manner (Tanos et al., 2012a,b). Furthermore, SAhRMs have been identified and found to modulate transcriptional activities independent of DRE binding (Muku et al., 2017; Murray et al., 2010). Here, we examined the capacity of selective AHR ligands, SGA360 and SGA315, to attenuate fatty acid synthesis in immortalized human sebocytes. We found that both ligands significantly lower the expression of several enzymes involved in the fatty acid synthesis pathway and reduce lipid production in human sebocytes. The negative regulation of fatty acid gene expression is elucidated by AHR was mediated through a decrease in transcriptional activity of the transcription factor SREBP-1. Moreover, topical application of AHR ligands on skin of mouse ears reduces the number and size of sebaceous glands. Taken together, these results clearly indicate an important role for AHR in the regulation of lipid synthesis in the skin, and its potential as a therapeutic target for the treatment of acne.
MATERIALS AND METHODS
Reagents
SEB-1 cells are an immortalized normal sebocyte line established in Dr. Thiboutot’s laboratory (Thiboutot et al., 2003). Cell viability assay was conducted using CellTiter 96 Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI). SGA360 and SGA315 were synthesized as previously described (Murray et al., 2010). β-Naphthoflavone (BNF) was obtained from Indofine (Hillsborough, NJ). TCDD was kindly provided by Dr. Steve Safe (Texas A&M University). Cytokines were purchased from PeproTech (Rocky Hill, NJ). Primary antibodies used for immunoblot analysis are listed in Supplementary Table 1.
Cell culture
SEB-1 cells were maintained in low glucose Dulbecco’s minimal essential medium (Sigma, St. Louis, MO) and Ham’s F-12 nutrient mixture (3:1), supplemented with 2.5% fetal bovine serum (FBS) (HyClone Labs, Logan, UT), 24 µg/ml adenine, 0.0452 µg/ml hydrocortisone, 10 ng/ml insulin, 1.2 × 10−10 M cholera toxin (Sigma), 3 ng/ml epidermal growth factor (Austral Biologicals, San Ramon, CA), 100 units/ml penicillin, and 100 μg/ml streptomycin (Sigma) in a humidified incubator at 37°C, with an atmospheric composition of 95% air and 5% CO2. Primary keratinocytes were isolated from neonatal pups as previously described and cultured at 36°C with 7% CO2 (Smith et al., 2017). Cells were exposed to BNF (5 µM), TCDD (10 nM), SGA360 (10 µM), SGA315 (10 µM), or carrier solvent dimethyl sulfoxide for indicated time points.
RNA isolation and reverse transcription
RNA samples were isolated from cultured SEB-1 cells using TRI Reagent according to the manufacturer’s instructions (Sigma Aldrich). cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA).
Real-time quantitative PCR
Real-time quantitative PCR assays were performed as previously described (Muku et al., 2017). PerfeCTa SYBR Green SuperMix for iQ (Quanta Biosciences, Gaithersburg, MD) was used in the reaction and data were analyzed using MyIQ software (Bio-Rad Laboratories, Hercules, CA). The qPCR primers utilized are listed in Supplementary Table 2.
Preparation of cell lysates
Lysates were prepared from SEB-1 cells in MENG (25 mM MOPS, 2 mM EDTA, 0.02% sodium azide, and 10% glycerol), 1% Igepal CA-630, protease and phosphatase inhibitors (Roche, Indianapolis, IN). Cell homogenates were then centrifuged at 14 000 × g for 10 min and proteins were analyzed. For detection of SREBP-1, SEB-1 cells were lysed in RIPA buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA, 1 mM EGTA, 140 mM NaCl, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1% Igepal CA-630) supplemented with protease and phosphatase inhibitors. Cell lysates were centrifuged at 14 000 × g for 15 min and protein concentration was determined by Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). Nuclear and cytosolic extracts were prepared as previously described (Muku et al., 2017). Briefly, cell pellets were resuspended in MENG + protease inhibitors and homogenized with a stainless-steel Dura-Grind dounce homogenizer (Wheaton Instruments, Millville, NJ). Cell homogenates were centrifuged at 1000 × g for 20 min. The supernatant was then subjected to centrifugation at 42 000 × g for 30 min to generate cytosolic extracts. The nuclear pellet was washed three times with MENG, extracted with MENG + 500 mM NaCl and the extract collected after centrifugation at 42 000 × g for 30 min.
Immunoblotting analysis
Cell lysates were resolved on 8% or 10% SDS-tricine polyacrylamide gels. Proteins were transferred to PVDF membrane and detected using antibodies listed in Supplementary Table 1. Primary antibodies were visualized with biotin-conjugated secondary antibodies (Jackson Immunoresearch, West Grove, PA) and subsequent incubation with 125I-streptavidin followed by autoradiography. Quantification was performed with the ImageJ software. However, for SREBP-1, SCAP, INSIG1, AMP-dependent kinase (AMPK), and pAMPK protein determination, the Simple Wes capillary system was utilized (Protein Simple, San Jose, CA).
