





Abstract
Since recombinant human soluble thrombomodulin (RH-TM) has anti-inflammatory properties through neutralizing high-mobility group box 1 protein (HMGB1), the protective effects of RH-TM were examined in anti-glomerular basement membrane (GBM) glomerulonephritis (GN) in Wistar–Kyoto rats.
Rats were injected with nephrotoxic serum (NTS) to induce anti-GBM GN on Day 0, and were given either RH-TM or vehicle from Day 0 to Day 6. Rats were sacrificed 7 days after NTS injection.
RH-TM-treated rats had decreased proteinuria and serum creatinine level. RH-TM significantly reduced the percentage of glomeruli with crescentic features and fibrinoid necrosis. In addition, RH-TM-treated rats had significantly reduced glomerular ED1+ macrophage accumulation as well as reduced renal cortical proinflammatory cytokine expression. Furthermore, RH-TM had a potent effect in reducing intercellular adhesion molecule-1 (ICAM-1) expression in kidneys and urine. RH-TM significantly reduced renal cortical mRNA levels for toll-like receptor -2 and -4, known as receptors for HMGB1, and their downstream adopter protein, myeloid differentiation primary respond protein 88 (MyD88).
We showed for the first time that anti-inflammatory effects, which were characterized by reduced glomerular macrophage influx concomitant with a marked reduction in proinflammatory cytokines, were involved in the mechanism of attenuating experimental anti-GBM GN by RH-TM. The observed effects might be attributable to the downregulation of ICAM-1 by reducing the HMGB1/TLR/MyD88 signaling pathway.
INTRODUCTION
Recombinant human soluble thrombomodulin (RH-TM) (Recomodulin®, Asahi Kasei Pharma Corp., Tokyo, Japan) was approved in 2008 and has been used for the treatment of disseminated intravascular coagulation in Japan. Thrombomodulin (TM) is a glycoprotein present on the membrane surface of endothelial cells in many organs including lung, liver and kidney. TM is known to form a high-affinity complex with thrombin and converts thrombin from a procoagulant to an anticoagulant enzyme. The thrombin–TM complex potently activates anticoagulant protease protein C, thereby inhibiting thrombus formation by inactivating factors Va and VIIIa. Apart from its role as an anticoagulant, TM has been demonstrated to possess beneficial roles in inflammation, fibrinolysis, apoptosis, cell adhesion and cellular proliferation [1–3]. The therapeutic potential of RH-TM for kidney diseases has been shown in rodent models of acute ischemic kidney injury [4, 5], thrombotic glomerulonephritis (GN) [6] and Shiga toxin-associated glomerular injury [7]. These reports showed RH-TM conferred renoprotection by improving microvascular blood flow and endothelial permeability, decreasing leukocyte adhesion to the endothelium [4], inactivating anaphylatoxins [6] and inhibiting further thrombin formation and hypercoagulant status [7].
Some studies have reported that the lectin-like domain of TM attenuates inflammation through the suppression of high-mobility group box 1 protein (HMGB1)-mediated proinflammatory responses [8–11]. HMGB1 is a nuclear architectural chromatin-binding protein released by immune cells including macrophages, monocytes, natural killer cells, dendritic cells, endothelial cells and platelets [12–15]. It is also passively released from necrotic or damaged cells and apoptotic cells [16, 17]. HMGB1 acts as a proinflammatory cytokine and can bind to cell surface receptors including the receptor for advanced glycation end products (RAGE), toll-like receptor 2 (TLR2) and TLR4, and mediates the release of proinflammatory cytokines. These receptors are involved in the myeloid differentiation primary-respond protein (MyD) 88-dependent pathway that ultimately leads to nuclear factor (NF)-κB activation. NF-κB regulates the expression of proinflammatory cytokines and chemokines [18]. HMGB1 is involved in the progression of severe inflammatory disease, such as septic shock, rheumatoid arthritis and atherosclerosis [13, 19, 20]. HMGB1 also induces renal ischemia-reperfusion injury (IRI), sepsis-induced acute kidney injury, anti-neutrophil cytoplasmic antibody-associated vasculitis and diabetic nephropathy [21–24].
Crescentic GN is characterized clinically by rapidly progressive renal failure and histologically by crescent formation, necrosis and inflammatory cell infiltration in glomeruli, and eventual glomerulosclerosis. In Wistar–Kyoto (WKY) rats, rabbit anti-rat glomerular basement membrane (GBM) antiserum (nephrotoxic serum, NTS) induces severe proliferative and necrotizing GN, with crescent formation resembling human crescentic GN. It is thought that the cardinal pathological features of crescentic GN in WKY rats are mediated largely by the infiltration of monocytes/macrophages and CD8+ cells [25, 26]. In addition, glomerular endothelial injury plays an important role in the pathogenesis of anti-GBM GN. Previous studies have reported that endothelial nitric oxide and vascular endothelial growth factor (VEGF) ameliorated experimental anti-GBM GN by inhibiting intercellular adhesion molecule-1 (ICAM-1) and leukocyte adhesion to the endothelium [27, 28].
These lines of evidence indicate that RH-TM may have the potential to ameliorate crescentic GN. Although corticosteroids and other immunosuppressive drugs are effective in patients with crescentic GN, increased susceptibility to infections and mortality by using these drugs makes it necessary to develop new treatment strategies. Therefore, the present study was designed to determine the protective effects of RH-TM and its mechanism in anti-GBM GN of WKY rats.
MATERIALS AND METHODS
Experimental protocol
The experimental protocol for this study was reviewed and approved by the Animal Care Committee of Showa University in Tokyo. Seven-week-old, female WKY rats weighing 135–150 g were purchased from Charles River Japan (Atsugi, Kanagawa, Japan) and used in all of the experiments. The animals were housed in the animal care facility of Showa University (25°C, 50% humidity, 12-h dark/light cycle) with free access to food and water. A total of 26 female WKY rats were injected intravenously with 20 μL of NTS, which was prepared as previously described [29], to induce anti-GBM GN on Day 0. The rats with nephritis were administered either RH-TM (1.5 mg/kg) or vehicle by intraperitoneal injection, from Day 0 to Day 6 twice daily. The first administration of RH-TM or vehicle was given 3 h before NTS injection on Day 0. The vehicle-treated groups received an equal volume of saline. Rats were sacrificed 7 days after NTS injection. Control WKY rats without NTS injection were given an equal volume of saline. At the end of the study, the rats were anesthetized, their blood was collected by cardiac puncture and their kidneys were collected. Right kidneys were snap-frozen in liquid nitrogen for biochemical and gene expression analysis, and left kidneys were fixed in 2% paraformaldehyde/phosphate-buffered saline (PBS) for histological analysis.
Proteinuria and creatinine determination
For the analysis of proteinuria, rats were individually housed in metabolic cages for 24-h urine collection. Urinary protein was determined using the Biuret method. Serum and urinary creatinine (Cr) levels were measured using an automated analyzer (Hitachi Corp., Tokyo, Japan) according to the manufacturer’s instructions.
Light microscopy analysis
Tissues fixed in 2% paraformaldehyde/PBS were embedded in paraffin using routine protocols. Paraffin-embedded materials were sectioned at 2 μm for routine staining with periodic acid–Schiff (PAS) and 1-μm-thick sections were used for periodic acid methenamine silver stain (silver). The glomerular tuft area was quantified in 50 full-sized glomeruli (PAS stain) using WinROOF image processing software (Mitani Corp., Tokyo, Japan). The number of crescentic or necrotizing glomeruli per 100 glomeruli from each rat was calculated and expressed as a percentage. The percentage of area occupied by crescents in each glomerulus was estimated and assigned one of the following scores: 0, absent; 1, <1/4; 2, between 1/4 and 1/2; 3, between 1/2 and 3/4; and 4, >3/4 of the whole glomerulus [30]. The mean score of 50 glomeruli was then calculated as the crescent score. All histological analyses were performed without knowledge of the origin of the slides.
Immunohistochemistry
The antibodies used in this study were: mouse anti-rat ED1 antibody (BMA, Augst, Switzerland) as a macrophage marker, mouse anti-rat CD8 antibody (Clone number: X8, Antigenix America, Huntington Station, NY, USA) and mouse anti-rat HMGB1 antibody (Abcam Ltd, Cambridge, UK). Biotinylated rabbit anti-mouse IgG and peroxidase-conjugated streptavidin (LSAB 2 kit/horseradish peroxidase) were purchased from Dako (Glostrup, Denmark). Immunohistochemical staining for ED1 (1:50 dilution) and CD8 (1:50 dilution) were performed as previously described [30]. Cells undergoing apoptosis were identified by in situ TdT-mediated dUTP nick end labeling (TUNEL) using the ApopTag plus peroxidase in situ apoptosis detection kit (Chemicon International Inc., Temecula, CA, USA). Two-color immunostaining was used to detect colocalization of ED1 with HMGB1. Sections were microwaved twice for 5 min for two-color immunostaining with ED1 (1:50 dilution)/HMGB1 (1:1000 dilution). CD8+ cells and TUNEL+ cells were estimated by counting the number of positive cells within 50 glomeruli and dividing the total number by 50. The extent of ED1 staining in each glomerulus was graded for 50 glomeruli on a 4-point scale: 0, absent; 1, weak; 2, moderate; and 3, severe [30]. The mean score was then calculated as the ED1 score. All histological analyses were performed without knowledge of the origin of the slides.
Immunofluorescence
The tissues were snap-frozen in liquid nitrogen and cut into 4-μm-thick sections. The deposition of rabbit IgG, rat fibrin and rat IgG in the kidney sections were evaluated using a previously described method [30]. The extent of fibrin deposition in each glomerulus was graded for 50 glomeruli using a 4-point scale: 0, absent; 1, <1/3; 2, between 1/3 and 2/3; and 3, >2/3 of the whole glomerulus. The mean score of 50 glomeruli was then calculated as the fibrin score.
Measurement of circulating anti-rabbit IgG antibody
The level of circulating anti-rabbit IgG antibody in rats with nephritis was measured by enzyme-linked immunosorbent assay (ELISA), as described previously [29, 30].
Quantification of serum and urinary cytokine levels
Serum HMGB1 (Shino-Test Corp., Kanagawa, Japan) and urinary ICAM-1 (R&D Systems, Minneapolis, MN, USA) protein levels were determined using commercially available ELISA kits according to each manufacturer’s instructions.
Real-time polymerase chain reaction
Gene expressions of rat tumor necrosis factor (TNF)-α, interleukin (IL)-1β, monocyte chemoattractant protein-1 (MCP)-1, IL-6, HMGB1, TLR2, TLR4, RAGE, MyD88, ICAM-1, VEGF-A and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were analyzed using real-time polymerase chain reaction (RT-PCR) in kidney tissues (cortex) as described previously [31]. mRNA expression was normalized using GAPDH as an endogenous control to correct for differences in the amount of total RNA added to each reaction.
Statistical analysis
Data were presented as means ± SEM. The Mann–Whitney test was performed to evaluate differences in group means, and values of P < 0.05 were considered statistically significant.
RESULTS
Effects of RH-TM on biochemical parameters in rats with nephritis
Figure 1a–c shows the urinary total protein levels, serum Cr levels and total kidney weights for each group. These three parameters were significantly higher in the WKY-Vehicle rats than in the WKY rats without nephritis [WKY-NTS (−)] on Day 7 (P < 0.01). Urinary protein in the WKY-Vehicle rats reached 68.60 ± 3.26 mg/day on Day 7. RH-TM treatment, which was started on Day 0, significantly improved the urinary protein levels by 34% on Day 7 (P < 0.01) (Figure 1a). The serum Cr levels were significantly reduced by 22% in the WKY-TM rats compared with the WKY-Vehicle rats on Day 7 (P < 0.001) (Figure 1b). RH-TM treatment also suppressed the kidney weight and kidney weight/body weight ratio of rats with nephritis by 18% (P < 0.0001) and 12% (P < 0.001), respectively (Figure 1c and d).
![Urinary total protein levels, serum Cr levels and total kidney weight in the study groups. (a) Urinary total protein levels, (b) serum Cr levels, (c) total kidney weight and (d) kidney weight/body weight ratio of WKY rats with nephritis treated with either vehicle (WKY-Vehicle) or RH-TM (WKY-TM) are shown. The horizontal dotted lines show the levels of proteinuria, serum Cr and kidney weight in WKY rats without nephritis [WKY-NTS (−)]. Data are means ± SEM. ##P < 0.01: WKY-NTS (−) versus WKY-Vehicle; **P < 0.01, ***P < 0.001, ****P < 0.0001: WKY-Vehicle versus WKY-TM.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ndt/34/5/10.1093_ndt_gfy201/1/m_gfy201f1.jpeg?Expires=1750301720&Signature=JkCFGyDReVBhvKs-jrPWJi67goo9JSfPV9En05ggFHdN6P7v0MKN9NC18HsB1ZX8rtJPOx8zmq6Ev9ZmR1~XvpLlZURwjY6DaIvfVhdZ078MTYaZdUc4y985FneEeqeI~1L28SQbTOGYR239KfYnZXTeygN5Ti5eqgyV~sr3VaTexU-sf0ivtmWPjDQxBHj~n1wHZs4ZTMbLNqXNUqqv1PnX2yeJnxWwHSu5IEzpBbfN4QAeLAYER29SeZHkJK5TMDuBJyBxlkSMhBjJoPJ7hvEfluvzr4M4026ydbZEooLVUwkvYqqs1PqqgZIyXk4JiUHhiQXLEuwkDXdcezjqDQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Urinary total protein levels, serum Cr levels and total kidney weight in the study groups. (a) Urinary total protein levels, (b) serum Cr levels, (c) total kidney weight and (d) kidney weight/body weight ratio of WKY rats with nephritis treated with either vehicle (WKY-Vehicle) or RH-TM (WKY-TM) are shown. The horizontal dotted lines show the levels of proteinuria, serum Cr and kidney weight in WKY rats without nephritis [WKY-NTS (−)]. Data are means ± SEM. ##P < 0.01: WKY-NTS (−) versus WKY-Vehicle; **P < 0.01, ***P < 0.001, ****P < 0.0001: WKY-Vehicle versus WKY-TM.
Immunofluorescence analysis
Prominent glomerular fibrin deposition was observed in the WKY-Vehicle rats on Day 7 (Figure 2a). The semi-quantitative evaluation of fibrin deposition showed that the fibrin scores were reduced in the WKY-TM rats compared with the WKY-Vehicle rats (P < 0.0001) (Figure 2b and c). Rabbit IgG was detected in an intense linear pattern along the glomerular capillaries in the WKY-Vehicle rats and WKY-TM rats. Rat IgG was also detected along the glomerular capillaries. There was no significant difference in rabbit IgG and rat IgG glomerular staining between the WKY-Vehicle rats and WKY-TM rats. Furthermore, there was no significant difference in the levels of serum anti-rabbit IgG antibody between the WKY-Vehicle rats and WKY-TM rats (O.D. value, 0.39 ± 0.008 versus 0.40 ± 0.003, NS, WKY-Vehicle rats versus WKY-TM rats). These findings suggest that RH-TM did not significantly affect the process of heterologous antibody deposition or autologous antibody production.
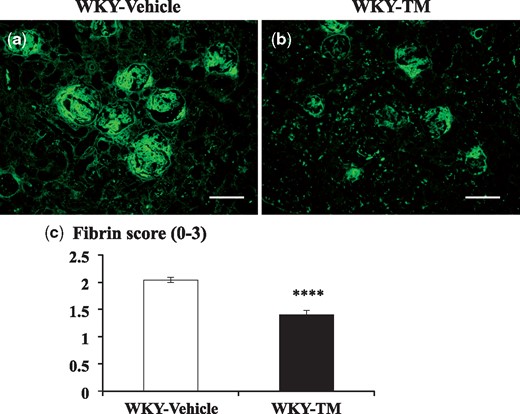
Fibrin staining by immunofluorescence microscopy in the study groups. Immunofluorescent staining for fibrin in a WKY-Vehicle rat (a) and a WKY-TM rat (b) on Day 7, and semi-quantitative assessment of fibrin deposition in each group (c) are shown. Each group contained 13 rats, and 50 glomeruli per rat were evaluated in a blinded fashion. Data are expressed as mean ± SEM. ****P < 0.0001: WKY-Vehicle versus WKY-TM. Original magnification, ×200. Scale bar, 100 μm.
Effects of RH-TM on renal histological findings in rats with nephritis
It was expected that RH-TM treatment had the potential to improve renal histological findings. Figure 3 shows representative images of silver-stained kidney sections on Day 7 in the study groups. The glomeruli on Day 7 showed severe fibrinoid necrosis and cellular crescent formation in the WKY-Vehicle rats (Figures 3a and d). RH-TM significantly reduced the degree of crescentic GN (Figure 3b and e), which was evaluated by measuring the glomerular tuft area and by scoring crescent and fibrinoid necrosis in glomeruli. The quantitative analysis of the glomerular tuft area is presented in Table 1. RH-TM treatment significantly reduced glomerular hypertrophy in rats with nephritis compared with vehicle treatment (P < 0.01) (Table 1). RH-TM treatment also significantly reduced the degree of crescent formation (crescent score: P < 0.01) and the percentage of crescent morphology (P < 0.001) and fibrinoid necrosis (P < 0.05) in glomeruli (Table 1).
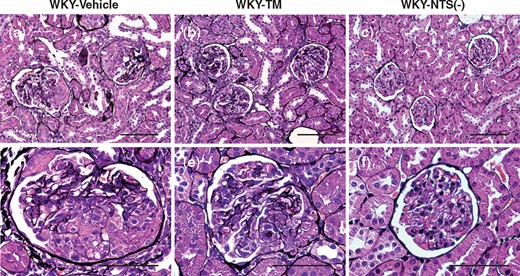
Light microscopy observations in the study groups. Representative images of sections stained with silver in a WKY-Vehicle rat (a, d), a WKY-TM rat (b, e) and a WKY-NTS (−) rat (c, f) on Day 7 are shown. The glomeruli on Day 7 show severe fibrinoid necrosis and cellular crescent formation (a, d). RH-TM treatment reduces glomerular hypertrophy, crescent formation and fibrinoid necrosis on Day 7 (b, e). Original magnifications, ×400 (a–c) and ×1000 (d–f). Scale bar, 100 μm (a–c) and 50 μm (d–f).
Assessment of histological findings on Day 7
| Group | WKY-Vehicle, n = 13 | WKY-TM, n = 13 | WKY-NTS (−), n = 6 |
|---|---|---|---|
| Glomerular tuft area (µm2) | 8572.69 ± 151.21 | 7021.58 ± 553.83** | 4412.38 ± 291.92 |
| Percentage of crescent | 94.15 ± 1.16 | 80.67 ± 3.50*** | 0 |
| Crescent score (0–4) | 1.96 ± 0.07 | 1.40 ± 0.15** | 0 |
| Percentage of necrosis | 64.92 ± 2.57 | 48.00 ± 6.76* | 0 |
| ED1 score (0–3) | 1.51 ± 0.05 | 1.19 ± 0.08** | 0.15 ± 0.07 |
| CD8+ cells/glomerular cross-section | 1.58 ± 0.24 | 1.39 ± 0.22NS | 0.61 ± 0.09 |
| TUNEL+ cells/glomerular cross-section | 0.82 ± 0.05 | 0.36 ± 0.20*** | 0.06 ± 0.01 |
| Group | WKY-Vehicle, n = 13 | WKY-TM, n = 13 | WKY-NTS (−), n = 6 |
|---|---|---|---|
| Glomerular tuft area (µm2) | 8572.69 ± 151.21 | 7021.58 ± 553.83** | 4412.38 ± 291.92 |
| Percentage of crescent | 94.15 ± 1.16 | 80.67 ± 3.50*** | 0 |
| Crescent score (0–4) | 1.96 ± 0.07 | 1.40 ± 0.15** | 0 |
| Percentage of necrosis | 64.92 ± 2.57 | 48.00 ± 6.76* | 0 |
| ED1 score (0–3) | 1.51 ± 0.05 | 1.19 ± 0.08** | 0.15 ± 0.07 |
| CD8+ cells/glomerular cross-section | 1.58 ± 0.24 | 1.39 ± 0.22NS | 0.61 ± 0.09 |
| TUNEL+ cells/glomerular cross-section | 0.82 ± 0.05 | 0.36 ± 0.20*** | 0.06 ± 0.01 |
Data are means ± SEM.
P < 0.05, ** P < 0.01, ***P < 0.001 versus WKY-Vehicle; NS, not significant.
Assessment of histological findings on Day 7
| Group | WKY-Vehicle, n = 13 | WKY-TM, n = 13 | WKY-NTS (−), n = 6 |
|---|---|---|---|
| Glomerular tuft area (µm2) | 8572.69 ± 151.21 | 7021.58 ± 553.83** | 4412.38 ± 291.92 |
| Percentage of crescent | 94.15 ± 1.16 | 80.67 ± 3.50*** | 0 |
| Crescent score (0–4) | 1.96 ± 0.07 | 1.40 ± 0.15** | 0 |
| Percentage of necrosis | 64.92 ± 2.57 | 48.00 ± 6.76* | 0 |
| ED1 score (0–3) | 1.51 ± 0.05 | 1.19 ± 0.08** | 0.15 ± 0.07 |
| CD8+ cells/glomerular cross-section | 1.58 ± 0.24 | 1.39 ± 0.22NS | 0.61 ± 0.09 |
| TUNEL+ cells/glomerular cross-section | 0.82 ± 0.05 | 0.36 ± 0.20*** | 0.06 ± 0.01 |
| Group | WKY-Vehicle, n = 13 | WKY-TM, n = 13 | WKY-NTS (−), n = 6 |
|---|---|---|---|
| Glomerular tuft area (µm2) | 8572.69 ± 151.21 | 7021.58 ± 553.83** | 4412.38 ± 291.92 |
| Percentage of crescent | 94.15 ± 1.16 | 80.67 ± 3.50*** | 0 |
| Crescent score (0–4) | 1.96 ± 0.07 | 1.40 ± 0.15** | 0 |
| Percentage of necrosis | 64.92 ± 2.57 | 48.00 ± 6.76* | 0 |
| ED1 score (0–3) | 1.51 ± 0.05 | 1.19 ± 0.08** | 0.15 ± 0.07 |
| CD8+ cells/glomerular cross-section | 1.58 ± 0.24 | 1.39 ± 0.22NS | 0.61 ± 0.09 |
| TUNEL+ cells/glomerular cross-section | 0.82 ± 0.05 | 0.36 ± 0.20*** | 0.06 ± 0.01 |
Data are means ± SEM.
P < 0.05, ** P < 0.01, ***P < 0.001 versus WKY-Vehicle; NS, not significant.
Effects of RH-TM on macrophage accumulation, CD8+ cell influx and apoptosis in rats with nephritis
To identify the cell phenotype of leukocytes affected by RH-TM, ED1+ and CD8+ cells in glomeruli were examined. Immunostaining for ED1 in the WKY-Vehicle rats showed intense glomerular staining, whereas significantly less staining was observed in the WKY-TM rats on Day 7 (P < 0.01) (Figure 4a–c; Table 1). The CD8+ cell number per glomerular cross-section on Day 7 was not significantly different between the WKY-TM rats and the WKY-Vehicle rats (Figure 4d–f; Table 1). RH-TM significantly reduced apoptosis in rats with nephritis on Day 7, evaluated by counting the TUNEL+ cell number per glomerular cross-section (P < 0.001) (Figure 4g–i; Table 1).
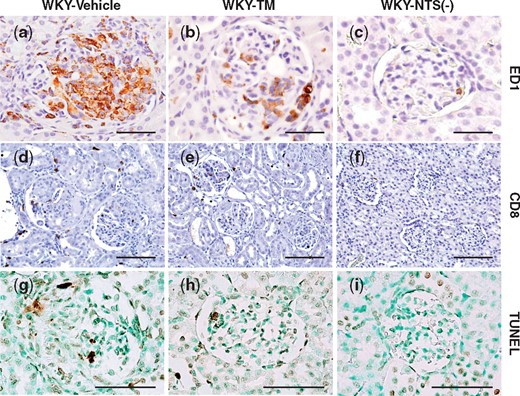
Immunohistochemical analysis of ED1, CD8 and TUNEL expression in the study groups. Representative pictures stained with immunohistochemistry for ED1 (a–c), CD8 (d–f) and TUNEL (g–i) of a WKY-Vehicle rat (a, d, g), a WKY-TM rat (b, e, h) and a WKY-NTS (−) rat (c, f, i) on Day 7 are shown. Original magnifications, ×1000 (a–c, j–i) and ×400 (d–f). Scale bar, 50 μm (a–c, j–i) and 100 μm (d–f).
Effects of RH-TM on renal cortical proinflammatory cytokine expression in rats with nephritis
Since RH-TM significantly reduced macrophages in glomeruli in immunohistochemistry experiments, proinflammatory cytokines, which are fundamental in the pathogenesis of crescentic GN, were examined by RT-PCR. The gene expression levels of the proinflammatory cytokines TNF-α, IL-1β, MCP-1 and IL-6 were much higher in the WKY-Vehicle rats than in WKY-NTS (−) rats. RH-TM significantly decreased gene expression levels of TNF-α (P < 0.001), IL-1β (P < 0.001), MCP-1 (P < 0.01) and IL-6 (P < 0.05) on Day 7 (Table 2).
Results of RT-PCR on Day 7
| Group | WKY-Vehicle, n = 13 | WKY-TM, n = 13 |
|---|---|---|
| TNF-α | 14.62 ± 1.01#### | 7.77 ± 0.44*** |
| IL-1β | 6.96 ± 0.57#### | 3.77 ± 0.70*** |
| MCP-1 | 47.14 ± 4.19#### | 23.261 ± 8.01** |
| IL-6 | 27.96 ± 2.66#### | 18.30 ± 4.50* |
| HMGB1 | 0.85 ± 0.02### | 0.96 ± 0.08NS2 |
| TLR2 | 8.52 ± 0.38#### | 4.69 ± 0.89*** |
| TLR4 | 2.81 ± 0.10#### | 1.57 ± 0.21**** |
| RAGE | 2.47 ± 0.31#### | 3.63 ± 0.56* |
| MyD88 | 1.00 ± 0.03NS1 | 0.85 ± 0.08* |
| Group | WKY-Vehicle, n = 13 | WKY-TM, n = 13 |
|---|---|---|
| TNF-α | 14.62 ± 1.01#### | 7.77 ± 0.44*** |
| IL-1β | 6.96 ± 0.57#### | 3.77 ± 0.70*** |
| MCP-1 | 47.14 ± 4.19#### | 23.261 ± 8.01** |
| IL-6 | 27.96 ± 2.66#### | 18.30 ± 4.50* |
| HMGB1 | 0.85 ± 0.02### | 0.96 ± 0.08NS2 |
| TLR2 | 8.52 ± 0.38#### | 4.69 ± 0.89*** |
| TLR4 | 2.81 ± 0.10#### | 1.57 ± 0.21**** |
| RAGE | 2.47 ± 0.31#### | 3.63 ± 0.56* |
| MyD88 | 1.00 ± 0.03NS1 | 0.85 ± 0.08* |
Data are means ± SEM. The values were normalized to the GAPDH values and then expressed relative to WKY-NTS (−).
###P < 0.001, ####P < 0.0001 versus WKY-NTS (−).
P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus WKY-Vehicle.
NS1, not significant if comparing values between WKY-Vehicle and WKY-NTS (–).
NS2, not significant if comparing values between WKY-TM and WKY-NTS (–).
Results of RT-PCR on Day 7
| Group | WKY-Vehicle, n = 13 | WKY-TM, n = 13 |
|---|---|---|
| TNF-α | 14.62 ± 1.01#### | 7.77 ± 0.44*** |
| IL-1β | 6.96 ± 0.57#### | 3.77 ± 0.70*** |
| MCP-1 | 47.14 ± 4.19#### | 23.261 ± 8.01** |
| IL-6 | 27.96 ± 2.66#### | 18.30 ± 4.50* |
| HMGB1 | 0.85 ± 0.02### | 0.96 ± 0.08NS2 |
| TLR2 | 8.52 ± 0.38#### | 4.69 ± 0.89*** |
| TLR4 | 2.81 ± 0.10#### | 1.57 ± 0.21**** |
| RAGE | 2.47 ± 0.31#### | 3.63 ± 0.56* |
| MyD88 | 1.00 ± 0.03NS1 | 0.85 ± 0.08* |
| Group | WKY-Vehicle, n = 13 | WKY-TM, n = 13 |
|---|---|---|
| TNF-α | 14.62 ± 1.01#### | 7.77 ± 0.44*** |
| IL-1β | 6.96 ± 0.57#### | 3.77 ± 0.70*** |
| MCP-1 | 47.14 ± 4.19#### | 23.261 ± 8.01** |
| IL-6 | 27.96 ± 2.66#### | 18.30 ± 4.50* |
| HMGB1 | 0.85 ± 0.02### | 0.96 ± 0.08NS2 |
| TLR2 | 8.52 ± 0.38#### | 4.69 ± 0.89*** |
| TLR4 | 2.81 ± 0.10#### | 1.57 ± 0.21**** |
| RAGE | 2.47 ± 0.31#### | 3.63 ± 0.56* |
| MyD88 | 1.00 ± 0.03NS1 | 0.85 ± 0.08* |
Data are means ± SEM. The values were normalized to the GAPDH values and then expressed relative to WKY-NTS (−).
###P < 0.001, ####P < 0.0001 versus WKY-NTS (−).
P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus WKY-Vehicle.
NS1, not significant if comparing values between WKY-Vehicle and WKY-NTS (–).
NS2, not significant if comparing values between WKY-TM and WKY-NTS (–).
HMGB1 immunostaining, serum HMGB1 levels and renal cortical expression levels of HMGB1, its receptors and their downstream adaptor protein MyD88
We performed immunostaining for HMGB1 to investigate the pathogenicity of HMGB1 in rats with nephritis. In the WKY-NTS (−) rats, HMGB1 staining was observed in podocytes, parietal epithelial cells and tubular epithelial cells (Figure 5a and b). Double immunostaining of ED1 with HMGB1 was performed to identify whether macrophages are the major local source of HMGB1 in rats with nephritis. HMGB1 was detected strongly at glomerular crescents in the WKY-Vehicle rats (Figure 5c). The HMGB1+ cells in crescents were scarcely double-stained with ED1+ macrophages in the WKY-Vehicle rats (Figure 5d). HMGB-1 binds to cell surface receptors including RAGE, TLR2 and TLR4, and mediates release of proinflammatory cytokines by the MyD88-dependent pathway. To determine the importance of HMGB1 as a target of RH-TM, we examined serum HMGB1 levels, and renal cortical mRNA expression levels of HMGB1, TLR2, TLR4, RAGE and MyD88. RH-TM significantly reduced renal cortical TLR2, TLR4 and MyD88 mRNA expression levels (Table 2), whereas serum HMGB1 levels and renal cortical HMGB1 mRNA expression was not altered in rats with nephritis (serum HMGB1 levels, 4.03 ± 0.68 versus 5.97 ± 1.36, NS, WKY-Vehicle rats versus WKY-TM rats) (Table 2).
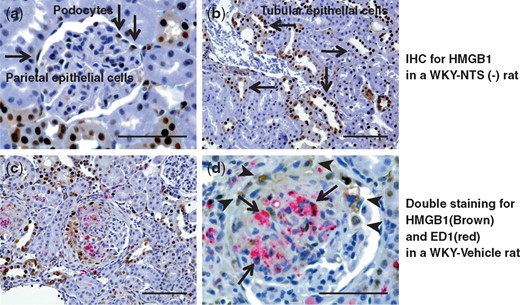
Immunohistochemical analysis of HMGB1 expression in the study groups. Representative images of sections immunohistochemically stained for HMGB1 in a WKY-NTS (−) rat (a, b) are shown. HMGB1 was stained in podocytes (a), parietal epithelial cells (a) and tubular epithelial cells (b) (arrows). Kidney sections were stained using two-color immunohistochemistry with ED1 stained red and HMGB1 stained brown in a WKY-Vehicle rat (c, d). Original magnifications, ×400 (b, c) and ×1000 (a, d). Scale bar, 50 μm (a, d) and 100 μm (b, c).
Effects of RH-TM on ICAM-1 and VEGF-A in rats with nephritis
To identify the effects of RH-TM on adhesion molecule and endothelial function, we measured the levels of renal cortical ICAM-1 and VEGF-A mRNA and urinary ICAM-1 (Figure 6). The renal cortical ICAM-1 expression levels and urinary ICAM-1 levels in the WKY-Vehicle rats were significantly higher compared with the WKY-NTS (−) rats (P < 0.0001). Furthermore, ICAM-1 levels were significantly reduced by RH-TM treatment (P < 0.001 and P < 0.05, respectively) (Figure 6a and b). In addition, RH-TM significantly recovered the decreased VEGF-A mRNA expression levels in rats with nephritis (P < 0.05) (Figure 6c).
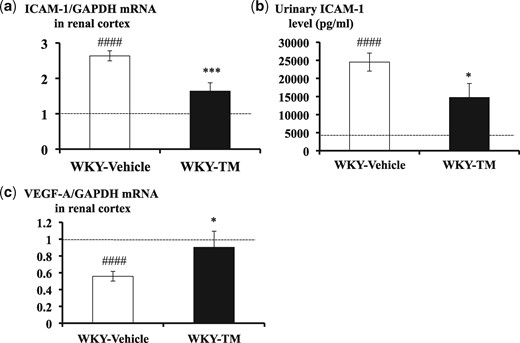
Renal cortical mRNA expression levels for ICAM-1 and VEGF-A, and urinary ICAM-1 levels in the study groups. The results of RT-PCR for renal cortical ICAM-1 (a), ELISA for urinary ICAM-1 (b) and RT-PCR for renal cortical VEGF-A (c) are shown. The ICAM-1 and VEGF-A mRNA values were normalized to GAPDH values and expressed as a relative quantification. The horizontal dotted lines show the average values observed in WKY-NTS (−) rats. Data are means ± SEM. ####P < 0.0001: WKY-NTS (−) versus WKY-Vehicle, *P < 0.05, ***P < 0.001: WKY-Vehicle versus WKY-TM.
DISCUSSION
In the present study, we demonstrated that the administration of RH-TM attenuated anti-GBM GN in rats when given 3 h before the administration of NTS. It is noteworthy that RH-TM had beneficial effects on virtually all aspects of the disease. Thus, there were significant reductions in urinary protein levels, serum Cr levels, kidney weight, glomerular hypertrophy, the degree of glomerular crescent and necrosis formation, and fibrin deposition in glomeruli.
RH-TM has been shown to ameliorate experimental renal IRI [4, 5], thrombotic glomerular injury [6] and Shiga toxin-associated glomerular injury [7]. However, little is known about the potential mechanisms of renoprotection by RH-TM treatment in kidney diseases. In the present study, the principal mechanism by which RH-TM mediates renoprotection was anti-inflammatory effects, which were evidenced by the suppressed glomerular macrophage accumulation and the concomitant reduction in proinflammatory cytokines. In addition, we showed that RH-TM significantly reduced ICAM-1 levels in kidney and urine, which is a critical factor for the induction of anti-GBM GN by recruiting macrophages [32, 33].
There are several reports that indicate the anti-inflammatory effects of RH-TM are attributable to the suppression of HMGB1-related mediators. Abeyama et al. [8] showed that RH-TM exerted anti-inflammatory effects by sequestering HMGB1, thereby preventing leukocyte activation in vitro, and attenuating UV irradiation-induced cutaneous inflammation and lipopolysaccharide (LPS)-induced lethality in vivo. Nagato et al. [9] showed that RH-TM inhibited proinflammatory cytokines and decreased plasma HMGB1 levels in experimental endotoxemia, which led to a marked reduction in liver dysfunction and mortality. Furthermore, Hagiwara et al. [11] showed that RH-TM prevented heatstroke by inhibiting HMGB1 and reducing proinflammatory cytokines in rats.
However, there have been no reports describing the anti-inflammatory effects of RH-TM through the suppression of HMGB1 in kidney disease. In the present study, HMGB1 was detected strongly in glomerular crescents in rats with nephritis, suggesting the involvement of HMGB1 in the pathogenesis of crescent formation. However, serum HMGB1 levels in rats with nephritis were not significantly different compared with rats without nephritis. We presume that serum HMGB1 released from necrotic tissue or secreted by macrophages in glomerular crescents peaked in earlier phase of nephritis and was already reduced on Day 7. According to the results for RT-PCR, TLR2 and -4 may be the principal receptors for HMGB1 in anti-GBM GN because gene expression levels for TLR2 or -4 but not RAGE were significantly downregulated by RH-TM. On the other hand, it has also been reported that HMGB1 activates the mitogen-activated protein kinase (MAPK)-NF-κB pathway, and that it plays an important role in inflammation [34]. NF-κB activation and cytokine production are both thought to be mediated by the MyD88-dependent pathway [35]. In this study, MyD88 was also significantly reduced by RH-TM treatment. Thus, RH-TM had the potential to attenuate nephritis through reducing the HMGB1/TLR/MyD88 signaling pathway.
It has been reported that HMGB1 could activate endothelial cells and monocytes, and increase their expression of ICAM-1 and their release of pro‐inflammatory mediators [14, 36, 37]. A recent report showed that antagonizing HMGB1 prevented the adhesion of cancer cells by downregulation of ICAM-1 in vitro [38]. Consistent with these reports, RH-TM had a potent effect in reducing ICAM-1 expression in kidneys and urine in the present study. It has been shown that adhesion of macrophages to glomerular endothelial cells through the lymphocyte function-associated Ag-1 (LFA-1)/ICAM-1 pathway is crucial for the initiation and subsequent progression of nephritis in this model [39]. ICAM-1 deficiency in mice with anti-GBM GN led to a significant reduction of glomerular macrophage influx and crescent formation [33]. In addition, Kushiro et al. [40] reported that administration of prostacyclin, known as an anticoagulant, significantly reduced glomerular ICAM-1 expression and macrophage infiltration, but not CD8+ cells, in this model of nephritis. This selective inhibition of macrophages is consistent with our result in which RH-TM did not affect CD8+ cell numbers in glomeruli. Furthermore, RH-TM significantly upregulated VEGF-A expression, which suggests the recovery of endothelial function. VEGF promotes survival, proliferation and differentiation of glomerular endothelial cells, and decreased expression of VEGF has been associated with various glomerulopathies including crescentic GN [41]. Accordingly, we believe that neutralizing HMGB1 is a possible anti-inflammatory mechanism by which RH-TM can protect against endothelial injury through inhibiting ICAM-1 expression and subsequent selective reduction of macrophage infiltration and proinflammatory cytokines secretion.
On the other hand, it has been reported that TM binds thrombin and facilitates thrombin-induced cleavage of the plasma zymogen protein C into activated protein C (APC), which inhibits coagulation, inflammation, apoptosis and vascular edema [42, 43]. Previous studies indicated that APC inhibited LPS-induced proinflammatory cytokine production in rats and in cultured monocytes [44, 45], and diminished the inflammation of endothelial cells by suppressing c-Fos-dependent induction of MCP-1 and ICAM-1 [46]. However, this anti-inflammatory mechanism may not be involved in this study, because a previous in vitro study demonstrated that RH-TM activated protein C in human and monkey plasma, but not in rat plasma [47].
In addition, it is notable that RH-TM potently reduced TUNEL+ apoptotic cells in glomeruli. This is consistent with a previous study which demonstrated that RH-TM significantly improved renal IRI concomitant with a reduction in apoptotic cells [5]. It has been reported that RH-TM can reduce serum starvation-induced apoptosis of endothelial progenitor cells via activating the phosphatidylinositol 3-kinase-protein kinase B/Akt survival pathway and suppressing the c-Jun N-terminal kinase death pathway in vitro [48]. Thus, it is possible that RH-TM reduced apoptosis by a mechanism involving direct effects on apoptosis-associated signaling pathways. Alternatively, reduced TUNEL+ apoptotic cells, which are largely induced in the infiltrated macrophages, podocytes or parietal epithelial cells in crescents in this model of nephritis [49], may simply have been a reflection of the attenuated crescentic GN by RH-TM.
In conclusion, we showed for the first time that anti-inflammatory effects, which were characterized by reduced glomerular macrophage influx concomitant with a marked reduction in proinflammatory cytokines, were involved in the mechanism of attenuating experimental anti-GBM GN by RH-TM. The observed effects might be attributable to the downregulation of ICAM-1 by reducing the HMGB1/TLR/MyD88 signaling pathway. These results identify RH-TM administration as a potential new therapeutic approach for patients with anti-GBM GN.
ACKNOWLEDGEMENTS
We thank Ms Tomoko Suzuki for the excellent technical assistance. RH-TM was a gift from Asahi Kasei Pharma Corp., who did not participate in the design, execution or funding of this study.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
Author notes
Shohei Tachibana and Masayuki Iyoda authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments