





Abstract
Resistance to thyroid hormone (RTH) syndrome is associated with mutations in the human thyroid hormone receptor-β (hTRβ), many of which show marked reduction in hormone binding. Here, we investigated the structural consequences of two RTH mutants (A234T and R243Q), residing in the flexible N-terminal portion of the ligand binding domain (LBD), which exhibit modestly reduced hormone binding with impaired release of corepressor. X-ray crystallography analyses revealed that these two RTH mutants modulate the position of this flexible region by either altering the movement of helix 1 (A234T) or disrupting a salt bridge (R243Q). The subsequent increased flexibility and mobility in regions after the two sites of mutation coincided with a disorganized LBD. Consistent with this finding, the ability of these mutant N-terminal regions (234–260) to recruit the remaining LBD was decreased in a ligand-dependent helix assembly assay. Collectively, these data suggest that structural information imparted by the flexible segment in the N-terminal LBD is critical for overall stability of the LBD. Thus, these structural analyses provide mechanistic insight into the etiology of RTH disease in human TRβ mutants that exhibit hormone binding with decreased ligand-dependent corepressor release.
THE THYROID HORMONE receptor β (TRβ) is a member of the large nuclear receptor family of transcription factors that can be activated by the thyroid hormone T3 to affect gene expression. As with other members of this superfamily, TRβ contains an N-terminal domain (A/B) activation function (AF)1 domain, a DNA-binding domain, (DBD), and a ligand binding domain (LBD), which undergoes a conformational change upon ligand binding (1). TRβ forms complexes with many coregulators in the unliganded state. Corepressors such as silencing mediator of retinoid and thyroid hormone receptor, nuclear receptor corepressor, and nuclear receptor interacting protein 13 interact with the LBD by utilizing some of the same binding surfaces as used for coactivator binding (2–5). The structural changes conferred upon hormone binding result in the dissociation of corepressors and subsequent recruitment of coactivators with a corresponding repositioning of helix 12 to pack tightly against the body of the receptor (6, 7). This dynamic structural change within the LBD results in formation of a hydrophobic groove that enables binding of coactivators via their nuclear receptor interaction domain (NR box) coactivator sequence LXXLL (8). Ligand-dependent conformational changes and subsequent coactivator binding are conserved among other nuclear receptors, as evidenced by the solved coactivator peptide/receptor structures for other receptors including the estrogen, retinoid X receptor, and peroxisome proliferator-activated receptor-γ receptors (9–11).
The human syndrome of resistance to thyroid hormone (RTH) is frequently associated with missense mutations that reside in the LBD of the hTRβ. Clinically, many of these patients exhibit normal thyroid function due to an excess production of thyroid hormone due to the lack of normal feedback mechanisms within the hypothalamic-pituitary-thyroid axis. Thus, an increase in circulating hormone levels is thought to compensate for the low hormone binding affinities exhibited by many of these mutant receptors (12). Indeed, the concentration of free hormone in a patient is often inversely correlated with the hormone binding affinity of mutant receptor (13). When RTH mutants are localized to the TRβ LBD, they cluster in three regions, including residues 310–353, 429–461, and 234–282, designated clusters 1, 2, and 3, respectively (14). Many mutations in the first and second clusters impair thyroid hormone binding directly because these residues form the hydrophobic ligand-binding pocket (Refs.1, 15 , and 16 and our unpublished data). Other mutations increase the dissociation rate of hormone from the receptor by affecting the overall stability of the LBD (our unpublished data), or instead, selectively inhibit coactivator binding without markedly reducing hormone binding because of their proximity to the hydrophobic coactivator-peptide cleft (7). Finally, some TRβ mutant receptors associated with RTH have been reported to show only a modest decrease in hormone binding affinity but are defective in corepressor release; two such mutants are A234T and R243Q (12, 17, 18). These mutations are located before helix 3 in the third cluster of RTH mutations between residues 234–282 and, when assayed in standard cell transfection assays, both RTH mutants require increased T3 hormone levels to activate standard reporters (15).
The nomenclature for the location of the A234T and R243Q RTH mutants has evolved over the years as more has been learned about this N-terminal LBD region. Originally, molecular studies assigned these two mutations to the TRβ hinge region that connects the LBD to the DBD and included up to helix 3 (19). Now, the hinge region is defined as a short domain that begins at the termination of the DBD and extends to helix 1 (202–215) based on structural arguments (19). Moreover, the ability of this N-terminal LBD region to assemble in trans with its cognate remaining LBD in a ligand-dependent manner reinforced the assignment of the region to the LBD (20). Aside from the ability of this N-terminal LBD region to recruit helices 2–12, cellular and biochemical studies also suggested that this region influences corepressor binding (3, 21, 22). Indirectly, these collective results suggest that this N-terminal region in the TRβ LBD is important for receptor integrity.
Here, to understand the underlying structural basis of RTH mutants that were reported to exhibit modestly reduced hormone binding affinities and defective corepressor release, we obtained high-resolution three-dimensional structures of the A234T and R243Q hTRβ mutants. The first A234T mutation resides at the carboxyl terminus of helix 1 leading into loop 1 and the second R243Q mutation is located within loop 2, just after helix 2 (Fig. 1A). Our analysis revealed an increased flexibility and mobility of the N-terminal LBD region in these two particular mutant TRβ receptors. Because these receptors exhibit delayed corepressor release, we infer that this N-terminal region of the LBD contributes to corepressor interaction by altering the change in the LBD structure induced by the ligand binding.
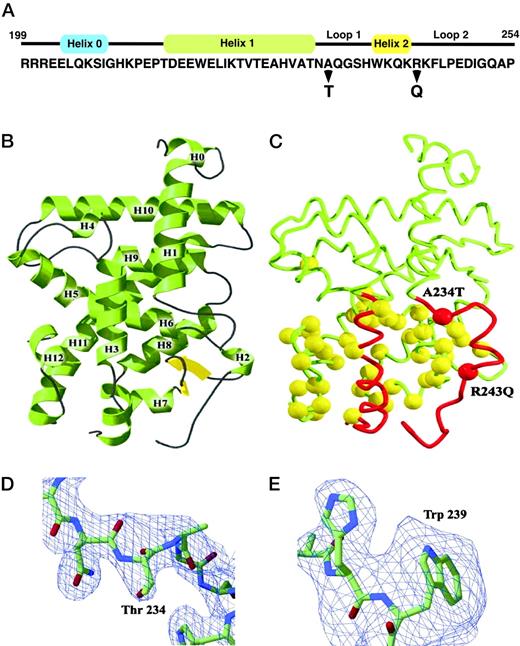
Mutant A234T and R243Q TRβ Receptor Crystal Structures A, Amino acid sequence of the N-terminal region of TRβ LBD is provided with secondary structure α-helices indicated above in colored rectangles. Location of both A234T and R243Q mutations are indicated with bold arrows. B, A ribbon drawing of the TRβ LBD is provided for the wild-type receptor with α-helices numbered from H1 to H12. C, Differences in the relative position of helix 1 through helix 3 region of TRβ LBD are shown for both A234T and R243Q mutants and are illustrated in red with the main chains depicted in yellow. The positions of both A234T and R243Q mutations are indicated. Views of the 2Fo-Fc σ electron density maps of the A234T (D) and R243Q (E) mutants contoured at 2σ are shown. In the A234T model, residues 231–237 were omitted during the calculation of the electron density map. Residues 235–242 were omitted from the 2Fo-Fc electron density map of the final R243Q structure to show the position of Trp 239. Stick representations of atoms are colored by chemical type: oxygen, red; nitrogen, blue; carbon, green.
RESULTS
X-ray crystallography was used to obtain the high-resolution three-dimensional structures of the two A234T and R243Q RTH TRβ mutants. LBDs of mutant receptor proteins were expressed and purified in the presence of the T3 analog, 3,3′,5-triiodo-l-acetic acid (TRIAC), using methodology previously employed for the wild-type hTRβ, as described in Materials and Methods. TRIAC was chosen based on previous x-ray diffraction optimization trials for yielding high quality, diffractable crystals. X-ray diffraction data measurements and coordinate refinement for both hTRβ mutants are summarized in Table 1. Both mutants display the classic nuclear receptor helical bundle consisting of a three-layer stack of 12 α-helices with a small region of β-sheet adjacent to the ligand-binding pocket (Fig. 1, B and C). Structures for both mutant receptors and wild-type TRβ are similar (Ref.1 and Fig. 1B) with A234T and R243Q structures exhibiting an overall root mean square deviation of 0.77 Å and 0.48 Å, respectively. Both mutant receptor LBD structures also include helix 0, which terminates the DBD region and is followed by a five-amino acid segment N-terminal to the beginning of the LBD (Fig. 1A and Ref.19).
Data Collection and Refinement Statistics
| Data Collection | |||||
|---|---|---|---|---|---|
| Data Set | Res (Å) | Reflections Measured | Coverage (%) | Rsyma | |
| Measured | Unique | ||||
| hTRβ A234T | 2.4 | 80196 | 14564 | 99.6 | 0.051 |
| hTRβ R243Q | 2.9 | 54104 | 13743 | 93.5 | 0.055 |
| Data Collection | |||||
|---|---|---|---|---|---|
| Data Set | Res (Å) | Reflections Measured | Coverage (%) | Rsyma | |
| Measured | Unique | ||||
| hTRβ A234T | 2.4 | 80196 | 14564 | 99.6 | 0.051 |
| hTRβ R243Q | 2.9 | 54104 | 13743 | 93.5 | 0.055 |
| Coordinate Refinement Summary | ||||||
|---|---|---|---|---|---|---|
| Data Set | Res (Å) | Reflections | Rb | Rfreeb | RMS Bonds (Å) | RMS Angles (degrees) |
| hTRβ A234T | 2.4 | 13085 | 21.3% | 25.5% | 0.012 | 1.99 |
| hTRβ R243Q | 2.9 | 12346 | 22.6% | 26.4% | 0.010 | 1.43 |
| Coordinate Refinement Summary | ||||||
|---|---|---|---|---|---|---|
| Data Set | Res (Å) | Reflections | Rb | Rfreeb | RMS Bonds (Å) | RMS Angles (degrees) |
| hTRβ A234T | 2.4 | 13085 | 21.3% | 25.5% | 0.012 | 1.99 |
| hTRβ R243Q | 2.9 | 12346 | 22.6% | 26.4% | 0.010 | 1.43 |
Rsym = ΣhΣi|Ih,i − 〈Ih〉|/Σ Ih for the intensity (I) of i observations of reflection h.
R factor = Σ|Fobs − Fcalc|/Σ|Fobs|. Rfree is calculated the same as the R factor, using 10% of the reflections that were set aside for cross-validation and not used in refinement.
Data Collection and Refinement Statistics
| Data Collection | |||||
|---|---|---|---|---|---|
| Data Set | Res (Å) | Reflections Measured | Coverage (%) | Rsyma | |
| Measured | Unique | ||||
| hTRβ A234T | 2.4 | 80196 | 14564 | 99.6 | 0.051 |
| hTRβ R243Q | 2.9 | 54104 | 13743 | 93.5 | 0.055 |
| Data Collection | |||||
|---|---|---|---|---|---|
| Data Set | Res (Å) | Reflections Measured | Coverage (%) | Rsyma | |
| Measured | Unique | ||||
| hTRβ A234T | 2.4 | 80196 | 14564 | 99.6 | 0.051 |
| hTRβ R243Q | 2.9 | 54104 | 13743 | 93.5 | 0.055 |
| Coordinate Refinement Summary | ||||||
|---|---|---|---|---|---|---|
| Data Set | Res (Å) | Reflections | Rb | Rfreeb | RMS Bonds (Å) | RMS Angles (degrees) |
| hTRβ A234T | 2.4 | 13085 | 21.3% | 25.5% | 0.012 | 1.99 |
| hTRβ R243Q | 2.9 | 12346 | 22.6% | 26.4% | 0.010 | 1.43 |
| Coordinate Refinement Summary | ||||||
|---|---|---|---|---|---|---|
| Data Set | Res (Å) | Reflections | Rb | Rfreeb | RMS Bonds (Å) | RMS Angles (degrees) |
| hTRβ A234T | 2.4 | 13085 | 21.3% | 25.5% | 0.012 | 1.99 |
| hTRβ R243Q | 2.9 | 12346 | 22.6% | 26.4% | 0.010 | 1.43 |
Rsym = ΣhΣi|Ih,i − 〈Ih〉|/Σ Ih for the intensity (I) of i observations of reflection h.
R factor = Σ|Fobs − Fcalc|/Σ|Fobs|. Rfree is calculated the same as the R factor, using 10% of the reflections that were set aside for cross-validation and not used in refinement.
To assess the structural changes in the two hTRβ receptor mutants, regions with the least change with the native structure were aligned by iterative elimination of divergent structural elements. In doing so, we identified regions of the largest structural shifts. Regions with the greatest differences were identified with a difference diagonal matrix plot, which highlights the shifted α-carbon atoms. Regions with the highest similarity were confined to residues 213–232, 278–318, 338–380, and 392–443. Using these clusters to perform comparative alignments, we found that the most significant structural shift was observed throughout loop 1, helix 2, and loop 2 (Fig. 1C) with residues 234–237 shifting a root mean square distance of 1.7Å. A smaller shift of 1.3Å was observed in the five-residue loop after helix 11 (445–449). Shifted regions are depicted in the molecule schematic for both receptor mutants (Fig. 1C). Interestingly, this region between helix 1 through loop 2 does not make direct contact with the ligand.
Electron density maps were mathematically generated to verify our molecular replacement solutions for both mutant LBDs. Maps for both structures were calculated with the mutations and the surrounding residues were omitted; the omitted residues included 231–237 for A234T and residues 235–242 for R243Q. The positioning of His 238 and Trp 239 in both mutants is supported by 2Fo-Fc electron density maps contoured at 2σ (Fig. 1, D and E). Although the mutated glutamine residue at 243 was not seen at the 2σ contour level, there was evidence of repositioning of Trp 239 (Fig. 1E). Taken together, these data suggest that the increased flexibility of the N-terminal TRβ LBD observed in the two mutations might increase the overall mobility of the remaining LBD.
Disorder in Loops 1 and 2 in the N-Terminal LBD of Mutant Receptors
We measured B values to determine whether the overall mobility is altered in the mutant receptors. This measurement provides information about the distribution of electron density at assigned positions and therefore the extent of mobility. Average B values in the A234T mutant structure were 6.7 Å2 higher than those obtained for the native structure of 48 Å2. This difference may reflect the particular quality of the crystal but could also be attributed to increased mobility in the A234T structure. Because absolute B values vary from crystal to crystal, we also compared scaled B values to assess any relative change between the native and mutant receptors. B values were linearly scaled using residues 264–440; these residues were chosen because they showed the least amount of positional shift and also had a similar overall distribution of B values in the mutant and native structures. Using this approach, B values for the region after helix 1 to the beginning of helix 3 showed elevated B values in both mutants (Fig. 2). We also noted that the loop after helix 11 also showed slightly higher relative B values (Fig. 2). The observations that mutant TRβ proteins exhibit small structural shifts with high B values suggest that there is a change in mobility rather than a change in position of the atoms.
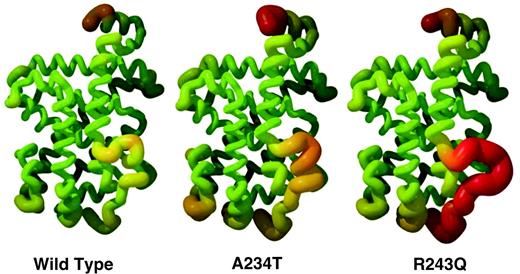
Mutated Receptors Show Increased Observed B Values The structural mobility is depicted in the wild-type and mutated TR models as measured by B values. In the helical representations of the three LBDs, trace thickness and color range represent experimental mean of the B value range. Sections with lower B value are thinner and colored in green; conversely, sections with higher B value are thicker and red in color. B values were scaled using residues 270–440 in each individual LBD structure.
Structural Changes in the A234T and R243Q Mutants
Crystal structures for both TRβ mutants revealed an altered positioning of the flexible segment just after helix 1. For the A234T receptor, the mutated residue is shifted away from the body of the receptor, and there is also a change in the termination of helix 1. The length of helix 1 was reduced by one residue due to the altered packing of Thr 234 located at the terminus of helix 1 (Fig. 3). Although the Thr 234 side chain occupies the same hydrophobic indentation on the body of the receptor as Ala 234 does in the native structure, loop 1 is pushed away from the body of the receptor due to the larger side chain of threonine. Thus, the threonine side chain is accommodated in the mutant receptor by rearrangement of the chain segment (Fig. 3).
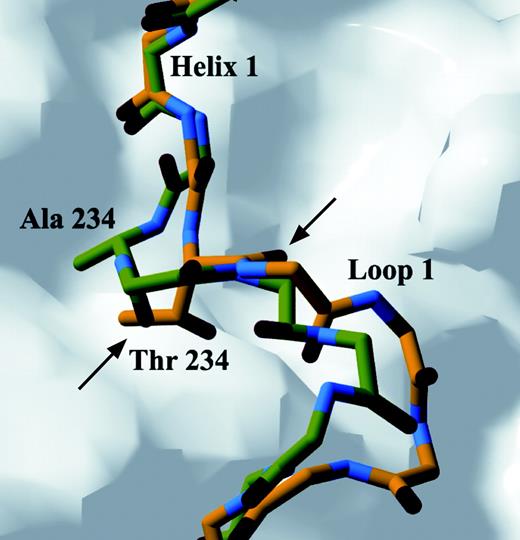
Structural Changes in Loop 1 and Helix 1 of A234T TRβ Mutant Conformations of A234T mutant (carbon atoms in yellow) and wild-type (carbon atoms in green) regions of TRβ LBD are superimposed. Substitution of Ala 234 by a Thr residue pushes loop 1 away from the native position. The position of the main chain of Thr 234 is significantly altered compared with Ala 234 in wild type as indicated by the black arrows, thus decreasing the length of helix 1 by one residue. Red and blue atoms represent oxygen and nitrogen, respectively. The body of the receptor is represented as a molecular surface calculated with a 1.4-Å probe.
For the R243Q mutation, we observed a disruption in helix 2, which in the native structure is a single α-helical turn (residues 239–243). These residues form a coil in the R243Q structure extending away from the receptor to form a new crystal contact between residues 235–242 and residues 291–286 in the symmetry-related molecule. Electron density in this region showed that only residues His 238 and Trp 239 are clearly defined (Fig. 1E). Refinement of the region by using His 238 and Trp 239 as an anchor revealed a large shift N-terminal to the mutation in loop 1 after helix 1 (Fig. 4). In contrast to loop 1, loop 2 is unchanged. Our structural data also showed that the normal salt bridge formed between Arg 243 and Asp 322 is absent in the R243Q mutant. Instead, the shorter side chains present in Gln 243 form a hydrogen bond with the backbone of Lys 242. The loop containing Trp 239 shifts a root mean square distance of 2.3 Å away from the native position and forms a crystal contact between Ala 235 and Glu 295 in the symmetry-related molecule.
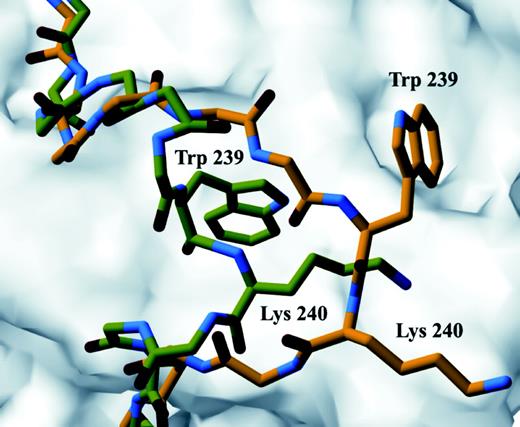
Structural Changes in Loop 1 in the R243Q TRβ Mutant Conformations of R243Q mutant (carbon atoms in yellow) and wild-type (carbon atoms in green) regions of TRβ LBD were superimposed. Loop 1 of R243Q mutant is reoriented away from the body of the receptor and forms a new crystal contact in R243Q as represented here with Trp 239 and Lys 240 changing orientation compared with wild type. Red and blue atoms represent oxygen and nitrogen, respectively. The body of the receptor is represented as a molecular surface calculated with a 1.4-Å probe.
TRβ Mutants Exhibit Decreased Helix LBD Assembly
To further assess the functional impact of increased mobility in the N-terminal region observed for both mutant receptors, we chose to use the helix assembly assay whereby helix 1 specifically binds to the remaining LBD in a ligand-dependent manner (20). For TRβ, helices 1 and 2 (204–260) bind in trans to residues 261–461 (helices 3–12) in the presence of either hormone or corepressor. A234T and R243Q mutations were introduced into the N-terminal LBD fragment (205–260) and assayed with hTRβ (261–461) in the presence of increasing concentrations of hormone (Fig. 5A). In comparison to wild-type hTRβ, the A234T N-terminal LBD fragments exhibited impaired assembly at all hormone concentrations, as measured by activation of the Gal4-luciferase reporter activity (Fig. 5B). These data are consistent with the low transcriptional activity reported for the A234T TRβ mutant at high hormone concentrations (14). Although assembly of the R243Q TRβ mutant was consistently lower than that of wild-type receptor, this reduction was much less significant than that observed for A234T. This difference likely reflects the locations of the A234T compared with the R243Q mutant with respect to the critical helix 1 that normally interfaces with the remaining LBD.
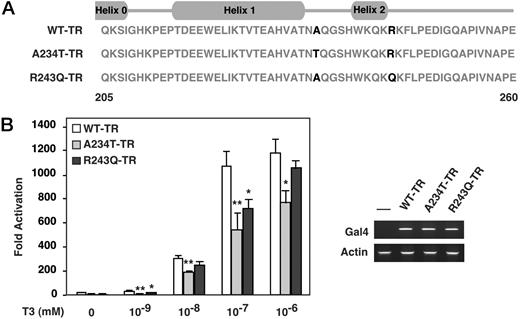
Impaired Helix Assembly in Both A234T and R243Q TRβ Mutants A, Amino acid sequence of the N-terminal domain of TRβ and mutants used in the assembly assays is provided with the positions of α-helices shown. B, Association of the N-terminal region of TRβ wild-type (WT) and A234T and R243Q mutant LBDs with the remainder of the LBD was tested in a mammalian two-hybrid experiment using human liver HEP-G2 cells. WT-TRβ, A234T-TRβ, and R243Q-TRβ mutants, respectively, fused to Gal4 binding domain were cotransfected with the activation domain VP16 fused to TRβ helices 2–12, and the Gal4-responsive luciferase reporter pGAL-RE-TK in Hep-G2 cells for 48 h. Twenty-four hours after transfection, increasing amounts of T3 (10−9, 10−8, 10−7, 10−6m) were added to cells for an additional 24 h. Assembly was significantly impaired in the A234T-TRβ mutant (P < 0.01 for 1 nm, 10 nm, 100 nm, and P < 0.05 for 1 mm, paired t test); statistical analysis was performed on three independent experiments. For the R243Q-TRβ mutant, assembly was statistically reduced at 1 nm and 100 nm, P < 0.05, but not at 10 nm or 1 mm, paired t test. Results from one transfection experiment are shown in panel B with error bars showing sd. Expression levels of Gal4-WT-TRβ, Gal4-A234T-TRβ, and Gal4-R243Q-TRβ mutants constructs used in panel B were monitored by RT-PCR using primers for the Gal4 DBD present in all constructs. Levels of actin transcripts are also shown to control for sample variation. RT-PCR results for all three constructs transfected are shown in the right lower panel.
Further evidence that these two N-terminal TRβ mutations lead to decreased stability of the LBD comes from hormone binding studies using in vitro-transcribed-translated TRβ full-length or LBD solution equilibrium binding (Table 2). Binding results obtained with full-length A234T and R243Q receptors show a modest reduction compared with the wild-type receptor and agree with previously published values of 20–60% reduction (17, 18). Hormone affinities for the mutant LBD proteins were consistently weaker (∼80% reduction, Table 2), suggesting that mutants outside of the immediate ligand binding pocket influence the overall stability of the LBD.
Relative T3 Binding to Hinge LBD and Full-Length (FL) Mutant TR Receptors
| Receptor | FL TRβ (% WT) | LBD TRβ (% WT) |
|---|---|---|
| hTRβ wild type | 1.000 ± 0.03 | 1.000 ± 0.11 |
| hTRβ A234T | 0.333 ± 0.09 | 0.24 ± 0.06 |
| hTRβ R243Q | 0.322 ± 0.09 | 0.21 ± 0.05 |
| Receptor | FL TRβ (% WT) | LBD TRβ (% WT) |
|---|---|---|
| hTRβ wild type | 1.000 ± 0.03 | 1.000 ± 0.11 |
| hTRβ A234T | 0.333 ± 0.09 | 0.24 ± 0.06 |
| hTRβ R243Q | 0.322 ± 0.09 | 0.21 ± 0.05 |
Shown are the Ka values for mutant hTRβ relative to wild-type hTRβ (100%). Values are average of four determinations and the ses are indicated.
Relative T3 Binding to Hinge LBD and Full-Length (FL) Mutant TR Receptors
| Receptor | FL TRβ (% WT) | LBD TRβ (% WT) |
|---|---|---|
| hTRβ wild type | 1.000 ± 0.03 | 1.000 ± 0.11 |
| hTRβ A234T | 0.333 ± 0.09 | 0.24 ± 0.06 |
| hTRβ R243Q | 0.322 ± 0.09 | 0.21 ± 0.05 |
| Receptor | FL TRβ (% WT) | LBD TRβ (% WT) |
|---|---|---|
| hTRβ wild type | 1.000 ± 0.03 | 1.000 ± 0.11 |
| hTRβ A234T | 0.333 ± 0.09 | 0.24 ± 0.06 |
| hTRβ R243Q | 0.322 ± 0.09 | 0.21 ± 0.05 |
Shown are the Ka values for mutant hTRβ relative to wild-type hTRβ (100%). Values are average of four determinations and the ses are indicated.
DISCUSSION
In this study, we obtained high-resolution structures for two RTH human mutations located in the N-terminal region of the LBD. Previous reports showed that these two RTH mutants have minimal effects on ligand binding but can influence ligand-dependent corepressor release. Here, we show that these mutations impair receptor function by influencing the local structural environment between the end of helix 1 and the start of helix 3, which ultimately leads to decreased stability of the LBD. Consistent with these observations, these mutations affect receptor function as evidenced by the decreased helix assembly assay and decreased hormone binding of isolated LBDs. Both the A234T and R243Q RTH N-terminal TRβ mutations result in a disorganized LBD, and thus, our findings now provide a structural explanation underlying the pathophysiology of a subset of RTH mutants that exhibit normal hormone binding with impaired corepressor release.
Mobility of the N-Terminal LBD Region
The two human TRβ mutations studied here are located in the N-terminal region of the LBD and, therefore, are thought not to participate directly in forming the hydrophobic ligand binding pocket and thus hormone binding. Structural analyses of many ligand-dependent nuclear receptors have now reassigned the original hinge region that exhibits low sequence conservation to the N-terminal portion of the LBD. For the hTRβ, this N-terminal region begins at residue 215 and contains helices 1 and 2, as well as loops 1 and 2 (Fig. 1A; Ref.19). Recent studies using a helix assembly assay have shown that this N-terminal region of the LBD is receptor specific for both ligand-dependent and ligand-independent receptors and thus represents a signature motif for each nuclear receptor (20, 23, 24).
The A234T and R243Q mutant proteins show significant structural shifts that modulate the conformation of the LBD. In the A234T mutant, the most significant shift was found in loop 1 (234–237) of the N-terminal part of the LBD in which the Thr 234 introduces a steric obstruction between helix 1 and the rest of the LBD of the receptor, thereby displacing the mutated residue away from the receptor. We observed increased mobility in the region after the A234T mutation based on our observed B values. Although experimental B values for the R243Q structure are less reliable than for the A234T mutant structure, our data suggest that this mutation in loop 2 promotes a new conformation of loop 1 and simultaneously unfolds helix 2 (235–242). Specifically, loop 2 (244–270) is more mobile in the mutant structure than in the native structure. Disruption of the potential salt bridge (Arg 243-Asp 322) and new crystal contact due to rearrangement of loop 2 most likely result in the conformation shift of both loop 1 and helix 2. Thus, both the A234T and R243Q hTRβ mutant structures show significant alterations in the stability and flexibility of the N-terminal domain of the LBD, which in turn destabilize the remaining LBD. Consistent with this notion, hormone binding affinities for the isolated LBD are decreased compared with their wild-type counterpart (Table 2). Taken together, the structural changes in both TRβ mutant receptors demonstrate how the mobility and repositioning of the signature motif region (helix 1 through loop 2) influences the overall structural stability of the LBD. These findings led us to hypothesize that structural changes within the N-terminal region LBD influence cofactor interactions and alter the dynamic of exchange corepressor/coactivator.
Previous studies using the helix assembly assay demonstrate that helix 1 binds specifically to its cognate remaining LBD in a ligand-dependent manner. The dependency on ligand suggests that the TRβ LBD adopts a liganded conformation to interact with helix 1 (20). In addition, structural analysis on liganded and unliganded peroxisome proliferator-activated receptor-γ showed that helix 1 is mostly unstructured in the absence of ligand and becomes structured when ligand is present in the hormone-binding pocket (25). From a structural standpoint, helix one is the least mobile element of the N-terminal LBD based on the low B values and forms the majority of the stabilizing interactions with the body of the receptor. Here, results from the helix assembly assay show that the N-terminal region of both TRβ receptor mutants is able to interact with the remaining LBD, albeit at high ligand concentrations. Thus, whereas the major interface between helix 1 and the body of the receptor are maintained in these receptor mutants, the high requirement of ligand to achieve sub-threshold levels of assembly, especially for the A234T mutant, suggest that this N-terminal region, which includes loop 1, modulates the overall stability of the LBD.
In studies to be published elsewhere, we have also determined x-ray crystal structures of two hTRβ LBDs frequently mutated in RTH (A317T and R316H) that show impaired hormone binding and require increased concentrations of ligand to overcome the defective function. In the A317T mutation, the ligand is repositioned, resulting in a packing defect and an increased receptor volume causing a bulging out of the opposite face of the receptor that binds coactivators and corepressors. The structural consequences of the R316H mutation destabilize the receptor in the ligand binding pocket and cause movement of the C-terminal helix of the DBD, helix 1, and the loop after helix 1, leading to higher overall structured mobility. However, the instability of the R316H mutation cannot be corrected by ligand, and most likely explains why this receptor is defective even when bound by ligand. These results contrast findings reported here with the A234T and R243Q mutant receptors, where effects on the integrity of the ligand binding pocket are secondary to structural changes in the N-terminal LBD region.
N-Terminal Mutations and Corepressor Release
Our structural data suggest that modulation of corepressor/coactivator interactions with the LBD rather than a change in receptor dimerization accounts for the lowered receptor activity displayed by both mutant receptors. We find no evidence that suggest major structural shifts or altered positioning of the dimer-forming surfaces present in helices 10 and 11 (26). These data are consistent with previous studies suggesting unchanged dimerization of both mutants (15). The N-terminal region of the LBD is involved in the hormone-induced conformational shift that causes the dissociation of corepressor and association of coactivator (27). For A234T and R243Q mutants, hormone levels required for corepressor release are increased 2- and 5-fold, respectively, when compared with wild-type receptor (18). These data suggest that the R243Q mutant exhibits greater defects in corepressor release. Interestingly, although the R243Q mutant is only mildly crippled in the helix assembly assay, this mutant receptor displays a higher degree of mobility in the N-terminal region or signature motif of the LBD. Thus, positional changes in the signature motif of each receptor may account for selective interaction with either coactivators or corepressors.
Studies of the interaction between the TRβ and corepressor indicate that residues buried under helix 1 in the liganded structure are involved in corepressor interaction (27). Mutational mapping studies of hTRβ indicate that three surfaces can affect binding of the receptor LBD with corepressor (22). Of these three surfaces, the most critical is found under helix 12, just next to helix 3, based on association assays. Part of this surface is inaccessible in the liganded structure. The other two surfaces are found adjacent to helix 1 and to helix 10 and are likely to participate in dimer formation and DNA binding by the DBD. Whereas mutations at every surface residue of helix 1 have no effect on corepressor binding, mutating residues important for stabilizing the packing of the helix on the body of the receptor, such as Trp 219, strongly affect corepressor binding. These data support a role for the N-terminal LBD region in controlling equilibriums between corepressor-receptor and receptor-hormone association rather than directly interacting with repressor. Given that helix 1 interacts poorly with the unliganded receptor (20), and that the transition to liganded receptor appears blocked in the A234T and R243Q mutants, we propose that these specific mutations affect the receptor’s conformational equilibrium and result in a shift to the repressed unliganded state.
Finally, the inability of A234T and R243Q RTH mutant receptors to undergo normal ligand-dependent conformational changes due to the altered flexibility in the N-terminal region or signature motif of the LBD may partially account for the physiological defects observed in these TRβ human mutants. Normally, elevated T3 levels in patients harboring these N-terminal RTH mutations would be predicted to repress TSH levels owing to the classic feedback inhibition loops by T3 on the hypothalamic-pituitary-thyroid axis. However, the majority of these untreated patients display normal TSH levels (14), often with enhanced TSH bioactivity (28), implying a lack of T3-dependent feedback (29–32). Similar to TRβ-dependent gene activation, negative regulation of the TSH gene, is also ligand dependent, whereas TR association with corepressor activates basal levels of TSH (33). The normal levels of TSH observed in patients harboring either the A234T or R243Q mutant receptor are consistent with recruitment of corepressor by these mutant receptors, despite their delayed release of corepressor. Additional structural studies on other classes of TRβ mutant receptors will continue to provide insights into the molecular basis underlying RTH endocrine disease.
MATERIALS AND METHODS
Expression and Purification of TRβ Receptor Protein
Mutant hTRβ LBDs spanning from Glu 202 to Asp 461 were subcloned into a His-tagged bacterial expression vector, pET28a (Novagen) at the unique PstI-BamHI sites; LBD fragments were derived from pCDNA hTRβ (gift of S. Refetoff). Mutant hTRβ LBDs were grown in Lennox broth (Sigma, St. Louis, MO) in Escherichia coli strain BL21DE3 pLysS (Novagen) at 22 C, and induced at an OD600 = 1.5 with 0.5 mm isopropyl β-d thiogalactoside for an additional 6 h. Harvested bacteria pellets were frozen in liquid nitrogen and stored at −80 C. Thawed pellets were lysed in 50 mm sodium phosphate (pH 8.0), 300 mm NaCl, 10% glycerol, 0.1% monothioglycerol, 1 mm phenylmethylsulfonyl fluoride, 1 mm benzamidine HCl, with 0.2 mg/ml lysozyme for 20 min at 0 C, sonicated, and centrifuged to remove debris. LBD protein was purified by Ni-affinity chromatography using Talon (CLONTECH Laboratories, Inc., Palo Alto, CA) equilibrated in sodium phosphate buffer, and eluted with a 0–300 mm imidazole gradient. Liganded TRIAC (Sigma) receptor was further isolated using TSK (tempered silica potassium)-phenyl HPLC (TosoHaas, Philadelphia, PA) as described (34). Yields for wild-type and mutant hTRβ were approximately 3 mg/liter of bacterial culture.
Crystallization
For crystallization, hTRβ LBDs were diluted into 20 mm HEPES (pH 8.0), 3 mm dithiothreitol and concentrated to 11.5 mg/ml by ultrafiltration (UFV2BGC10, Millipore Corp., Bedford, MA). The N-terminal His tag was not removed before crystallization. Crystals for the A234T mutant bound to TRIAC were obtained in the same conditions as previously noted for wild-type liganded hTRβ E202–461 (1). Refinement of this starting crystallization condition resulted in a well buffer of 700 mm sodium acetate and 100 mm sodium cacodylate, pH 7.6. Crystals were flash frozen in liquid nitrogen after incremental increases in the glycerol concentration through a series of soaks. Cryo solvent contained 900 mm sodium acetate, 100 mm sodium cacodylate (pH 7.6), and a range of glycerol concentrations from 5–25% in five equal steps. These crystals diffracted to 2.4Å at the Stanford Synchrotron Radiation Laboratory 7-1 beam line. Crystals for the R243Q mutant bound to TRIAC were produced in well buffer of 700 mm sodium acetate and 100 mm sodium cacodylate, pH 7.6. Microseeding was needed to obtain optimal crystals using hTRβR243Q/T3 crystals crushed with the seed bead kit (Hampton Research, Riverside, CA). Cryo solvent was the same as the mother liquor with the addition of 300 mm sodium acetate and a range of glycerol from 5% to 25% in five equal steps. Crystals of the R243Q mutant diffracted to 2.9Å at the Stanford Synchrotron Radiation Laboratory 7-1 beam line.
Data Measurements and Structural Refinement
An x-ray data set for A234T was measured using 1-degree oscillations over a rotation of 117°. The data set was found to be 99.7% complete to 2.4Å resolution. Crystals of A234T with TRIAC bound exhibited the same hexagonal bipyramidal morphology found in wild-type receptor. Crystals were space grouped P3121 (a = 68.82 Å, c = 131.07 Å). Reflections were indexed and scaled using DENZO and SCALEPACK (35). A molecular replacement solution was found with Crystallography and Nuclear Magnetic Resonance System (CNS) 1.0 (36) rotational search using the wild-type hTRβ/TRIAC structure with ligand and mutant side chain omitted. The structure was refined with CNS using multiple rounds of simulated annealing and manual rebuilding with the Quanta98 software package. Electron density maps and coordinates were manipulated with the Collaborative Crystallography Project Number Four in Protein crystallography package (37). The resulting structure had an Rcryst of 21.8% and an Rfree of 25.7%. A data set collected for R243Q crystals using 1-degree oscillations over 90° exhibited the same space group and morphology as the native data set. Crystal constants were slightly different (a = b = 68.84; c = 130.53). The data set for this mutant was reduced with DENZO and SCALEPACK. An initial molecular replacement solution was found with CNS 1.0 using a rotational search of the native hTRβ with ligand and mutation site omitted. The structure was refined using the same method described for A234T receptor. The final structure exhibited very high B values. This is consistent with both the high mosaic quality of the crystal (∼1.0) and the calculated Wilson B value of 85.9. The resulting structure was refined to an R factor of 23.9% and a RFree of 28.7%. Statistics from these refinements are listed in Table 1.
Plasmid Construction for Assembly Assay
Fusion constructs used in the assembly assay were constructed as follows: For Gal4 fusion containing the N-terminal fragments of wild-type hTRβ receptor fused to the Gal4 DBD, PCR amplification was used to generate the DNA fragment corresponding to amino acids from 196–260; corresponding wild-type or mutant fragments were then subcloned into the Gal4 DBD pM vector (CLONTECH Laboratories, Inc.) at the EcoRI/XbaI sites. The A234T and R243Q were generated with the Quickchange site-directed mutagenesis kit according to the manufacturer protocol (Stratagene, La Jolla, CA). For creating the VP16 fusion protein containing wild-type hTRβ from helices 2 to 12, PCR amplification was used to generate the appropriate TRβ fragment (236–460), which was then subcloned into the pVP16 vector (CLONTECH Laboratories, Inc.) using EcoRI/SalI. Plasmids were verified by DNA sequencing. Human liver carcinoma HepG2 cells were grown in DMEM supplemented with 10% bovine serum. One day before transfection, cells were plated on 24-well dishes at 50% density in DMEM supplemented with 10% charcoal-stripped serum. Transfections were performed using Fugene (Roche Clinical Laboratories, Indianapolis, IN) for 24 h. Typically, 200 ng of reporter constructs, 50 ng of Gal4 expression vector, and 100 ng of β-galactosidase expression vector (control for transfection efficiency) were used in all transfection experiments. Cells were harvested 24 h following transfection of DNA and assayed for both luciferase activity (Luciferase assay kit, PharMingen, San Diego, CA) and β-galactosidase activity. All experiments were performed at least three times in triplicate.
Ligand-Binding Kinetic Assays
Wild-type, A234T, R243Q full-length and LBD TRβ receptors were expressed from the CMX and pET28 plasmids, respectively, using the TNT T7 Quick in vitro translation kit (Promega Corp., Madison, WI). The affinities of binding of T3 to all receptors were determined using a saturation binding assay. Briefly, 15 fmol of each in vitro translated protein were incubated overnight at 4 C with varying concentrations of l-3,5,3′-[125I]T3 (NEN Life Science Products, Boston, MA) in a 100 μl volume of E400 buffer (400 mm NaCl, 20 mm KPO4, pH 8, 0.5 mm EDTA, 1.0 mm MgCl2, 10% glycerol) and 1 mm monothioglycerol and 50 μg calf thymus histones (Calbiochem, La Jolla, CA). The receptor-bound [125I]T3 was isolated by gravity flow through a 2-ml course Sephadex G25 (Pharmacia Biotech, Piscataway, NJ) column and quantified using a γ-counter (COBRA, Packard Instruments, Meriden, CT). Binding curves were fit by nonlinear regression and dissociation constant (Kd) values were calculated using the one-site saturation binding model contained in the Prism 3.0 program (GraphPad Software, Inc., San Diego, CA).
Acknowledgments
We would like to thank Dr. S. Refetoff for providing the hTRβ cDNA plasmids containing the A234T and R243Q mutations.
This work was supported by NIH Grants DK-41842 and DK-09516 (to J.D.B.) and a P01 DK-58390 (to R.J.F. and H.A.I.), as well as an American Heart Postdoctoral Fellowship (to M.D.).
J.D.B has proprietary interests in, and serves as a consultant and Deputy Director to, Karo Bio AB, which has commercial interests in this area of research.
Abbreviations:
- CNS,
Crystallography and Nuclear Magnetic Resonance System;
- DBD,
DNA-binding domain;
- hTRβ,
human thyroid hormone receptor-β;
- LBD,
ligand-binding domain;
- RTH,
resistance to thyroid hormone;
- TRIAC,
3,3′,5-triiodo-l-acetic acid.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}