





Abstract
Polintons are double-stranded DNA, virus-like self-synthesizing transposons widely found in eukaryotic genomes. Recent metagenomic discoveries of Polinton-like viruses are consistent with the hypothesis that Polintons invade eukaryotic host genomes through infectious viral particles. Nematode genomes contain multiple copies of Polintons and provide an opportunity to explore the natural distribution and evolution of Polintons during this process. We performed an extensive search of Polintons across nematode genomes, identifying multiple full-length Polinton copies in several species. We provide evidence of both ancient Polinton integrations and recent mobility in strains of the same nematode species. In addition to the major nematode Polinton family, we identified a group of Polintons that are overall closely related to the major family but encode a distinct protein-primed DNA polymerase B (pPolB) that is related to homologs from a different group of Polintons present outside of the Nematoda. Phylogenetic analyses on the pPolBs support the evolutionary scenarios in which these extrinsic pPolBs that seem to derive from Polinton families present in oomycetes and molluscs replaced the canonical pPolB in subsets of Polintons found in terrestrial and marine nematodes, respectively, suggesting interphylum horizontal gene transfers. The pPolBs of the terrestrial nematode and oomycete Polintons share a unique feature, an insertion of an HNH nuclease domain, whereas the pPolBs in the marine nematode Polintons share an insertion of a VSR nuclease domain with marine mollusc pPolBs. We hypothesize that horizontal gene transfer occurs among Polintons from widely different but cohabiting hosts.
Introduction
Polintons/Mavericks (hereafter referred to as Polintons) were discovered as double-stranded DNA (dsDNA) transposons that encode a self-synthesizing, protein-primed DNA polymerase B (pPolB) and a retroviral-element-like integrase (INT) (hence, the name Polintons) (Kapitonov and Jurka 2006; Pritham et al. 2007). Thus far, primarily identified and characterized in silico, Polintons are among the larger known DNA transposons found widely across unicellular and multicellular eukaryotes, ranging from 13 to 25 kb with 100- to 1,500-bp terminal inverted repeats (TIRs) and with 5- to 8-bp target site duplications (TSDs) (Kapitonov and Jurka 2006; Feschotte and Pritham 2007; Jurka et al. 2007; Pritham et al. 2007; Krupovic and Koonin 2015). In addition to pPolB and INT, Polintons typically encode a core set of conserved genes encoding homologs of dsDNA virus proteins involved in virion morphogenesis, such as adenovirus-like maturation protease (PRO), genome-packaging ATPase, and major and minor capsid proteins (MCPs and mCPs, respectively) (Feschotte and Pritham 2005; Gao and Voytas 2005; Krupovic et al. 2014; Krupovic and Koonin 2014; Koonin et al. 2015; Krupovic and Koonin 2015, 2016). Polintons generally occupy a low fraction of their host genome (Novick et al. 2011; Haapa-Paananen et al. 2014; da Silva et al. 2018; Shao et al. 2019; Klai et al. 2020; Barreat and Katzourakis 2021; Chase et al. 2022; Bellas et al. 2023); however, there are genomes with much higher occurrence, such as the excavate Trichomonas vaginalis where Polintons expanded to occupy more than 30% of the genome (Feschotte and Pritham 2007).
While Polintons were first thought to be transposable elements, recent experimental and metagenomic discoveries of Polinton-like virophages and Polinton-like viruses suggest that Polintons found in multicellular eukaryotic genomes are capable of transmission between host genomes as infectious viral particles (Fischer and Suttle 2011; Yutin et al. 2015; Fischer and Hackl 2016; Bellas and Sommaruga 2021; Starrett et al. 2021; Roitman et al. 2023; Roux et al. 2023). A combination of inferred protein characteristics (the presence of genes encoding homologs of the MCP and mCP and the ATPase and PRO involved in capsid maturation) strongly supports this possibility (Krupovic et al. 2014; Krupovic and Koonin 2015). A dual capability as viruses and transposons (Krupovic et al. 2014; Krupovic and Koonin 2015; Chase et al. 2022) would afford Polintons an ability to propagate both within and between genomes.
Nematode genomes provide an excellent opportunity to observe transmissible genetic elements (Bessereau 2006; Laricchia et al. 2017) from diverse environments. The nematode phylum includes both parasitic and free-living species, with the latter having successfully adapted to diverse environmental niches including soil, rotten fruits, freshwater, and seawater (De Ley 2006; Vanreusel et al. 2010; Frezal and Felix 2015; Majdi and Traunspurger 2015; Nigon and Felix 2017; Hodda 2022). Because nematodes interact with diverse types of organisms (viruses, bacteria, protists, animals, and plants) (Felix et al. 2011; Schulenburg and Felix 2017; Zhang et al. 2017; Felix and Wang 2019; Pulavarty et al. 2021), the phylum is likely to have been exposed to a broad cross section of potentially infectious or transmissible elements. Thus, nematode genomes serve as a potentially broad detector for elements capable of interspecies transfer and acquisition. Although several RNA viruses have been reported to infect or to be associated with nematodes (Brown et al. 1995; Bekal et al. 2011; Felix et al. 2011; Franz et al. 2012; Bekal et al. 2014; Ruark et al. 2017; Lin et al. 2018; Vieira and Nemchinov 2019; Williams et al. 2019), only limited information about possible dsDNA virus infection and evolution in nematodes is available (Poinar et al. 1980; Hess and Poinar 1985).
In silico analyses have identified a handful of Polintons from nematode species (Kapitonov and Jurka 2006; Pritham et al. 2007; Starrett et al. 2021; Widen et al. 2023). These nematode Polintons encode a core set of Polinton proteins including pPolB, INT, ATPase, PRO, mCP and MCP, and nematode Polinton-specific protein PC1 (Kapitonov and Jurka 2006), which was recently proposed to function as a fusogen (Widen et al. 2023). The presence of a fusogen suggests that nematode Polintons may form an enveloped viral particle (Vance and Lee 2020). However, the natural distribution and evolution of Polintons across nematode genomes have not been extensively studied.
In this study, we use the diversity of available nematode genome sequences to investigate the natural variation and history of Polintons in this phylum. Of particular note, we observed an apparently modular history of the elements, with 2 distinct classes of pPolBs in otherwise similar nematode Polintons. Additional analysis of these 2 classes outside of Nematoda suggests that Polintons have acquired and exchanged pPolB genes during evolution. The distribution of pPolB homologs in the Earth biome suggests that Polintons act as gene transfer vehicles.
Results and Discussion
Two Groups of Polintons Encoding Distinct DNA Polymerases in Nematodes
To explore the distribution and evolution of Polintons in nematodes, we searched for full-length Polintons across 354 nematode genomes from 180 species and identified 266 Polintons in 66 genomes of 29 species (supplementary table S1, Supplementary Material online; see Materials and Methods). While surveying the identified Polinton sequences, we noticed that a group of nematode Polintons encompassed a pPolB that was only weakly similar to the typical pPolB encoded by other nematode Polintons, in contrast to all other core Polinton genes and TIRs that were highly conserved. We denoted the distant pPolB group “pPolB2” to distinguish it from the typical nematode Polinton pPolBs (hereafter “pPolB1”) (Fig. 1a).
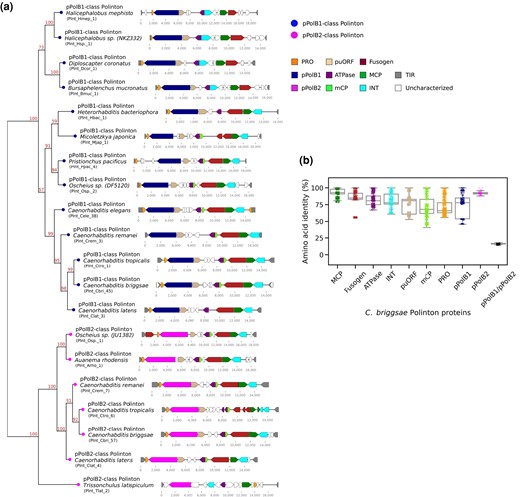
Two distinct DNA polymerase B families in Caenorhabditis polintons. a) A phylogenetic tree (left) was constructed by applying a ML-based method (IQ-TREE) to a multiple sequence alignment of pPolB proteins from Polintons of 19 nematode species. The genetic architectures of these elements are shown on the right. These Polintons were chosen for initial sequence alignment based on uninterrupted DNA polymerase and other Polinton protein coding regions. Bootstrap supporting values are shown on the top position of tree branches. Abbreviations for inferred polinton components are as follows: PRO for adenovirus-like maturation protease, puORF for Polinton-uncharacterized ORF, ATPase for a packaging ATPase, MCP and mCP for major and minor capsid proteins, INT for a retroviral-like-element integrase, and TIR for terminal inverted repeat. "Uncharacterized" are novel (potentially either spurious or functional) ORFs that have not been previously described in other Polintons. Labels in parentheses denote unique identifiers (UIs) for each Polinton (see supplementary table S1, Supplementary Material online). b) A dot plot and a background box plot show percent (%) amino acid identities between each Polinton protein found in 49 C. briggsae Polintons.
The unexpected identification of 2 distinct groups of pPolBs in the nematode Polintons prompted us to examine the molecular diversification of Polinton proteins in nematodes in greater detail. To compare the extent of protein sequence similarity among different Polinton proteins, we constructed the corresponding multiple sequence alignments and calculated the percentage amino acid identity for each pair of homologous proteins from Polintons of Caenorhabditis briggsae (C. briggsae was chosen because this species was found to encompass the greatest number of both pPolB1- and pPolB2-class Polintons). The resulting comparisons showed 92% median identity for MCP, 85% for Fusogen, 81% for ATPase, 80% for INT, 66% for PRO, 78% for Polinton-uncharacterized open reading frame (puORF), 68% for mCP, 77% for pPolB1, and 91% for pPolB2 (Fig. 1b), indicating that these proteins are highly conserved among C. briggsae Polintons.
In a sharp contrast, pairwise comparisons between pPolB1 and pPolB2 showed a median of 16% amino acid sequence identity. The Polinton pPolBs belong the protein-primed DNA polymerases B family that contain conserved exonuclease motifs (Exo I, II, and III) required for proofreading activity and polymerase motifs (Pol A, B, and C) required for DNA polymerization activity (Blanco and Salas 1995, 1996; Redrejo-Rodriguez et al. 2017; Kazlauskas et al. 2020). Examination of the multiple sequence alignment of representative pPolB1 and pPolB2 proteins from Polintons of 19 nematode species with the prototypical Phi29 phage pPolB indicated that all 3 exonucleases and all 3 polymerase motifs are conserved in both pPolB1 and pPolB2 (supplementary figs. S1 and S2a, Supplementary Material online), suggesting that both groups of the nematode Polintons possess fully functional DNA polymerases.
Given the low sequence similarity between pPolB1 and pPolB2, we also compared the structural models of the 2 proteins obtained using AlphaFold2 (Jumper et al. 2021) through ColabFold (Mirdita et al. 2022). Structural alignment and superposition of C. briggsae pPolB1 and pPolB2 structures show evidence of a close structural similarity between the 2 groups of pPolBs, in spite of the low sequence identity, in line with the prediction that both are active DNA polymerases (supplementary fig. S2b to d, Supplementary Material online; Fig. 2a). In addition to the conserved structural core of pPolB1 and pPolB2, the structural alignment also revealed potential pPolB1- and pPolB2-specific domains (Fig. 2a). In particular, we identified a pPolB2-specific domain that is conserved across all nematode pPolB2 proteins except the pPolB2 from 3 Polintons in a marine species, Trissonchulus latispiculum (Fig. 2a; supplementary fig. S1, Supplementary Material online). A protein domain search using HHpred (Gabler et al. 2020) identified this domain as a putative HNH endonuclease (Shen et al. 2010) (Fig. 2b to e). An additional structural alignment between terrestrial C. briggsae pPolB2 and marine T. latispiculum (Tlat) pPolB2 revealed a Tlat pPolB2-specific domain, which was subsequently identified as a VSR endonuclease domain (Fig. 2g to j). Apparently, this pPolB2 family acquired an additional endonuclease domain during its evolution.
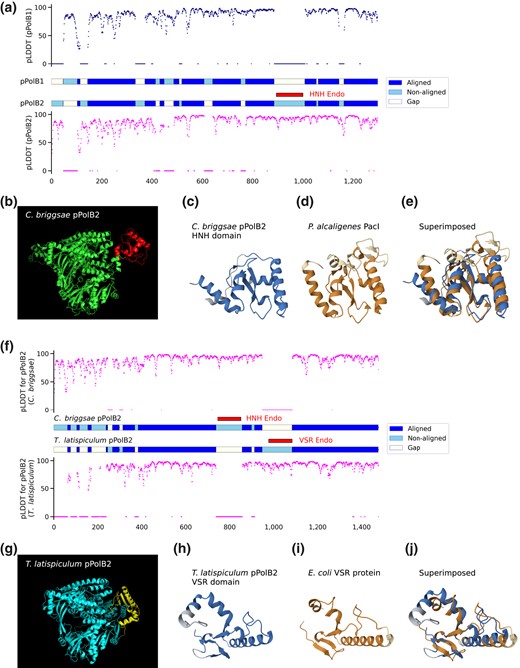
HNH and VSR endonuclease domains are found in terrestrial and marine nematode pPolB2s, respectively, but not in pPolB1. a) Aligned, nonaligned, and gap parts of pPolB1 and pPolB2 (middle panel) from sequence-free structural alignment are shown with pLDDT (predicted local distance difference test) score predicted at the single amino acid level by AlphaFold2. Overall aligned parts for both pPolB1 and pPolB2 showed high pLDDT score, suggesting confident prediction of the structures. b) The predicted structure of C. briggsae pPolB2 (green) with the HNH endonuclease domain (red). c to e) The predicted structure of pPolB2-specific HNH domain (c) and the crystal structure of P. alcaligenes PacI HNH endonuclease (d) (PDBid: 3M7K) (Shen et al. 2010) are superimposed e) (RMSD: 3.03, TM-score: 0.55) to show potentially conserved structural folds. f) Aligned and non-aligned parts of C. briggsae pPolB2 and T. latispiculum pPolB2 (middle panel) from sequence-free structural alignment are shown with pLDDT score. g) The predicted of T. latispiculum pPolB2 (cyan) with the VSR domain (yellow). h to j) The predicted structure of T. latispiculum pPolB2 VSR endonuclease domain (h) and the crystal structure of Escherichia coli VSR endonuclease (i) (PDBid: 1VSR) (Tsutakawa et al. 1999) are superimposed (j) (RMSD: 3.06, TM-score: 0.63) to show potentially conserved structural folds. Dark and light colors for both orange and blue (c to e and h to j) indicate aligned and nonaligned positions, respectively.
Taken together, these observations suggest an ancient separation between pPolB1 and pPolB2 polymerase families, with an additional split between 2 divisions within the pPolB2 class (represented by terrestrial and marine pPolB2 homologs, respectively). In the following sections, we investigate the potential origins and evolution of the 2 groups of nematode Polintons.
Interspecies and Intraspecies Copy Number Variations of pPolB1 and pPolB2 Classes of Polintons in Nematode Genomes
An extensive search for Polintons in diverse nematode genomes revealed variations in the interspecies and intraspecies distributions and copy numbers of the pPolB1 and pPolB2 groups of Polintons. The pPolB1-class and pPolB2-class Polintons were detected in 25 and 12 nematode species, respectively (out of a total of 180 species) (Fig. 3; supplementary table S1, Supplementary Material online). Polintons of both classes were detected in several nematode genera (Pristionchus, Oscheius, and Caenorhabditis), with varying copy numbers; notable copy number variations were also observed between species within a genus, in particular, Caenorhabditis, for which multiple genomes are available (Fig. 3). Generally, the pPolB1-class Polintons were substantially more abundant than the pPolB2-class ones, with several species carrying only pPolB1 Polintons and only 4 possessing pPolB2 Polintons (Fig. 3; supplementary table S1, Supplementary Material online). These observations appear to be compatible with independent spread of the pPolB1 and pPolB2 classes of Polintons among nematodes.
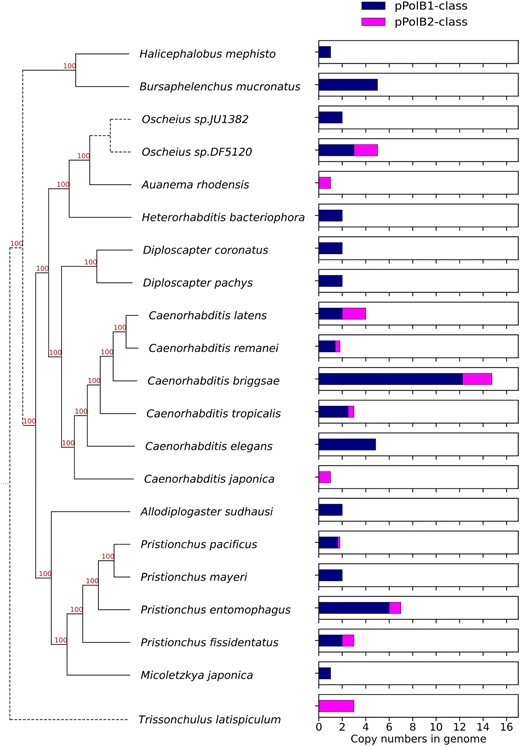
Interspecies copy number variations of nematode Polintons. Bar graphs (right panel) showing average copy numbers per genome of full-length pPolB1-class and pPolB2-class Polintons in selected Nematoda genomes where the species can be placed on a current phylogenetic tree based on recent analyses (Ahmed et al. 2022; Lee et al. 2023). Note that several species that have not yet been placed on the tree are not shown here; all information for species (placed and not) is shown in supplementary table S1, Supplementary Material online.
We also observed distinct patterns and copy numbers of Polintons within each species, most clearly, in a set of Caenorhabditis species for which genome sequences are available for multiple wild-type strains (supplementary fig. S3a to d, Supplementary Material online). Because genomes for 3 wild-type strains of C. briggsae have been assembled at chromosome level (Stein et al. 2003; Stevens et al. 2022) with the largest copy numbers of both pPoB1- and pPolB2-class Polintons in nematodes (Fig. 3; supplementary fig. S3d, Supplementary Material online), we further analyzed the genomic neighborhoods of the Polintons, to test whether transposition occurred before or after the diversification of the wild isolates. Only 2 pPolB1-class Polintons shared the same positions in all 3 C. briggsae strains and 6 pPolB1-class Polintons shared the positions in AF16 and QX1410 strains (supplementary fig. S3e, Supplementary Material online), suggesting that these Polintons already occupied the same positions in the genomes of the respective C. briggsae ancestors. In contrast, the remaining pPolB1-class Polintons and all pPolB2-class Polintons were found at unique positions (supplementary fig. S3e, Supplementary Material online), suggesting transposition events that occurred after the strain divergence. These intraspecies variations imply that Polintons were actively integrating into Caenorhabditis genomes both before and after the diversification of the characterized strains, with each genome reflecting a distinct natural history of exposure to the 2 classes of Polintons.
Transfer of pPolB Genes Among Polintons
The discovery of 2 distinct groups of pPolBs in nematode Polintons could be explained through one of two alternative scenarios. Under the first scenario, there are 2 distinct classes of nematode Polintons with only an ancient relationship, in which case, all proteins would show low sequence conservation, in the same range as the pPolBs. The second scenario involves a relatively recent pPolB swap, in which case, the other proteins would be far more similar than the pPolBs.
The bulk protein sequence comparisons described above favor the second scenario, but to investigate the evolution of the 2 groups of Polintons further, we examined the relationships among the Polintons within a single species (C. briggsae) by calculating sequence similarities for all possible pairwise combinations of Polintons. This analysis showed strong positive correlations between the pairwise similarities of all proteins other than pPolB, regardless of whether the compared Polintons encoded pPolB1 or pPolB2 (Fig. 4a and b; supplementary fig. S4a and b, Supplementary Material online). In within-class comparisons, strong correlations between the similarities among pPolBs and those among other Polinton proteins (Fig. 4c) suggest that pPolB1 and pPolB2 proteins can be maintained within Polintons through substantial evolutionary spans. By contrast, in between-class comparisons, the similarities among pPolB1 and pPolB2 did not correlate with those among other proteins (Fig. 4c). These observations strongly suggest that in one of the nematode Polinton classes, pPolB was relatively recently replaced by a distantly related homolog.
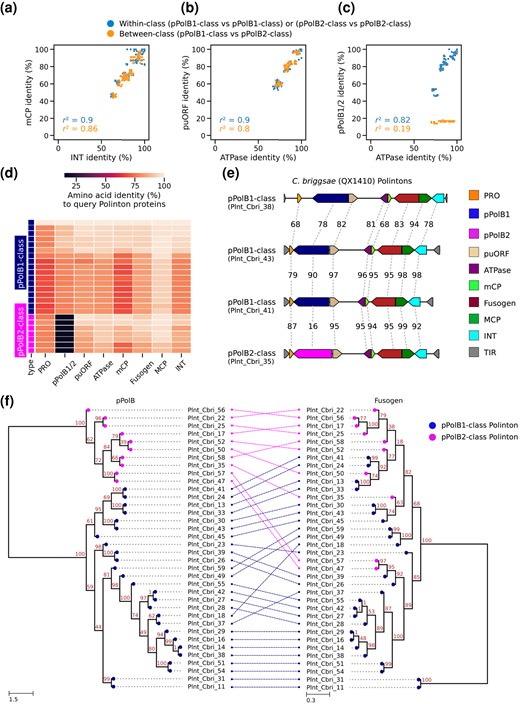
Two distantly related groups of pPolBs in C. briggsae Polintons. a to c) Scatter plots showing percent amino acid identity of 1 protein for each axis from C. briggsae Polinton species; mCP and INT a), puORF and ATPase b), and pPolB1/2 and ATPase c). Each dot indicates a pair of 2 C. briggsae Polintons, whose proteins indicated at the X and Y axes were aligned, respectively, for calculating percent identity. Dot colors indicate whether 2 Polintons in a pair for comparison are within the same pPolB group or from different pPolB groups. d) Heatmap showing percentage amino acid identity of proteins. Each row shows an individual C. briggsae Polinton, while each column represents one of the conserved Polinton proteins. All comparisons were performed against a query C. briggsae Polinton (Plnt_Cbri_18). e) Genetic architectures of 4 C. briggsae Polintons (QX1410 strain, a wild isolate) shown with % amino acid identities for each Polinton protein. Plnt_Cbri_41 and Plnt_Cbri_43 are relatively close to each other, showing >90% identity, while Plnt_Cbri_43 and Plnt_Cbri_38 are more distant (evidenced by lower % identities). The continuous nature of the homology outside of the pPolB region provides evidence for gradual divergence in these regions during the ancestry of the current elements. By contrast, the Plnt_Cbri_35 Polinton showed strong similarity in all proteins except pPolB (particularly to PInt_Cbri_41), with dramatic divergence for pPolB, suggesting a swapping event of the pPolB gene during the derivation of this Polinton. f) A tanglegram showing the relationship between 2 phylogenetic trees constructed from multiple sequence alignment of C. briggsae pPolB1/2 (left panel) and Fusogen (right panel) proteins. The colors of the nodes indicate pPolB1-class and pPolB2-class. Dashed lines between 2 trees denote the correspondence of pPolB and Fusogen proteins found from the same Polinton. The tree exemplifies that close relationships between non-pPolB proteins can be accompanied by very distant relationships between the corresponding pPolBs.
We further searched for examples of pPolB1 and pPolB2 genes being embedded in otherwise closely similar Polinton sequences. One such example, providing a useful illustration, was observed in C. briggsae strain QX1410 (Fig. 4d and e), where Polintons Plnt_Cbri_41 and Plnt_Cbri_35 both showed high similarity to the canonical Plnt_Cbri_43 sequence in all non-pPolB proteins (Fig. 4d), whereas the pPolBs showed dramatic divergence (only 16% sequence identity) (Fig. 4e). A more general view of these relationships is apparent from the phylogenetic trees of the pPolBs and the rest of the Polinton proteins (supplementary fig. S5a to h, Supplementary Material online). In the pPolB tree, the 2 groups of pPolBs formed 2 well separated clades whereas the other Polinton proteins associated with either pPolB1 or pPolB2 were interspersed in the respective trees (Fig. 4f; supplementary figs. S4c and S5a to h, Supplementary Material online). In addition to C. briggsae, we also observed a possible inter-Polinton exchange of pPolB genes in an Oscheius species (DF5120) as indicated by the observation that an MCP protein from pPolB2-class Polinton clustered with a pPolB1-class Polinton MCP in the phylogenetic tree (supplementary fig. S6a and b, Supplementary Material online). These observations are compatible with the proposed exchange of distinct pPolB genes between Polintons.
Distribution of pPolB1 and pPolB2 Homologs Across Eukaryotes Suggests Distinct Evolutionary Trajectories of pPolB1 and pPolB2
To characterize the evolutionary trajectories of the 2 pPolB types on a larger evolutionary scale, we explored the distribution of pPolB1 and pPolB2 homologs across the available eukaryotic genome assemblies (7,897 assemblies from 64 phyla; 1.63 Terabases available through NCBI genome database as of late April 2023). To focus on Polintons, we restricted our analysis to pPolB genes linked to INT homologs (see Materials and Methods). We identified 20,702 INT-associated pPolB1 and 1,331 pPolB2 homologs distributed across 960 and 164 genomes of diverse eukaryotes, respectively (supplementary tables S2 and S3, Supplementary Material online). These pPolBs found from all taxon groups contained conserved exonuclease and polymerase motifs for pPolB1 and pPolB2 groups (supplementary fig. S7, Supplementary Material online), suggesting that these pPolBs are functional Polinton polymerases.
Distinct patterns of pPolB1 and pPolB2 distribution at the phylum level were identified (Fig. 5a). Mimicking the case of nematodes, pPolB1-containing Polintons showed a wide spread across 30 phyla. pPolB1 was found to be most abundant in invertebrates, with multiple copies found in 17 of the 23 invertebrate phyla. By contrast, pPolB2 showed a sparse distribution, being represented mostly in 2 unicellular eukaryote phyla, Metamonada and Oomycota, and 5 invertebrate phyla, Annelida, Brachiopoda, Echinodermata, Mollusca, and Nematoda. The highest pPolB2 copy numbers were detected in Metamonada, particularly, in T. vaginalis (388 copies) and Tritrichomonas foetus (259 copies), consistent with a previous report on the high abundance of Polintons in T. vaginalis (Feschotte and Pritham 2007).
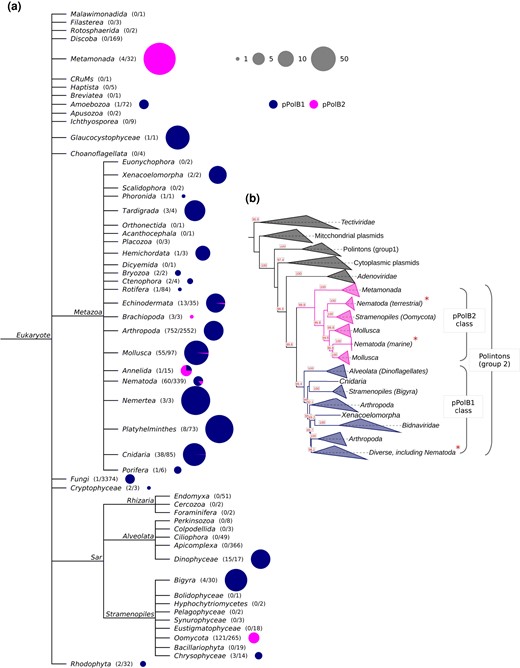
Distinct distribution and evolutionary trajectories of pPolB1 and pPolB2 groups in the biosphere. a) A NCBI taxonomy tree with Eukaryotic taxa. The numbers in parentheses indicate the number of genomes having at least 1 copy of pPolB and the number of total genomes subjected to the Polinton pPolB search (see Materials and Methods for details). The sizes and colors in the pie charts indicate the copy number of Polinton pPolBs per pPolB-containing genomes and the classes of pPolB. b) A phylogenetic tree was constructed from multiple sequence alignment of pPolB proteins found in diverse taxa including viruses of prokaryotes, eukaryotes, DNA plasmids, and Polintons using the IQ-TREE-inferred ML method. Bootstrap supporting values are shown on the bottom positions of tree branches. All pPolBs identified in this study were found within Group 2 Polinton pPolBs where pPolB1 and pPolB2 classes form two major monophyletic groups. Asterisks indicated the Nematoda pPolB1 and pPolB2 proteins identified in this study.
A broader phylogenetic analysis that included a set of representatives from the main groups of pPolBs encoded by viruses of prokaryotes and eukaryotes as well as Polintons and DNA plasmids further illuminated the evolutionary trajectories of pPolB1 and pPolB2 (Fig. 5b). Each of the 2 groups of pPolBs was found to be monophyletic (Fig. 5b; supplementary fig. S8, Supplementary Material online). pPolB1 and pPolB2 formed distinct clades in the tree, and their relative positions suggested that these 2 DNA polymerase forms diverged early on. Given the greater diversity and broader distribution among the nematodes of pPolB1 compared with pPolB2 (Fig. 3; supplementary fig. S3a to e, Supplementary Material online), it appears most likely that the ancestral nematode Polintons encoded pPolB1, which was replaced in 1 nematode branch. That replacement might have occurred via horizontal gene transfer from oomycete Polintons, given the tree topology and the highest sequence similarities between oomycete pPolBs and nematode pPolB2s (Fig. 5b; supplementary fig. S8, Supplementary Material online). Furthermore, protein domain search showed that pPolB2 of oomycete Polintons shares the HNH endonuclease domain with the homolog from the nematode Polintons (Fig. 6; supplementary figs. S9 and S10a, Supplementary Material online), to the exclusion of other pPolB2s. Thus, the HNH domain is a unique synapomorphy (shared derived character) that links the oomycete and nematode Polintons encoding pPolB2, strongly supporting the horizontal gene transfer scenario with the oomycete being the likely donor. The HNH domain could potentially have been captured by the oomycete pPolB2 from a Group I self-splicing intron (Hausner et al. 2014), before finding its way into the nematode genomes.
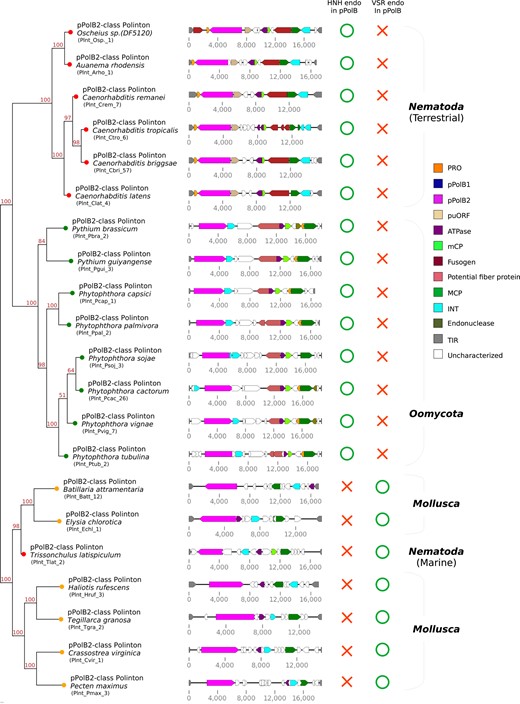
Phylogenetic relationships of Polinton pPolB2 proteins from 3 distinct phyla. A phylogenetic tree (left) has been built by using IQ-TREE-inferred ML on a multiple sequence alignment of pPolB2 protein sequences found from Nematoda, Oomycota, and Mollusca (see supplementary fig. S9, Supplementary Material online for full alignment data). Bootstrap supporting values are shown on the top position of tree branches.
While analyzing protein profiles of oomycete Polintons, we noted that oomycete Polintons encode a conserved protein showing similarity to beta-helix-fold tail fiber proteins of tailed bacteriophages (Fig. 6), raising a possibility that these proteins mediate recognition and binding of oomycetes Polintons and oomycetes hosts. Notably, unlike nematode Polintons, oomycete Polintons do not encode membrane fusion proteins and hence are unlikely to be enveloped. Whether potential envelopment is an exclusive property of the nematode Polintons compared with Polintons from other organisms remains to be investigated.
Given the apparent relatively recent incursion of pPolB2 into nematode Polintons, direct interphylum horizontal transfer of the pPolB2 gene between oomycete and nematode Polintons, perhaps, via virus particles formed by Polintons appears likely. Under this scenario, a nematode would have been infected by the oomycete Polinton virus particles, with subsequent recombination with a resident nematode Polinton such that only the pPolB gene was transferred. Alternatively, the immediate source of the pPolB2 homologs in nematodes might have been an intermediate species. In either case, the pPolB2-encoding Polinton family was fixed in the nematodes and spread to a variety of nematode species, again, possibly, through Polinton virions.
A Distinct Group of pPolB2 Polintons in a Marine Nematode Species
A recent study of marine nematode genomes (Lee et al. 2023) allowed us to further examine Polintons in an ecological niche for which genome data were initially limited. In addition to land-dwelling nematodes (both free and parasitic), there are many nematode species that dwell in aquatic habitats (De Ley 2006; Majdi and Traunspurger 2015; Hodda 2022). Notably, analysis of the recently sequenced marine nematode genomes identified a Polinton-containing genome (T. latispiculum; Lee et al. 2023) that encompasses no pPolB1-class Polintons but carries a distinct group of 3 pPolB2-class Polintons (Fig. 1a; supplementary table S1, Supplementary Material online). Sequence and structural comparison of the terrestrial C. briggsae pPolB2 and a marine T. latispiculum pPolB2 showed that T. latispiculum pPolB2 lacked the HNH nuclease domain but instead contained a VSR (very short patch repair) endonuclease domain (Fig. 2f and g; supplementary figs. S9 and S10b, Supplementary Material online), which was not found in any other nematode pPolB2s (supplementary fig. S1, Supplementary Material online).
The pPolB2s of the marine nematode Polinton are highly diverged from the pPolB2s of the terrestrial nematode species. Among the known Polintons, the marine nematode pPolB2s showed the highest similarity to pPolB2s of mollusc Polintons as indicated by the phylogenetic tree topology (Figs. 5b and 6; supplementary fig. S8, Supplementary Material online). Polintons of a mud snail (Batillaria attramentaria) and a sea slug (Elysia chlorotica) encode the closest relatives of the marine nematode pPolB2 with the shared VSR endonuclease domain, an apparent synapomorphy of this subgroup of pPolB2 (Fig. 6; supplementary fig. S8, Supplementary Material online). The presence of the VSR domain along with the phylogenetic tree topology supports an additional, independent interphylum horizontal gene transfer, in this case between Polintons of nematodes and mollusc Polintons that inhabit the same marine environments. The interphylum transfer in this lineage appears to be limited to pPolB as evidenced by the topologies of the phylogenetic trees for other highly conserved Polinton proteins (MCP and INT are shown as examples in supplementary fig. S10c and d, Supplementary Material online), which showed the expected phylum-specific clustering suggestive of vertical evolution.
Given that both gene exchange events detected in this work involved the pPolB family, still unknown characteristics of this gene family might favor selection for gene exchange. Alternatively, gene exchange among Polintons might be a general event, if relatively rare, with a coincidental occurrence of these 2 exchanges with the same gene family. Finally, we note the possibility of ongoing genetic exchange of genes or fragments between Polintons within Nematoda, with the relatively close similarity between elements potentially both favoring such events and rendering detection based on sequence comparison more challenging. As current data were insufficient to track such events, they remain a possibility for further investigation.
Conclusions
In this work, we present the unexpected finding that nematode Polintons form 2 distinct groups that differ by encoding highly diverged pPolBs. The pPolB1 group is by far more widely spread and more abundant among the nematodes than the pPolB2 group suggesting that pPolB2 comparatively recently displaced pPolB1 in 2 distinct lineages of nematode Polintons at least one of which subsequently spread across nematode species. Phylogenetic analysis of a broad selection of pPolBs suggested that pPolB2s originated from oomycete and mollusc Polintons independently of pPolB1. The DNA polymerase displacement then might have occurred by horizontal transfer of the pPolB2 gene, possibly, via infection of nematodes by oomycete and mollusc Polinton virus particles, with subsequent recombination with a resident nematode Polinton, resulting in the displacement of the DNA polymerase gene alone. The 2 independent incursions of pPolB2 subfamilies into the terrestrial and marine nematodes, from the oomycete and marine mollusc Polinton families, respectively, indicate that the transfers are consistent with the native ecosystems of different groups of nematodes. In this context, we note that terrestrial nematodes can serve as hosts for natural oomycete infection, which could have facilitated the transfer (Osman et al. 2018; Grover and Barkoulas 2021), whereas marine nematodes most likely shared environments, such as sediment, with prevalent marine molluscs. Although Polinton virions so far have not been observed, the conservation of the MCPs and mCPs, the packaging ATPase, and the maturation PRO across the entire diversity of Polintons strongly suggest that such particles exist. Most likely, these virions mediate the Polinton spread, with the precise characteristics of genomic Polinton elements in a species group reflecting the historical environmental niche rather than the phylogeny. The findings presented in this paper are compatible with this prediction.
Materials and Methods
Search of Polintons Through Nematoda Genomes
Assembled genomes of Nematoda were downloaded from the NCBI genome assembly database by using the NCBI data sets (https://www.ncbi.nlm.nih.gov/datasets/) command-line tools. The taxon keyword “Nematoda” was used to download fasta format genome data. Genome sequences were subjected to a tBLASTn search (with 0.001 as an e-value cutoff and 50 as a minimum bitscore). Query sequences (pPolB, INT, MCP, ATPase, and Pro) for tBLASTn search were obtained from ORF prediction by ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) on C. briggsae Polinton-1 (Polinton-1_CB, WBTransposon00000832) (Kapitonov and Jurka 2006) that was downloaded from the WormBase (https://wormbase.org/). We filtered out candidate genetic blocks that had all 5 query proteins within 20 kb and gathered all of these segments with upstream and downstream buffers of 10 kb. Next, the candidates were further filtered out by the presence of TIRs that were detected using Inverted Repeat Finder (https://tandem-test.bu.edu/irf/irf.download.html) (Warburton et al. 2004). The 6 bases upstream and downstream of each candidate TIR were used to test whether a TSD was present. Polinton candidate sequences were further validated by the presence of at least 5 Polinton proteins through ORF prediction on candidate sequences and protein-protein BLAST (BLASTp, bitscore cutoff: 100) of the ORFs against query Polinton proteins. To ensure that the Polintons are from nematode (and not from other contaminating species), we scanned all NCBI-available genomes (WGS; all assemblies for the Whole Genome Shotgun sequencing projects available as of Feb 14, 2022) for closely matching sequences using the PebbleScout resource (https://pebblescout.ncbi.nlm.nih.gov/) (Shiryev and Agarwala 2023). The search across preindexed WGS nucleotide resources (17.33 terabases) detected only sequences from Nematoda genomes, arguing against any significant signal from contamination by unrelated species (this search does not rule out the possibility that other related nematodes have contaminated some assemblies; while unlikely, this situation would not substantially affect any of the conclusions of this work). A total of 266 Polintons from 66 nematode genomes (29 species) were identified and summarized in supplementary table S1, Supplementary Material online. Parallel approaches were taken to the identification of mollusc and oomycete Polintons (used for Fig. 6), using a primary search for pPolB sequences with refinement for the presence of linked coding regions for the additional Polinton proteins. PebbleScout analyses similar to those with the nematode Polinton set were consistent with the phyla assignments for these Polintons.
Multiple Sequence Alignment and Phylogenetic Tree Analysis
Multiple sequence alignment for nematode pPolB proteins, C. briggsae Polinton proteins, and pPolB proteins found from each taxon group was performed using ClustalO (Sievers et al. 2011; Sievers and Higgins 2018). Phylogenetic trees were generated with maximum likelihood (ML) method by using IQ-TREE (Kalyaanamoorthy et al. 2017; Hoang et al. 2018; Minh et al. 2020) and mid-point rooted and visualized by ETE3 Python package (Huerta-Cepas et al. 2016). Percent identity between protein sequences was calculated based on the pairwise comparison of sequences within the ClustalX environment (Larkin et al. 2007). Conserved motifs were searched from the multiple sequence alignment results, and the enrichment of amino acids was visualized by using the LogoMaker Python package (Tareen and Kinney 2020).
We noted that a subset of Polintons contained fragmented ORFs, mainly due to nonsense mutations and/or frame-shifting small indels, suggesting pseudogenization. In the alignments used for phylogenetic analysis, we included only those sequences that appeared to be intact based on minimum amino acid length cutoffs from a survey of known Polintons (cutoffs used were as follows: pPolB: 900; INT: 360; ATPase: 190; puORF: 300; Fusogen: 730; mCP: 110; MCP: 390; and PRO: 110).
For phylogenetic analyses of pPolBs from multiple taxa, pPolB protein sequences in each taxon group were clustered and a representative sequence was selected for each cluster by using CD-HIT (Fu et al. 2012). Then, pPolBs were subjected to multiple sequence alignment by using ClustalO (Sievers et al. 2011; Sievers and Higgins 2018). Gap regions in the alignment data were trimmed off with the trimAl gappyout option (Capella-Gutierrez et al. 2009), and then phylogenetic trees were reconstructed by using ML method inferred by IQ-TREE v2.0.3 with ultrafast bootstrapping (1,000 iterations) (Kalyaanamoorthy et al. 2017; Hoang et al. 2018; Minh et al. 2020) and with LG + F + R10 model as the best fitting model. The nodes of the phylogenetic tree were collapsed if all the pPolB leaves of the node were from the same taxon groups. The phylogenetic trees were mid-point rooted and visualized by using ETE3 Python package (Huerta-Cepas et al. 2016).
For the more global phylogenetic analysis, a data set of pPolB sequences was retrieved from Krupovic and Koonin (2014). The data set included pPolB sequences from bacterial tectiviruses, linear mitochondrial and cytoplasmic plasmids, adenoviruses, and a limited number of bidnaviruses and Polintons. This data set was supplemented with a greatly expanded set of pPolB sequences of Polintons, bidnaviruses, and adintoviruses (Starrett et al. 2021). The data set was then clustered to 50% identity over 80% of alignment length using MMseqs2 (Steinegger and Soding 2017; Mirdita et al. 2019). The sequences were aligned using MAFFT with the G-INS-1 option (Katoh et al. 2019). Low-information content positions were removed using trimAl with the gappyout option (Capella-Gutierrez et al. 2009). The ML phylogenetic tree was calculated using IQ-TREE v2.0.6 (Minh et al. 2020) with the best-fitting substitution model determined for the data, which was LG + F + R10.
Protein Structure Prediction and Alignment
Structure prediction steps of C. briggsae pPolB1 or pPolB2 proteins were executed using MMSeqs2 (Steinegger and Soding 2017; Mirdita et al. 2019) and AlphaFold2 (Jumper et al. 2021) on ColabFold v1.5.2 (Mirdita et al. 2022) (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb). Pairwise structural alignment of predicted pPolB1 and pPolB2 structures was performed with FATCAT (Ye and Godzik 2004; Li et al. 2020) (https://www.rcsb.org/alignment) with default parameters. The structure was visualized using the PyMOL Molecular Graphics System, Version 2.5 Schrödinger, LLC.
Polinton pPolB Search Through NCBI Genome Assemblies
We downloaded 7,897 genomes from 64 phyla of eukaryotes (except Viridiplantae or Chordata) available through the NCBI genome database as of late 2023 April (assembly source: GenBank). To explore the distribution of nematode pPolB1 and pPolB2 homologs in the genomes, each fasta format genome file was indexed for tBLASTn search, and Polinton pPolB1 and pPolB2 protein sequences were searched through the genome using tBLASTn. To provide a stringent search, we filtered out small matches by setting a match length cutoff (650 amino acids for both pPolB1 and pPolB2). Then, the matched sequences with 20 kb both upstream and downstream were used to find potential ORFs for pPolB and other Polinton proteins. We further filtered candidates based on the length of predicted pPolB ORFs (a minimum ORF length: 900 amino acids). Subsequently, hmmsearch (Eddy 2011) (http://hmmer.org/) was performed to detect homology between predicted ORFs and Polinton proteins. Profiles for the Polinton proteins (INT, MCP, and ATPase) were built from the multiple sequence alignments of each Polinton protein obtained from RepBase Polintons (https://www.girinst.org/repbase) (Jurka 2000; Jurka et al. 2005). To ensure that pPolB gene candidates are indeed Polinton polymerases, we selected pPolBs that codetected with an INT protein nearby (see supplementary table S3, Supplementary Material online for pPolB-Polinton information). For each taxon group, copy numbers of pPolB1 and pPolB2 were normalized by the number of genomes that had at least a single copy of pPolB and visualized by using the Python ETE3 package (Huerta-Cepas et al. 2016).
Supplementary Material
Supplementary material is available at Molecular Biology and Evolution online.
Acknowledgments
We thank Karen Lynn Artiles, Lamia Wahba, Nimit Jain, Massa Shoura, Matthew James McCoy, Orkan Ilbay, Jingxun Chen, Usman Enam, Emily Greenwald, Ivan Nikolay Zheludev, Drew Galls, Janie Soo-hyun Kim, Colette Benko, David Lipman, Sergey Shiryev, and Richa Agarwala for helpful discussion.
Funding
This study was supported by the National Institute of General Medical Sciences (NIGMS) grant (R35GM130366 to A.Z.F.) and by the Long-term Postdoctoral Fellowship (LT000329/2019-L) from the Human Frontier Science Program and the Bernard Cohen Postdoctoral Fellowship from Stanford University School of Medicine (to D.-E.J.). S.S. is supported by the Stanford Cardiovascular Institute Summer Undergraduate Fellowship. E.V.K. is supported by the Intramural Research Program (ZIALM000073) of the National Library of Medicine, National Institutes of Health (USA).
Data Availability
The genome sequence data underlying this article are available via the NCBI genome assembly database (https://www.ncbi.nlm.nih.gov/assembly/). All the genomes used for the Polinton search are listed in supplementary tables S1 and S3, Supplementary material online.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}