Lipid staining
SEB-1 cells were seeded at 12-well plates and incubated with ligands for 48 h. At the end of the incubation period, cells were washed with PBS and fixed in 4% formaldehyde. The cells were washed with distilled water three times and stained with Oil red O solution for 1 h at room temperature. The cells were then washed in distilled water and stained with hematoxylin followed by additional washes in distilled water. The dye was eluted using 100% isopropanol, and optical density (OD) was measured at 500 nm. For the fluorescent staining of lipids, the DyRect Live-Cell Neutral Lipid Imaging Kit (Marker Gene Technologies, Eugene, OR) was used and protocol was followed according to the manufacturer’s instructions.
Stable isotope incorporation into fatty acids
Upon treatment of SEB-1 cells with SGA360 (10 µM) or SGA315 (10 µM) in media containing lipid-depleted serum, sebocyte growth media was switched to labeling media using glucose-free DMEM supplemented with sebocyte growth medium components (excluding F-12 nutrient mix). For isotope labeling experiment, universally labelled 13C (U-13C) glucose and U-13C acetate were used, whereas natural abundance glucose and acetate were employed for control groups. Cells were incubated in described media in the presence of ligands for an additional 24 h. Following PBS wash, scraping, and pelleting cells, lipids were extracted for further detection.
Ultra performance liquid chromatography–mass spectrometry analysis of fatty acids
Lipids were extracted according to the previously described protocol with minor modifications (Cajka and Fiehn, 2016). Briefly, samples were extracted twice with isopropanol:water:ethyl acetate (30:10:60, vol/vol/vol) and internal standard. The combined supernatant was dried under nitrogen gas and reconstituted with isopropanol:acetonitrile:water (45:35:20) for ultra performance liquid chromatography–mass spectrometry analysis with 10 µl injection volume (Bielawski et al., 2009). Samples were separated by reverse phase HPLC using a Vanquish UHPLC system (Thermo Fisher Scientific) with a Waters (Milford, MA) CSH C18 column (150 mm × 1 mm; 1.7 µm particle size) maintained at 55°C and a 20 min aqueous/acetonitrile/isopropanol gradient, at a flow rate of 80 µl/min. Solvent A was water:isopropanol (40:60) with 10 mM ammonium acetate and solvent B was isopropanol:acetonitrile (90:10) with 10 mM ammonium acetate. The initial condition was 70% A and 30% B, increasing to 43% B at 2 min, 55% B at 2.1 min, 65% B at 12 min, 85% B at 18 min, and 99% B at 20 min, held at 99% B until 25.0 min before returning to the initial conditions at 25.1 min. The eluate was then delivered into an Orbitrap Fusion Lumos Tribrid Mass Spectrometer using a Heated Electrospray Ionization Source (Thermo Fisher Scientific). The mass spectrometer was operated in negative ion mode; the capillary voltage was set at 2.5 kV, with an radio frequency (RF) lens at 30%. The mass spectrometer was operated in data-dependent mode with a survey scan at maximum resolution (1 000 000) from 200 to 1000 m/z, followed by up to 20 MS/MS product ion scans at a resolution of 7500, using HCD collision energy of 25% with a 6% collision energy spread. The parent ion was isolated using the quadrupole with an isolation window of 1 m/z. Analytes of oleic acid and stearic acid were identified with authentic reference standards. Data were processed by MS-DIAL after raw files conversion by Abf Converter (Lai et al., 2018; Tsugawa et al., 2015).
Statistical analysis
Data were analyzed using one-way ANOVA in GraphPad Prism (v.6.01) software to determine statistical significance between treatments. p-Values <.05 were considered statistically significant (*p < .05; **p < .01;***p < .001; ****p < .0001).
RESULTS
Selective AHR Ligands Downregulate Expression of Genes Involved in the Fatty Acid and Cholesterol Synthesis Pathways in Human Sebocytes
The immortalized sebocyte cell line SEB-1 used in this study, has previously been shown to be androgen responsive, produce lipids, and express sebocyte markers, and thus, was utilized in our mechanistic studies (Thiboutot et al., 2003). In order to mimic the acne pathology at the molecular level (ie, elevated sebum levels), we incubated cells overnight in media containing lipid-depleted serum so that the absence of any external sterol source would induce the expression of genes required for lipid synthesis. To determine the ability of AHR ligands to repress the expression of genes directly involved in fatty acid synthesis, SEB-1 cells were treated with AHR ligands for 16 h. Distinct classes of AHR ligands, namely BNF (agonist), SGA360 (SAhRM), and SGA315 (SAhRM/partial agonist) repressed the mRNA levels of FASN, SCD1, and ELOVL6, all of which function in the fatty acid synthesis pathway (Figure 1A). In addition, we determined that selective ligand-activated AHR downregulates the expression of cholesterol synthesis genes, ACAT2, FDPS, FDFT1, HMGCR, IDI1, MVD, MVK, and SQLE in SEB-1 cells (Figure 1B).
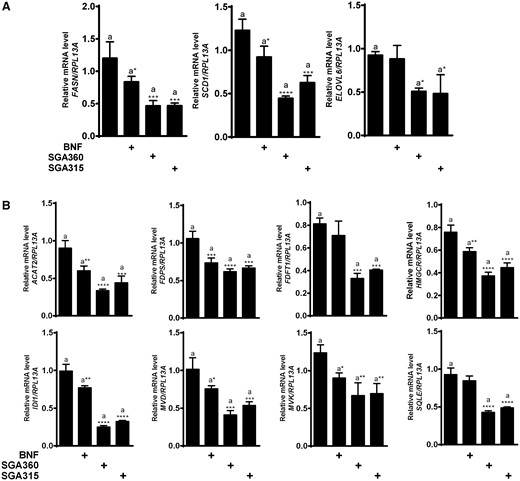
Exposure to aryl hydrocarbon receptor (AHR) ligands attenuates mRNA levels of fatty acid and cholesterol synthesis genes. After 16 h of treatment with BNF (5 µM), SGA360 (10 µM), or SGA315 (10 µM) in media containing delipidated serum, mRNA levels of indicated genes were examined by qRT-PCR analysis. Expression analysis of genes in the fatty acid synthesis pathway (A) and expression analysis of genes in the cholesterol synthesis pathway (B). Data are presented as mean ± SD, n = 3; significance was determined by one-way ANOVA.
SEB-1 cells utilized in this study have been demonstrated to exhibit sebaceous phenotype. However, production of certain sebum components squalene and wax esters are diminished in immortalized sebocytes compared with levels found in human sebum (Smith and Thiboutot, 2008). Furthermore, human sebum is largely comprised of triglycerides and fatty acids. For these reasons, we decided to focus on the capacity of AHR ligands to attenuate the fatty acid synthesis pathway.
Fatty Acid and Cholesterol Synthesis Genes Are Repressed upon Ligand Exposure in an AHR-Dependent Manner
Attempts to knockdown expression of the AHR in SEB1 cells yielded insufficient reduction in AHR levels. Therefore, to demonstrate that the attenuation of gene expression is mediated by the AHR, we utilized primary mouse keratinocytes isolated from newborn wild-type or Ahr-deficient mice. In wild-type keratinocytes, selective AHR modulators SGA360 and SGA315 significantly decreased the mRNA levels of cholesterol synthesis genes Hmgcr, Idi1, and Scd1 similar to human sebocytes. However, ligand treatment of Ahr-deficient keratinocytes failed to exhibit repression of Hmgcr, Idi1, indicating an AHR-mediated regulation of lipogenic genes that is not due to nonspecific effects (Figure 2A). Furthermore, selective AHR ligands SGA360 and SGA315 were able to inhibit the expression of FDFT1 and SCD1 genes in SEB-1 cells in a dose-dependent manner (Figure 2B).
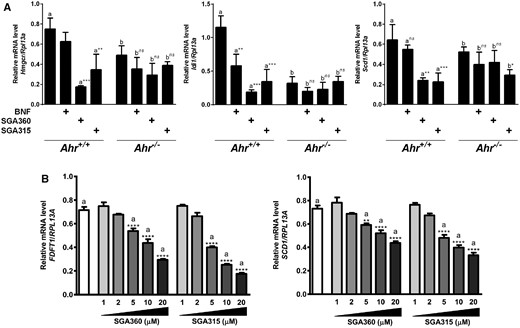
Selective AHR ligand-mediated repression of gene expression is mediated through AHR. A, Wild-type or Ahr-deficient primary mouse keratinocytes were treated with AHR ligands SGA360 (10 µM) or SGA315 (10 µM) for 16 h, and gene expression was analyzed by qRT-PCR. B, SEB-1 cells were treated with the indicated concentrations of SGA360 or SGA315 for 8 h in media containing delipidated serum, and mRNA levels were determined by qRT-PCR analysis. Data are presented as mean ± SD, n = 3; significance was determined by one-way ANOVA.
SGA360 Attenuates Proinflammatory Markers in SEB-1 Cells
In addition to lipogenesis, inflammation is a major contributing factor to acne pathogenesis. Thus, we examined the previously reported capability of the selective AHR modulator SGA360 in repressing inflammatory gene expression using human SEB-1 cells (Muku et al., 2017). Treatment of SEB-1 cells with IL1B and IL6 in SEB-1 prominently upregulated the mRNA expression of COX2, IL1B, and IL6, whereas pretreatment of SEB-1 cells with SGA360 resulted in a significant decrease in basal and cytokine-induced gene expression of all three genes. SGA360 exhibited a greater repressive capacity toward the COX2 gene in both basal and induced states when compared with IL1B and IL6 (Figure 3). Collectively, these results suggest that selective AHR modulation inhibits both lipogenic and inflammatory gene expression in human sebocytes.
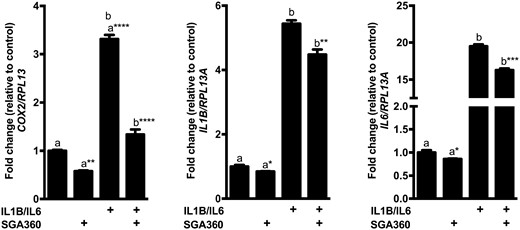
Selective AHR activation attenuates expression of proinflammatory genes in SEB-1 cells. SEB-1 cells were pretreated with SGA360 (10 µM) for 1 h, followed by human proinflammatory cytokines IL1B (10 ng/ml) and IL6 (10 ng/ml) for 6 h. Gene expression was assessed by qRT-PCR analyses. Data are presented as mean ± SD, n = 3; significance was determined by one-way ANOVA.
AHR Attenuates Protein Levels of Fatty Acid and Cholesterol Synthesis Enzymes in Human Sebocytes
Next, we investigated whether distinct classes of AHR ligands, TCDD, SGA360, and SGA315, are capable of repressing the protein levels of cholesterol and fatty acid synthesis enzymes in the immortalized SEB-1 cell line. We applied AHR ligand treatment following overnight incubation of the cells in delipidated media in order to induce the expression of enzymes involved in the cholesterol and fatty acid synthesis pathways and mimic the in vivo protein expression profile in acne patients. Consistent with AHR-mediated decrease in mRNA levels, 48 h-ligand treatment diminished protein expression of the fatty acid synthesis enzymes FAS and SCD1 (Figure 4A), as well as cholesterol synthesis enzymes HMGCR, FDFT1, MVD, FDPS, and IDI1 in SEB-1 cells (Figure 4B). Similar to our findings at the mRNA level, immunoblot analysis demonstrated that SGA360 and SGA315 were more capable of repressing the expression of enzymes compared with the agonist TCDD. Importantly, the attenuation of fatty acid and cholesterol expression was not a consequence of cytotoxicity upon treatment with AHR ligand exposure (Figure 4C).
Consistent with reduced expression of fatty acid synthesis enzymes mRNA and protein levels, AHR ligands SGA360 and SGA315 were able to attenuate lipid synthesis in SEB-1 cells as examined qualitatively by Oil Red O staining and fluorescent live cell lipid staining (Figs. 5A–D). Furthermore, de novo lipid synthesis was quantified by 13C-acetate incorporation into 13C-labeled C18 fatty acids and observed a similar significant decrease in synthesis of 13C-labeled C18 fatty acids by SGA360 and SGA315 relative to control cells (Figure 5E).
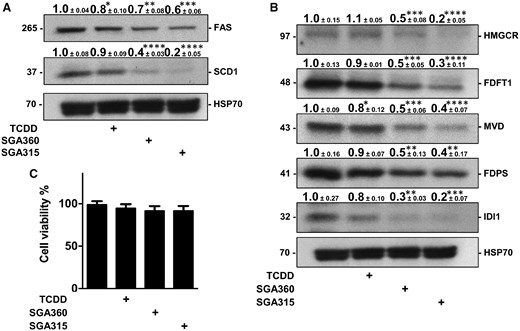
AHR activation diminishes protein levels of fatty acid synthesis and cholesterol enzymes. A, B, After 48 h of treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD; 10 nM), SGA360 (10 µM), or SGA315 (10 µM) in media containing delipidated serum, protein levels of indicated enzymes in SEB-1 cells were examined by quantitative immunoblot analyses. C, Cell viability was determined by MTT assay upon 48 h of ligand exposure. Data are presented as mean ± SD, n = 3; significance was determined by one-way ANOVA.
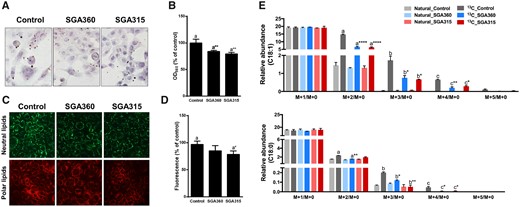
Selective AHR modulators are able to decrease lipid synthesis in SEB-1 cells. SEB-1 cells were treated with SGA360 (10 µM) or SGA315 (10 µM) for 48 h in the presence of linoleic acid and insulin. Lipids were stained by Oil Red O (A, B) or using live cell lipid imaging kit (C, D). E, Upon 48 h of ligand exposure (10 µM) in delipidated media, SEB-1 cells were incubated for an additional 24 h in stable isotope containing media (see Materials and Methods section) in the presence of ligands.U-13C-incorporated oleic acid and stearic acid were measured by liquid chromatography–mass spectrometry. Data are presented as mean ± SD, n = 3; significance was determined by one-way ANOVA.
Distinct Classes of AHR Ligands Exhibit Different Effects on AHR Translocation and Target Gene Expression
Upon binding to agonists, AHR translocates to the nucleus and forms an AHR/ARNT dimer that binds to DREs in target genes. However, this does not typically occur in the case of antagonist binding. To examine whether selective ligand binding leads to AHR nuclear retention, we performed immunoblotting assays on subcellular fractions isolated from SEB-1 cells treated with vehicle, TCDD (10 nM), BNF (5 μM), SGA360 (10 μM), or SGA315 (10 μM) for 16 h. Cytosolic AHR protein levels were comparable in vehicle and SGA315-treated samples, and higher than TCDD or BNF treatment as agonists prompted nuclear translocation of AHR and subsequent proteolytic turnover, although SGA360 treatment increased the cytosolic AHR levels (Figure 6A). Nuclear AHR levels in SGA315 treated samples were moderately higher compared with the control group, whereas TCDD and BNF increased AHR levels in the nucleus to a greater extent than SGA315. Similar to known AHR antagonists, SGA360 did not cause nuclear retention of the AHR (Figure 6A). In addition, qRT-PCR analysis revealed that the cytochrome P-4501A1 (CYP1A1) expression was significantly induced by the agonist BNF, but not affected by SGA360 treatment. SGA315 exhibited a modest but statistically significant CYP1A1 upregulation, which was ∼7-fold lower compared with BNF-mediated induction (Figure 6B).
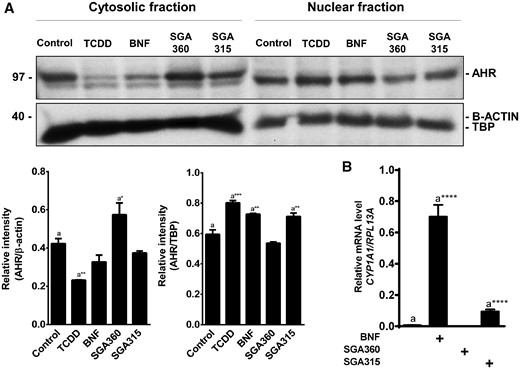
Translocation of AHR to the nucleus and target gene expression upon exposure to different classes of ligands. Upon 16 h of treatment with TCDD (10 nM), BNF (5 μM), SGA360 (10 μM), or SGA315 (10 μM), (A) protein levels of AHR were examined in subcellular fractions by immunoblot analyses, or (B) CYP1A1 expression was assessed by qRT-PCR. Data are presented as mean ± SD, n = 3; significance was determined by one-way ANOVA.
Ligand Activation of AHR Reduces the Size of Sebacous Glands in the Ears of Wild-Type Mice
Our in vitro findings prompted an investigation of the in vivo effects of topical treatment with different AHR ligands in vivo with regard to sebaceous gland morphology. First, we examined whether a lack of AHR expression effects sebaceous gland size, which can be accomplished through examination serial H&E histological sections and measuring the diameter of each gland at the midpoint when it is largest. In this regard, Ahr–/– mice exhibited significantly larger sebaceous glands when compared with Ahr+/+ mice. In addition, ears of Ahr–/– mice had an increased number of sebocytes within each gland and a higher number of individual sebaceous glands were present compared with histological ear sections from Ahr+/+ mice (Figure 7A), indicating a critical role for endogenous AHR activity in sebaceous gland physiology. Next, we examined whether topical application of AHR ligands TCDD and SGA360 is capable of decreasing the size of sebaceous glands in the ears of Ahr+/+ and Ahr–/– mice. Serial histological analysis from mouse ears topically administered with TCDD or SGA360 for 14 days demonstrated that the sebaceous glands in the ears of Ahr+/+ mice treated with TCDD or SGA360 were smaller compared with control mice, with a more pronounced effect observed following TCDD treatment. We did not observe this effect in Ahr–/– mice, indicating that sebaceous gland atrophy is mediated in an AHR-dependent manner. Similarly, the number of sebocytes per gland was significantly lower in TCDD-treated ears, and modestly reduced in SGA360-applied ears compared with control ear tissue samples in Ahr+/+ mice, but not in Ahr–/– mice (Figure 7B).
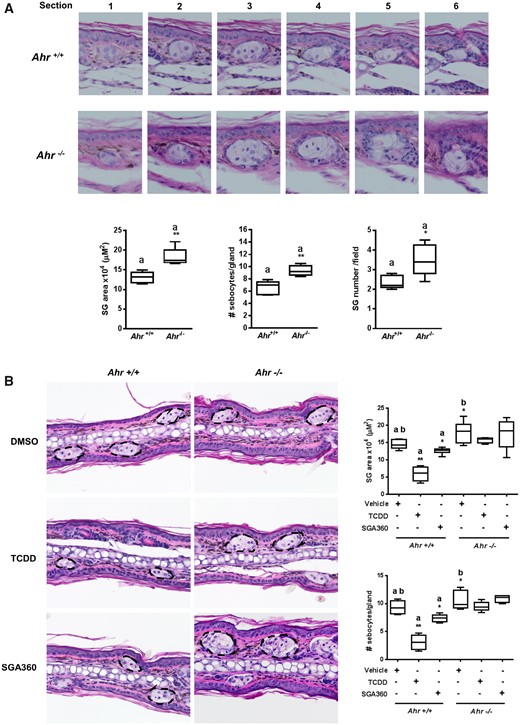
Topical application of AHR ligands reduces the size of sebaceous glands in mouse ears. A, After 14 days of topical application of TCDD or SGA360, mouse ears were sectioned, and H&E stained. B, Serial ear sections were taken to determine the largest middle section of glands in order to measure a cross-sectional area. Data are presented as mean ± SD, n = 3; significance was determined by one-way ANOVA.
AHR Attenuates Levels of Transcriptionally Active mSREBP-1
In order to gain mechanistic insight into the AHR-mediated repression of enzymes that are part of the fatty acid biosynthesis process, we examined the effect of selective AHR ligands on SREBP-1, the principal transcriptional regulator of fatty acid synthesis. SGA360 and SGA315 did not significantly affect the mRNA levels of SREBP-1a in SEB-1 cells. However, SREBP-1c transcripts were significantly reduced upon ligand treatment (Figure 8A). Selective ligands SGA360 and SGA315 significantly reduced the protein levels of the transcriptionally active mSREBP-1, although pSREBP-1 levels remained unaltered by ligand treatment (Figure 8B). Interestingly, the expression of factors involved in SREBP-1 processing, namely SCAP, S1P, and S2P, was not significantly altered, but INSIG1 was modestly increased at the protein level upon ligand treatment (Figs. 8C and D).
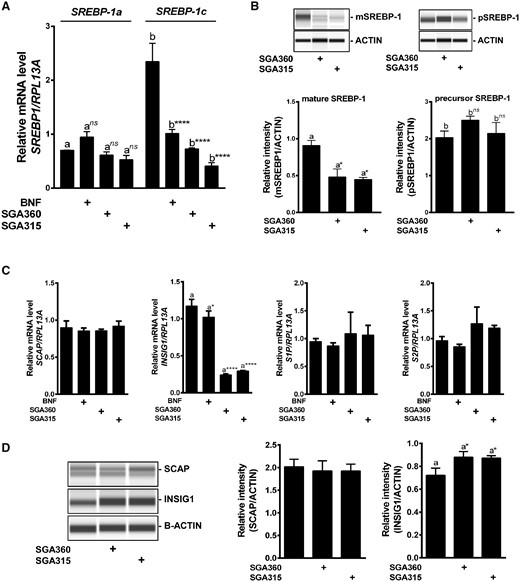
Selective AHR activation leads to a decrease in the transcriptionally active mSREBP-1 protein level without altering factors involved in SREBP proteolytic processing. SEB-1 cells were treated with SGA360 (10 µM) or SGA315 (10 µM) for 24 h in delipidated media, and SREBP-1 expression was examined at (A) mRNA or (B) protein level. C, Upon 24 h of ligand exposure, mRNA levels of SCAP, INSIG1, S1P, and S2P were assessed by qRT-PCR and (D) protein expression of SCAP and INSIG1 was analyzed by WES capillary system. Data are presented as mean ± SD, n = 3; significance was determined by one-way ANOVA.
Selective Activation of AHR Increases the Protein Turnover of the mSREBP-1
Next, we examined the effect of selective AHR ligands on the half-life of mSREBP-1 protein. The addition of the protein synthesis inhibitor, cycloheximide, resulted in a decrease in protein levels over the course of 8 h as expected. However, treatment with SGA360 and SGA315 led to a significantly higher rate of protein turnover of cleaved mSREBP-1 compared with the control group (Figure 9A). Cotreatment with AHR ligands and the calpain inhibitor ALLN largely reversed the AHR-mediated inhibition of the mSREBP-1 protein (Figs. 9B and C), suggesting that SGA360 and SGA315 decreased the mSREBP-1 protein, at least partly through inducing proteolytic degradation.
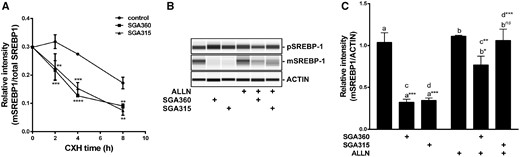
The liganded AHR increases the turnover of the transcriptionally active mSREBP-1 protein and ALLN treatment attenuates ligand mediated decrease in mSREBP1. A, SEB-1 cells were cotreated with cycloheximide (1 µg/ml) and SGA360 (10 µM) or SGA315 (10 µM) over the course of 8 h, mature SREBP-1 protein levels were examined by WES. B, Upon 24 h of treatment with ligands (10 µM) and ALLN (10 µg/ml), SREBP-1 protein levels were examined by WES and (C) the relative protein levels are shown in panel B. Data are presented as mean ± SD, n = 3; significance was determined by one-way ANOVA.
Phosphorylated AMPK Levels Inversely Correlate with the Mature SREBP-1 Protein upon Selective Ligand Exposure
The previously reported role of the AMPK in inhibition of the SREBP-1 cleavage process (Li et al., 2011) led us to investigate the effects of selective AHR ligand treatment on AMPK. Selective activation of AHR by SGA360 and SGA315 led to a significant increase in phosphorylated AMPK at the Thr172 residue (pAMPK), although AMPK levels remained unaltered in SEB-1 cells (Figure 10A). Either high glucose or high serum levels have previously been shown to attenuate AMPK activity. Phosphorylation of AMPK proved to be refractory to AHR ligand-mediated activity under high glucose or high serum conditions (Figure 10B). Importantly, an increase in the pAMPK protein levels inversely correlated with the amount of mature SREBP-1 levels (Figure 10C). These results suggest that an AMPK axis might be involved in the AHR-mediated inhibition of SREBP-1 activity.
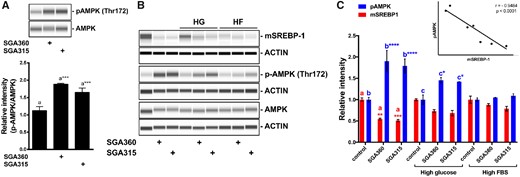
Phosphorylated AMPK levels inversely correlate with the mature SREBP-1 protein in the presence of selective AHR ligands. A, SEB-1 cells were treated with SGA360 (10 µM) or SGA315 (10 µM) in media containing lipid-depleted FBS for 24 h, AMPK and pAMPK (T172) protein levels were determined by WES. B, SGA360 (10 µM) or SGA315 (10 µM) was administered to SEB-1 cells in the presence of high glucose (25 mM) or high delipidated serum (20% FBS). C, Protein expression of SREBP-1, AMPK, and pAMPK (T172) was examined by WES analyses, and quantified. Regression analysis comparing pAMPK and mSREBP-1 protein levels. Data are presented as mean ± SD, n = 3; significance was determined by one-way ANOVA.
DISCUSSION
The involvement of AHR in lipid metabolism has been largely studied with regard to TCDD exposure in rodents. Microarray analysis demonstrated that TCDD exposure led to altered expression of genes involved in lipid synthesis, among many other pathways in liver and other tissues (Nault et al., 2013). Lower serum triglyceride levels and a decrease in adipose tissue and body weight were observed in TCDD-exposed rodents (Angrish et al., 2013). Furthermore, genome-wide AHR enrichment analysis identified gene groups associated with lipid metabolism in rodents (Dere et al., 2011). However, the underlying mechanism of attenuated lipogenic gene expression has not been identified. Our group previously showed that AHR agonist BNF administration significantly attenuates lipogenic gene expression in a transgenic mouse model that expresses the DRE-binding mutant in liver (Tanos et al., 2012a). Moreover, AHR deficiency in mice and human cells is associated with increased basal expression of these enzymes, further indicating an endogenous role of AHR in lipid homeostasis in the absence of any exogenous ligand.
The hallmark of dioxin toxicity in humans is chloracne (Suskind, 1985). However, chloracne is a misleading term; although it manifests as an acne-like eruption of comedones and cysts, patients with chloracne have smaller sebaceous glands, decreased lipogenesis, and reduced sebum levels on their skin. In contrast, elevated sebum production by enlarged sebaceous glands is a characteristic phenotype of acne vulgaris (Williams et al., 2012). Histological analyses of the skin lesions from Victor Yushchenko, who was exposed to TCDD at a single oral dose of 20 μg/kg, revealed a complete loss of sebaceous glands and formation of cystic lesions with epidermal-like differentiation (Saurat et al., 2012). Therefore, Saurat and Sorg proposed to term epidermal cysts formed upon dioxin exposure “metabolizing acquired dioxin-induced skin hamartomas (MADISH)” (Saurat and Sorg, 2010). The most likely mechanisms of TCDD-mediated reduction of sebaceous gland size involve attenuation of lipid synthesis, altered differentiation of sebocytes or effects on the stem cell population (Bock, 2017; Hu et al., 2016). Sebaceous glands have been found to be the initial responders to AHR agonists applied topically or systemically in mouse skin, as evidenced by CYP1A1 induction shortly after ligand exposure. Subsequently, multipotent progenitor cells expressing Leucine-rich repeats and immunoglobulin-like domains protein 1 have been identified as the specific cell type that exhibited high AHR agonist responsiveness (Fontao et al., 2017). Thus, TCDD leads to a dysregulation of sebaceous gland homeostasis and consequently to chloracne through sustained activation of AHR. Notably, similar to TCDD, the AHR full agonist BNF caused sebaceous gland atrophy and downregulated lipogenic genes in mice (Fontao et al., 2017). These observations led us to question whether AHR can be used as a therapeutic target for acne vulgaris using nontoxic ligands.
Acne is consistently one of the most prevalent skin conditions in the general population. Although it does not directly cause fatality, the direct and indirect costs of treatment, as well as the loss of productivity due to low quality of life and low self-esteem lead to a loss of over 3 billion dollars per year in the United States (Bhate and Williams, 2013). Depending on the severity of lesions, there are several topical and systemic acne treatments available that vary in mode of action. The most commonly used acne therapies for mild to moderate acne include benzoyl peroxide, topical retinoids, and antibiotics (Haider and Shaw, 2004). Topical retinoids are effective in both comedonal and inflammatory acne to some extent. They can induce local irritation and increase sensitivity to UV light. Benzoyl peroxide has potent bactericidal activity against C. acnes. Topical antibiotics act directly on Propionibacterium acnes and reduce inflammation. Oral isotretinoin is the most effective medication for clearing of severe acne; however, teratogenicity and adverse effects associated with isotretinoin limit its use. Thus, novel therapeutic agents with fewer side-effects are needed for acne treatment (Cong et al., 2019).
In this project, we propose that ligand stimulation of AHR by SGA360 and SGA315 is capable of attenuating the expression of genes directly involved in cholesterol and fatty acid biosynthesis, and subsequently decreasing the cellular lipid levels in human sebocytes. We demonstrated that this AHR-mediated regulation is dependent on the SREBP-1, the principal transcriptional regulator of fatty acid synthesis pathway. Mechanistical insights indicate that ligand activation of AHR represses the expression of fatty acid synthesis genes through decreasing the transcriptional activity of SREBP-1. At least part of the mechanism(s) underlying the attenuated SREBP-1 activity by selective activation of AHR is the induction of proteolytic degradation of the transcriptional active mSREBP-1 and obstruction of SREBP-1 processing independent of SCAP, INSIG1, and Golgi proteases S1P or S2P. AHR has previously been demonstrated to exhibit an E3 ubiquitin ligase activity (Ohtake et al., 2007, 2009). Furthermore, a physical interaction between AHR and SREBP-1 has been reported in naive CD4+ T cells, inhibiting AHR-mediated Il17 transcription (Cui et al., 2011). Thus, it is plausible that ligand-activated AHR functions as an E3 ubiquitin ligase upon physically interacting with SREBP-1, leading to its degradation. Interestingly, we have found that selective AHR ligands lead to an increase in phosphorylated AMPK protein levels. Furthermore, a decrease in the ratio of phosphorylated AMPK to AMPK inversely correlated with increased mSREBP-1 levels upon ligand treatment. Previous studies reported a role for AMPK in inhibiting proteolytic activation of SREBP-1 through direct phosphorylation (Li et al., 2011). Thus, our findings are consistent with previous reports and suggest that selective activation of AHR might influence the AMPK/SREBP axis. Nongenomic functions of AHR have been extensively studied and discovered to involve the activation of certain protein kinases. TCDD exposure causes a rapid activation of proto-oncogene tyrosine-protein kinase Src and extracellular signal-regulated kinases through elevated intracellular concentration of calcium and subsequent activation of calcium-dependent cytosolic phospholipase A2 (cPLA2), in immortalized normal mammary epithelial cell line MCF10A (Dong and Matsumura, 2008; Mazina et al., 2004). Moreover, low dose TCDD-treated rats and guinea pigs exhibit sustained elevation of protein kinases PKA, PKC, epithelial growth factor receptor (EGFR)-associated tyrosine kinases and Src kinase in liver plasma membrane (Bombick et al., 1985; Bombick and Matsumura, 1987; Madhukar et al., 1988). Several studies made the connection between AMPK and Src kinase and suggested that AMPK is activated by c-Src. Inhibition of c-Src abolished phosphorylation of AMPK that was enhanced by antidiabetic drug metformin in bovine aortic endothelial cells (Zou et al., 2004). Similarly, siRNA-mediated knockdown of c-Src led to a decrease in phosphorylated AMPK levels in cancer cell lines (Mizrachy-Schwartz et al., 2011). Furthermore, c-Src was found to physically interact with AMPK, and this interaction was increased under hypoxia-reoxygenation conditions (Zou et al., 2003). Although nongenomic pathways upon AHR activation have been examined using the potent agonist TCDD, it is plausible that selective ligands might lead to similar changes, thereby regulating the interplay between protein kinases and SREBP-1. Taken together, our data suggest that AHR-mediated inhibition of SREBP-1 involves proteolytic degradation as well as interference with cleavage processing of SREBP-1. Therefore, future studies are necessary to elucidate the main factor(s) and mechanism(s) involved in this process.
One question that we wanted to address is why selective AHR ligands exhibit greater potential to repress fatty acid synthesis than a full agonist. We have previously established that BNF treatment of transgenic mice that express the AHR mutant A78D, which fails to bind to DRE, are able to repress the expression of genes directly involved in fatty acid and cholesterol synthesis (Tanos et al., 2012a,b). These studies demonstrate that agonist-mediated nuclear translocation renders nuclear AHR sensitive to ubiquitin-dependent proteolytic inactivation. We have also demonstrated that SGA360 enhances AHR levels after 24 h exposure in keratinocytes (van den Bogaard et al., 2015). Conversely, agonists such as TCDD and indolo[3,2b]carbazole dramatically reduce overall AHR levels. The ability of the liganded AHR to influence mSREBP1 proteolytic turnover is most likely through a protein/protein interaction event. Thus, the level of SGA360/AHR complex would be high over a relatively long-time frame such as 24 h and is likely to be more effective in influencing mSREBP1 turnover compared with an AHR agonist. This concept is consistent with the data presented here that BNF is less effective in repressing fatty acid synthesis. Another question that should be addressed pertains to the location of AHR-mediated enhanced proteolysis of the mSREBP1 in the cell. We have not directly addressed this question in this report. However, SGA360 does not induce AHR translocation and in fact appears to enhance cytoplasmic localization (Muku et al., 2017). This would suggest that the ability of the AHR/SGA360 complex to influence mSREBP1 turnover most likely occurs in the cytoplasm. However, considering that the AHR undergoes nucleocytoplasmic shuttling, the presence of AHR in the nucleus cannot be excluded (Petrulis and Perdew, 2002).
TCDD treatment has been demonstrated to cause sebaceous gland shrinkage in human skin specimens ex vivo (Ju et al., 2011). On the other hand, sustained TCDD exposure leads to toxicity by altering global gene expression. Considering that a TCDD treated AHR mutant mouse that expresses a receptor that does not bind to DRE fails to exhibit thymic or liver toxicity would suggest that TCDD-mediated toxicity is primarily a DRE-driven response (Bunger et al., 2008). Thus, from a therapeutic standpoint, SAhRM/partial agonists likely have a greater potential to be used for acne treatment relative to full agonists (eg, TCDD). Especially considering that SAhRM with partial agonists activity do not induce CYP1A1 expression as much as a full agonist, nor block the endogenous AHR activity, which was found to be essential for normal keratinocyte differentiation (van den Bogaard et al., 2015). Therefore, the use of selective ligands attenuates the risk of CYP1A1-mediated bio-transformation of carcinogens, as well as toxicity, although maintaining endogenous AHR-mediated physiological processes.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
ACKNOWLEDGMENT
We thank Marcia H. Perdew for excellent editorial assistance
FUNDING
This work was supported by the National Institutes of Health Grants ES004869 and ES028244. This work was also supported by the USDA National Institute of Food and Federal Appropriations under Project PEN04607 and Accession number 1009993.
REFERENCES
Hu, T., Wang, D., Yu, Q., Li, L., Mo, X., Pan, Z., Zouboulis, C.C., Peng, L., Xia, L., and Ju, Q. (2016) Aryl hydrocarbon receptor negatively regulates lipid synthesis and involves in cell differentiation of SZ95 sebocytes in vitro. Chem. Biol. Interact.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments