





Abstract
The RNA-dependent RNA polymerase (RdRp) of all known double-stranded RNA viruses is located within the viral particle and is responsible for the transcription and replication of the viral genome. Through an RT-PCR assay, we determined that purified virions, in vitro translated RdRp proteins, and purified recombinant RdRp proteins of partitiviruses also have reverse transcriptase (RT) function. We show that partitivirus RdRps 1) synthesized DNA from homologous and heterologous dsRNA templates; 2) are active using both ssRNA and dsRNA templates; and 3) are active at lower temperatures compared to an optimal reaction temperature of commercial RT enzymes. This finding poses an intriguing question: why do partitiviruses, with dsRNA genomes, have a polymerase with RT functions? In comparison, 3Dpol, the RdRp of poliovirus, did not show any RT activity. Our findings lead us to propose a new evolutionary model for RNA viruses where the RdRp of dsRNA viruses could be the ancestor of RdRps.
Introduction
A majority of viruses infecting eukaryotes including humans, other animals, fungi, and plants have RNA genomes. These genomes may be double-stranded (ds) or single-stranded (ss). Viruses with a persistent lifestyle, described in fungi and plants, including many crop plants (Roossinck 2010, 2012), are often asymptomatic, have low titers, and are not transmitted horizontally, but remain in their hosts for thousands of years through nearly 100% vertical transmission. They lack cell-to-cell movement and distribute in their hosts by cell division (Roossinck 2016). Most persistent viruses, with the exception of members of the Endornaviridae family, have dsRNA genomes, and include the families Partitiviridae, Totiviridae, Chrysoviridae, and Amalgaviridae, with numerous members that infect either fungi or plants (Roossinck 2019). Persistent viruses have slow evolutionary rates in comparison with acute viruses (Molina et al. 2011; Safari and Roossinck 2018; Peyambari et al. 2019), likely due to long associations with the host resulting in strong purifying selection pressure, and the stamping machine mode of replication characteristic of dsRNA viruses (Safari and Roossinck 2014). In addition, dsRNA genomes are not subjected to the posttranscriptional modifications/damage that result in mutations in ssRNA molecules (Fordyce et al. 2013). Finally, these differences may be due to higher fidelity of the polymerases of dsRNA viruses, although this is poorly studied.
Nucleic acid polymerases are classified into four groups based upon the specificity for template and (deoxy) nucleoside triphosphate (NTP): 1) DNA-dependent DNA polymerase (DdDp); 2) RNA-dependent DNA polymerase (reverse transcriptase, RT); 3) DNA-dependent RNA polymerase (DdRp); and 4) RNA-dependent RNA polymerase (RdRp).
The RT enzyme, responsible for the synthesis of DNA from viral RNA, was first discovered in RNA tumor viruses (Baltimore 1970; Temin and Mizutani 1970). The RT enzyme plays an important role in retroviruses, pararetroviruses, retrotransposons, and mobile genetic elements in prokaryotes known as group II introns(Saldanha et al. 1993; Hohn and Rothnie 2013; Hughes 2014; Hu and Seeger 2015). A series of conserved motifs, required for enzymatic function, are identified in RTs (Xiong and Eickbush 1990) and some of these are conserved in all polymerases showing RNA template specificity (Poch et al. 1989). The enzymes most closely related to RTs are viral RdRps with essential roles in the transcription and replication of RNA viruses. In encapsidated viruses with dsRNA genomes, the RdRp is packaged into virions with the genome segments, and it is thought to be at a ratio of one RdRp molecule per packaged segment (Buck and Kempson-Jones 1973, 1974). No structures of any RdRps of persistent viruses with dsRNA genomes have been described. However, in other dsRNA viruses (i.e., ϕ6, reovirus, rotavirus, birnavirus, and picobirnavirus) the crystal structures of RdRps revealed that they adopt the right-handed polymerase form containing fingers, palm, and thumb domains that are common among distantly related RNA and DNA polymerases (Butcher et al. 2001; Tao et al. 2002; Pan et al. 2007; Lu et al. 2008; Collier et al. 2016).
With a few exceptions, including bacteriophages ϕ6, ϕ8, ϕ13, and ϕ12 (Makeyev and Bamford 2000; Yang et al. 2001; Yang, Gottlieb, et al. 2003; Yang, Makeyev, et al. 2003), RdRps of dsRNA viruses are not active in the absence of capsid proteins, perhaps to ensure that dsRNA molecules are only produced inside the capsid. This strategy prevents exposure of the dsRNA in the cytoplasm and avoids activation of the host cell’s antiviral response (Starnes and Joklik 1993; Patton et al. 1997; Boyce et al. 2004; Patton et al. 2006). Particle-associated RdRp molecules are responsible for both replication and transcription of dsRNA viruses. Through sequence analysis of persistent viruses with dsRNA genomes, a conserved domain was observed with similarity to the conserved RT domain.
Although these RdRps are classified into the RT-like superfamily, persistent viruses are not known to have RT activity, and there is no apparent reason why dsRNA viruses would require an RT, or preserve an unnecessary RT domain over a long evolutionary history.
Among all types of viruses, dsRNA viruses have been reported as endogenous viral elements integrated into the genomes of different eukaryotes including fungi and plants. Of these, viruses in the family Partitiviridae are dominant, followed by those of the families Totiviridae and Chrysoviridae (Liu et al. 2010; Chiba et al. 2011; Liu et al. 2012); however, no mechanism has been described for how a cytoplasmic RNA virus sequence can become integrated into nuclear DNA.
These observations, taken together with our sequence analysis, lead us to the hypothesis that RdRps of persistent viruses with dsRNA genomes may function as RTs.
To the best of our knowledge, there has been no previous analysis of the potential RT activity of these enzymes. In this study, we used RdRps of partitiviruses to demonstrate RT activity of these enzymes. The family Partitiviridae contains members with simple bisegmented dsRNA genomes that infect plants, fungi, and protists; the smaller segment usually encodes the coat protein (CP), and the larger segment typically codes for the RdRp.
Results
RT Activity of Purified Virus Particles
Partitiviruses are thought to package only one copy of the RdRp inside each particle that contains one of the two genomic RNAs (Buck and Kempson-Jones 1974). In this study, we used pepper cryptic virus 1 (PCV1), a persistent plant virus that infects nearly all cultivars of Jalapeño peppers (Safari et al. 2019). Purified PCV1 virus particles were used as the polymerase source in an RT reaction to experimentally demonstrate the activity of the RdRp (fig. 1A).
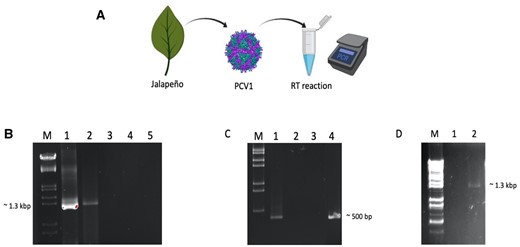
Reverse transcription activity of PCV1 virions using homologous and heterologous dsRNA templates. (A) A schematic representation of the assay using PCR to detect cDNA. (B) cDNA products of PCV1 dsRNA detected by PCR using RdRp primers; M: DNA marker, 1, MMuLV-RT used as enzyme, 2, PCV1 virions used as enzyme in RT reaction, 3, negative control (no enzyme), 4, negative control (no template), 5, negative control (isolated fraction from uninfected Jalapeño pepper used as enzyme in RT reaction). (C) RT-PCR analysis using ZMCV1 dsRNA as heterologous RNA template with ZMCV1-RdRp primers; M, DNA marker, 1, PCV1 virions used as enzyme, 2, negative control (no enzyme), 3, negative control (no template), 4, MMuLV used as enzyme in RT reaction. (D) RT-PCR analysis using PCV1 virions as the source of RNA template and PCV1-RdRp primers; M, DNA marker, 1, PCV1 virions were boiled and the RT reaction was without RT enzyme, 2, virions were boiled and then PCV1 virions were added to RT reaction as RT enzyme.
In the RT reaction with PCV1 virus particles as enzyme, the incorporation of dNTPs to generate cDNA was detected and confirmed by PCR from different RNA templates including homologous PCV1 dsRNA (fig. 1B), heterologous Zea mays chrysovirus 1 (ZMCV1) dsRNA (Peyambari et al. 2019) (fig. 1C) and boiled PCV1 virions as the source of RNA (fig. 1D). The amplified product is similar in size to that from the RT reaction catalyzed by the commercially available RT from murine leukemia virus (MMuLV) (fig. 1B, lane 1 vs. 5 and fig. 1C, lane 1 vs. 2). Controls for these reactions, including no enzyme (fig. 1B, lane 2 and fig. 1C, lane 3), no template (fig. 1B, lane 3 and fig. 1C, lane 4), and boiled virus particles without enzyme (fig. 1D, lane 1) showed no similar amplification product at the expected size. More importantly, an isolated fraction from uninfected Jalapeño peppers also did not exhibit any RT activity (fig. 1B, lane 4), hence the RT activity was unique to the purified PCV1 virions. After removal of RNAs by RNase A treatment, the products of the RT reactions were amplified by PCR, demonstrating that the products are DNA, and the enzyme is not template specific.
RT Activity of In Vitro Translated PCV1 RdRp
Persistent viruses have notoriously low titers, and PCV1 has been estimated to produce less than 500 progeny virus/cell (M. Safari, personal communication). We used PURExpress®, a cell-free transcription and translation system, to obtain an optimal amount of RdRp protein for further study. The RT activity of in vitro translated RdRp was evaluated using a similar strategy of PCR to detect the RT products (fig. 2A). The in vitro expressed PCV1 RdRp protein yielded a band with similar size to MMuLV-RT (fig. 2B, lane 1 and 2 vs. lane 5). The optimal incubation temperature of the RT reaction for in vitro translated PCV1 RdRp protein was room temperature (fig. 2B) and increasing the temperature to 42°C abolished the activity (fig. 2C).
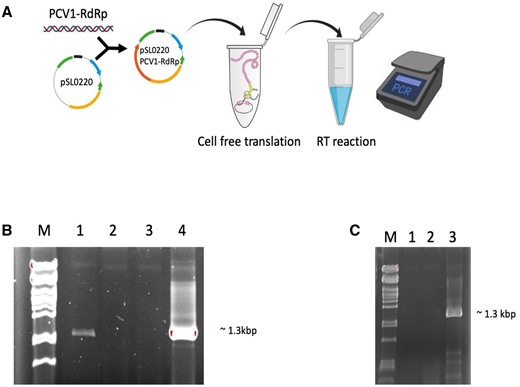
In vitro translated PCV 1 RdRp is active as an RT. (A) a schematic of in vitro translation of PCV1 RdRp and determination the RT activity of protein through RT-PCR. (B) RT-PCR analysis using PCV1 dsRNA as RNA template and PCV1-RdRp primers; M, DNA marker, 1, in vitro translated PCV1 RdRp as enzyme, 2, negative control (no enzyme), 3, negative control (no primer), 4, MMuLV used as enzyme. (C) RT-PCR analysis using PCV1 dsRNA as RNA template and PCV1-RdRp primers, RT reactions at 42°C; M, DNA marker, 1, negative control (no enzyme), 2, in vitro translated PCV1 RdRp used as RT enzyme, 3, MMuLV used as enzyme.
Expression and Purification of Recombinant RdRps
To analyze the RT activity of RdRps of PCV1 and eight other partitiviruses, codon-optimized sequences of RdRp genes were expressed in frame with the maltose basic protein (MBP) tag in E. coli and purified by amylose affinity chromatography. The amino acid sequences of RdRp proteins did not differ from wild-type sequences. The expression plasmids were introduced into E. coli Shuffle B strain, induced with isopropyl β-D-thiogalactopyranoside (IPTG), and incubated at 16°C. Incubation at higher temperatures (25–37°C) in some cases led to increases in protein expression, but most of the protein was insoluble, and in some cases expression was inhibited.
The cells produced recombinant proteins with expected size that were partially soluble after purification using amylose resin via N-terminal MBP tag.
In elution buffer that lacks glycerol, partitivirus RdRps were stable and active for at least four weeks at 4°C (data not shown). When frozen in 50% glycerol-containing buffer at −20°C, the enzymes were stable and active for months (data not shown).
We assessed the PCV1-RdRp recombinant protein product by mass spectrometry, and the results indicated that the correct protein was expressed, processed, and purified.
RT Activity of Recombinant RdRps
To access whether the RT activity of RdRp is specific to PCV1 or common to other partitiviruses, eight additional RdRp proteins fused with N-terminal MBP were expressed in E. coli and purified.
Reverse Transcription Polymerase Chain Reaction
Using PCR-based RT assays, all nine RdRps could synthesize cDNA from the transcript of pepper cryptic virus 2 (PCV2) RdRp as a ssRNA template, with similar efficiency (fig. 3A–H). Further enzymatic assays of Atkinsonella hypoxylon virus (AhV) and PCV1 RdRps showed that the enzymes are able to use dsRNA as a template (fig. 3I). This indicated that partitivirus RdRps have RT activity, and can efficiently transcribe both homologous and heterologous ds/ss RNA templates. The presence of the viral coat protein (CP) as cofactor for the RT activity of the enzyme was not required. The RT activity and fidelity of RdRp protein of Amasya cherry disease-associated mycovirus (ACDMV) was verified by DNA sequencing of the PCR product, confirming the activity of ACDMV RdRp and accurate amplification of the PCV2 transcript. The optimal reaction temperature for this RdRp activity was in the range of room temperature (around 24°C) to 45.7°C (data not shown).
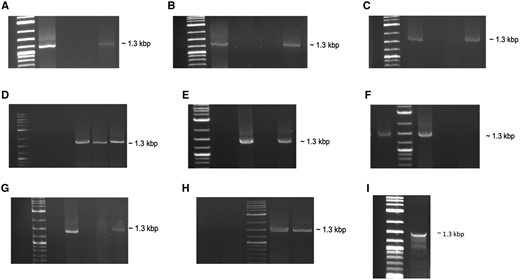
RT activity of various recombinant partitivirus RdRp enzymes. RT reactions were carried out using PCV2 transcript (ssRNA) as template. (A) AhV-RdRp as RT enzyme; M, DNA marker, 1, ProtoScript II used as enzyme, 2, negative control (no enzyme), 3, negative control (no template), 4, AhV-RdRp recombinant protein used as enzyme. (B) ACDMV-RdRp as RT enzyme; M, DNA marker, 1, ProtoScript II used as enzyme, 2, negative control (no enzyme), 3, negative control (no template), 4, ACDMV-RdRp recombinant protein used as enzyme. (C) HubeiPV53-RdRp as RT enzyme; M, DNA marker, 1, HubeiPV53-RdRp recombinant protein used as enzyme, 2, negative control (no enzyme), 3, negative control (no template), 4, ProtoScript II used as enzyme. (D) PdPV-RdRp and AfPV2 RdRp as RT enzymes; M, DNA marker, 1, negative control (no enzyme), 2, negative control (no template), 3, ProtoScript II used as enzyme, 4, PdPV-RdRp recombinant protein used as enzyme, 5, AfPV2-RdRp recombinant protein used as enzyme. (E) ClCV-RdRp as RT enzyme; M, DNA marker, 1, negative control (no enzyme), 2, ProtoScript II used as enzyme, 3, negative control (no template), 4, ClCV-RdRp recombinant protein used as enzyme. (F) PCV1-RdRp as RT enzyme; 1, PCV1-RdRp recombinant protein used as enzyme, M, DNA marker, 2, ProtoScript II used as enzyme, 3, negative control (no enzyme), 4, negative control (no template). (G) FpPV2-RdRp as RT enzyme; M, DNA marker, 1, ProtoScript II used as enzyme, 2, negative control (no enzyme), 3, negative control (no template), 4, FpPV2-RdRp recombinant protein used as enzyme. (H) PCV2-RdRp as RT enzyme; 1, negative control (no enzyme), 2, negative control (no template), M, DNA marker, 3, ProtoScript II used as enzyme, 4, PCV2-RdRp recombinant protein used as enzyme. (I) RT reaction was carried out using PCV1 dsRNA as template and AhV-RdRp as RT enzyme; M, DNA marker, 1, AhV-RdRp recombinant protein used as RT enzyme. All virus siglas are defined and GenBank accession numbers are given in supplementary table 2, Supplementary Material online.
Quantitative Reverse Transcription Polymerase Chain Reaction
To further confirm the RT activity of RdRp, RT-qPCR was used to monitor the activity and efficiency of RdRp proteins from ACDMV and Hubei partiti-like virus 53 (HubeiPV53). Fifty nanogram of human T-cell leukemia total RNA was used as RNA template and two RdRp proteins and two commercial RTs (ProtoScript II and LunaScript Super mix kit) were used to perform the RT step. Although RT activity of both RdRps is lower than commercial RTs, the ACDMV and HubeiPV53 enzymes are active with the mean Cq value of 34.2, and 32.2 for the beta-actin gene (ACTB), respectively. To exclude nonspecific products or primer dimer contamination, the end-point products of qPCR were resolved in a gel. The specific bands were clearly similar to the positive controls (fig. 4A, lane 10–15 vs. lane 1–3 and fig. 4B, lane 1–9 vs. lane 10–12).
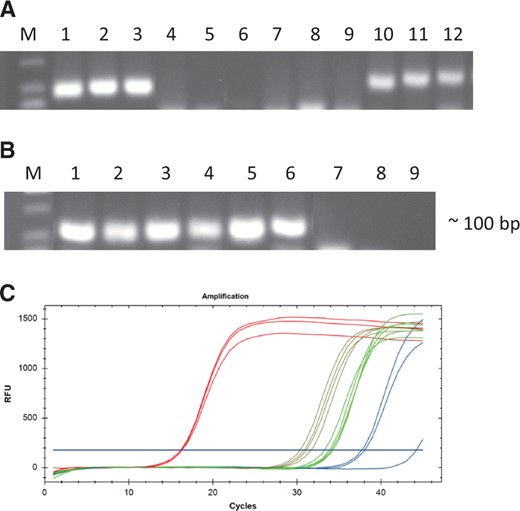
Evaluation of the RT activity of recombinant ACDMV and HubeiPV53 RdRp proteins by RT-qPCR. (A,B) RT-qPCR analysis using Human T-cell leukemia total RNA as template and ACTB primer; (A) M, DNA marker, 1–3, ProtoScript II used as enzyme in RT reaction, 4–6, negative control (no enzyme), 7–9, negative control (no template), 10–12, ACDMV-RdRp recombinant protein used as enzyme in RT reaction. (B) M, DNA marker, 1–3, HubeiPV53-RdRp recombinant protein used as enzyme in RT reaction, 4–6, cDNA synthesized by LunaScript Super mix kit, 7–9, negative control (no primer). (C) Amplification profiles of several commercial RT enzymes (red), compared to ACDMV RdRp (green) and HubeiPV53 RdRp (brown) as RT enzyme and negative controls (blue). The shift in Cq when the amplifications included ACDMV RdRp and HubeiPV53 RdRp as RT enzyme indicate a low abundance of amplifiable cDNA.
In addition, analysis of melting curves showed no nonspecific products or primer dimer synthesis. The same results were obtained with the glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH).
RT Activity of 3Dpol
As reported previously, poliovirus RdRp 3Dpol can catalyze dNTPs incorporation to extend an RNA primer (Arnold et al. 1999). We used 3Dpol (a gift of Dr Craig Cameron) as a control to test its ability to use dNTPs to extend DNA primers and function as an RT enzyme. The extension of 5′-FAM-labeled RNA and DNA primers by 3Dpol was monitored in the presence of both dNTPs and NTPs (fig. 5A). The 3Dpol was able to incorporate both NTPs and dNTPs to extend the 5′-FAM-RNA primer (fig. 5B). With dNTPs the extension of the RNA primer was not complete, and the 5′-FAM-DNA primer was not extended in the presence of 3Dpol using either NTPs or dNTPs (fig. 5C). Furthermore, nucleotide incorporation was not catalyzed by various concentrations of 3Dpol in an RT-PCR reaction using transcript of PCV2 RdRp as a template and a DNA primer (fig. 5D). Hence 3Dpol, an ssRNA RdRp, cannot fully function as an RT, although conditions to optimize this activity were not exhaustively tested.
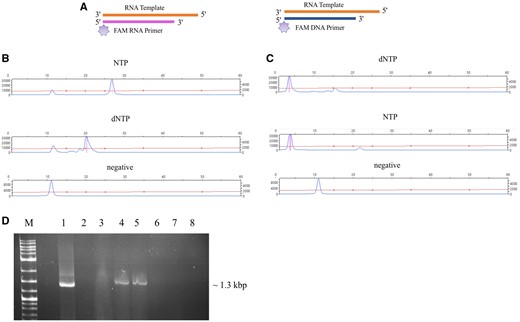
Nucleotide selection and primer extension by 3Dpol. (A) Schematic representation of FAM labeled RNA and DNA primer extension by 3Dpol. RNA template is shown in orange, RNA primer is shown in pink and DNA primer in blue. FAM labels are shown as light purple stars. (B) CE results of the extension of FAM-RNA primer; reactions contained 3Dpol, RNA template, FAM labeled RNA primer, and NTPs or dNTPs. (C) CE results of the extension of FAM-DNA primer; reactions contained 3Dpol, RNA template, FAM labeled DNA primer, and NTPs or dNTPs. (D) RT-PCR analysis using PCV2 transcript (ssRNA) as template and recombinant protein of FpPV2-RdRp and 3Dpol as RT enzymes; 1, ProtoScript II used as enzyme in RT reaction, 2, negative control (no enzyme), 3, negative control (no template), 4, FpPV2-RdRp (1 μM) recombinant protein used as enzyme in RT reaction, 5, FpPV2-RdRp (2 μM) recombinant protein used as enzyme in RT reaction, 6, 3Dpol (1 μM) recombinant protein used as enzyme in RT reaction, 7, 3Dpol (2 μM) recombinant protein used as enzyme in RT reaction, 7, 3Dpol (3 μM) recombinant protein used as enzyme in RT reaction, M, DNA marker.
DNA Phase of PCV1
To determine whether the RT activity of polymerases in this family has a role in the virus life cycle, the cytosol of Jalapeño pepper was examined for the presence of a DNA phase of PCV1. Control plasmid was amplified using specific primers by qPCR, but no amplification was observed in the cytosol fraction. Eleven-fold serial dilutions of the control plasmid were detected by qPCR (limit of detection was 4.5 target DNA molecules), but no evidence of PCV1-DNA was found. However, it is possible that the RT function of the viral polymerases are only activated as a response to stressful conditions.
Discussion
We show conclusively that partitivirus RdRp proteins have RT activities, in the intact virion, in vitro translated RdRps, and recombinant RdRps. Hence the virus-associated enzyme is both a reverse transcriptase and an RNA polymerase.
RT activity was detected in PCV1 virions, but not in material prepared by an identical extraction protocol from uninfected Jalapeño plants. Previous studies on partitiviruses showed RdRp activity associated with purified virions of several fungal partitiviruses (Buck 1978; Ratti and Buck 1978; Pan et al. 2009), confirming that the RdRps are packaged along with the CPs and RNA genomes. Whereas RT activity in vitro was low, cDNA was detectable using PCR. The RdRp proteins of partitiviruses are active after incubation times of 2–3 h or longer.
RdRps of partitiviruses were not only able to use homologous dsRNA templates but also capable of initiating DNA synthesis equally on heterologous dsRNAs and ssRNA templates in the absence of other viral or cellular proteins. We confirmed that the products of RT reactions using RdRp of partitiviruses as RT enzyme were RNase resistant by treating reactions with RNase A. The accuracy of DNA synthesis by partitivirus RdRps was verified by determining the sequence of the PCR product that was synthesized using transcript of PCV2-RdRp as template and purified ACDMV RdRp protein as the RT enzyme.
The ability of RdRps to incorporate dNTPs in the presence of an RNA primer or in a primer-independent activity has been described previously (Arnold et al. 1999; Hung et al. 2002). In this study, the ability of partitivirus RdRps to utilize dNTPs to synthesize DNA from an RNA template was compared with 3Dpol activity by a series of DNA primer extension experiments. Incorporation of dNTPs by partitivirus RdRps were more efficient than 3Dpol and could proceed to completion using a DNA primer.
The most widespread nonretroviral RNA virus sequences that have been found integrated into the genomes of eukaryotic organisms are related to the CP and RdRp genes of the families Partitiviridae and Totiviridae, which infect plants and fungi. This endogenization process required three events 1) viral RNA replication, 2) reverse transcription of viral RNA, and 3) integration of viral cDNA into host chromosomes. However, the mechanism by which viruses convert their RNA genome to DNA and introduce it to host genomes remains unknown (Liu et al. 2010; Chiba et al. 2011). Here we speculate that the RT function of partitivirus polymerases may be involved in their DNA synthesis to be introduced into plant genomes. However, no viral DNA was detected in the cytosol of infected Jalapeño pepper and the question is still open.
The analysis of dsRNA as a hallmark of RNA virus infection has gained importance in recent years with the rise of virus discovery work. However, dsRNA has been proven to be a difficult substrate for RT enzymes, as commercially available RT is a single-strand-specific enzyme. Several methods are available to convert dsRNA to ssRNA for use in RT reactions including boiling dsRNA, using chemicals, or using higher temperatures for RT reaction to keep the RNA single-stranded. The latter also dramatically reduces the fidelity of the RT enzyme, introducing mutations that can be mistaken for single nucleotide polymorphisms (SNPs) with biological significance. The partitivirus RdRps can convert dsRNA to cDNA without any manipulation, and at room temperature or less.
RNA viruses include the largest part of the global eukaryotic virome. Reconstruction of the evolutionary history of RNA viruses has gained a lot of attention in recent years due to major discoveries in this area. All RNA viruses share one universal gene with several conserved motifs that encodes RdRp, which is a natural target of evolutionary analysis. A phylogenetic tree of thousands of viral RdRps indicated the monophyly of the RdRps and revealed patterns into the evolution of different branches of RNA viruses in which dsRNA viruses evolved from (+) ssRNA viruses, while (−) ssRNA viruses are descended from a distinct group of dsRNA viruses (Wolf et al. 2018). However, in this analysis, RT was presumed to be ancestral and hence was used as a root for the tree. The discovery of RT activity in the dsRNA RdRp may require rethinking this supposition. Considering dsRNA RdRp as a root for the RNA tree may reshape our understanding of RNA virus evolution.
Materials and Methods
Virus Purification
Pepper cryptic virus 1 (PCV1) particles were purified from 500 g of plant tissue (Jalapeño pepper) by homogenization of plant leaves in a blender with 500 ml of 0.1 M sodium phosphate buffer pH 7.6 containing 0.5% thioglycolic acid, and 500 ml of chloroform. The resulting slurry was clarified by low-speed centrifugation for 15 min. The aqueous portion was filtered through Miracloth and subjected to ultracentrifugation through a 10% sucrose cushion. The pellets were resuspended in 0.03 M sodium phosphate buffer pH 7.6 containing 2% Triton X-100 and stirred at 4°C overnight. The solution was clarified by low-speed centrifugation, followed by a second ultracentrifugation as above. The final pellets were resuspended in 0.01 M sodium phosphate buffer pH 7.6 and allowed to incubate at 4°C overnight. The purified viral preparation was analyzed by 1% agarose gel in Tris-Glycine buffer. Short-term storage of purified virus was at 4°C; for long-term storage, sterile glycerol was added to a final concentration of 50%, and the preparation was stored at −80°C. The purified virus was used for evaluating the enzymatic activity of PCV1 particles.
Virus purification was also performed on an isogenic line of Jalapeño that is virus-free to provide a control to ensure that the activity was completely attributed to the virus.
RNA Templates
The RNA templates used in the RT assay are either dsRNA or ssRNA. The dsRNAs were extracted from 5 g maize or pepper plant leaves infected with Zea mays chrysovirus 1 (ZMCV1) or PCV1, respectively, using cellulose chromatography as described previously for mycoviruses (Peyambari and Roossinck 2018).
For ssRNA templates, we used the transcript of pepper cryptic virus 2 (PCV2) RdRp. Transcription templates were prepared by amplifying PCV2 RdRp by forward and reverse primers containing T7 promoter and T7 terminator, respectively, using Q5 High-Fidelity 2X Master Mix (New England Biolabs). The PCR product containing T7 promoter region upstream of the sequence was transcribed using the HiScribe T7 Quick High Yield RNA Synthesis Kit (New England Biolabs) and the Standard RNA Synthesis protocol for 2 h at 37°C. The synthesized RNA was treated by DNase I, then purified using LiCl precipitation.
The Human T-cell (Jurkat) leukemia total RNA that was used in RT-qPCR reaction was purchased from Ambion, Thermo Fisher Scientific (USA).
RT Activity of PCV1 Virion
To analyze the RT activity of PCV1 RdRp, dsRNA templates were incubated with purified virus particles. The RT reaction, as previously described by Roossinck et al. (2010), contained 5 μl (∼2 μg) of template (ZMCV1 and/or PCV1 dsRNA, or PCV1 virion), 0.5 mM Tris-EDTA (pH 8.0), 2 μM primer (supplementary table 1, Supplementary Material online), and nuclease-free water up to a final volume of 12 μl. In the case of dsRNA and PCV1 virion as templates, the RT reaction was boiled for 2 min and followed by rapid cooling on ice to denature dsRNA and maintain it as ssRNA. Then 8 μl of RT mix containing 4 μl 10× MMuLV buffer (supplied by the manufacturer), 1 μl dNTPs (10 mM each), 2 μl dithiothreitol (DTT) was added to the reaction. In the control reaction, 1 μl MMuLV (New England Biolabs) and in PCV1 reaction, 2–3 μl of purified particles were added to the RT mixture. The MMuLV reaction was transferred to 42°C for 2 h, while the PCV1 reaction was held at room temperature for the same amount of time. After 2 h incubation, samples were treated with 1 μl (10 mg/ml) of boiled ribonuclease A (Sigma, USA), and incubated at room temperature for 15 min. The samples then were heated to 85°C for 2 min, and the primers were removed using a cycle pure kit (Omega, Bio-Tek, USA) according to the manufacturer’s instructions. Following the cDNA reaction, the cleaned cDNA was amplified by PCR. The PCR reaction was contained Taq DNA polymerase (ThermoFisher Scientific), 10× PCR buffer (30 mM MgCl2, Idaho Technologies), 0.2 mM dNTPs, and 2 μM forward and reverse primers (supplementary table 1, Supplementary Material online). The PCR reactions were completed in capillary tubes with an Idaho Technologies rapid cycler for 35 cycles (94°C denaturation for 0 s, 48°C annealing for 0 s, and 72°C extension for 45 s). The RT-PCR products were separated on a 1.2% agarose gel.
Plasmid Constructs
The RdRp genes of partitiviruses in this study were codon optimized to optimal expression in E. coli (Genscript, Piscataway, NJ, USA). The codon-optimized synthetic RdRp genes of eight partitiviruses were inserted by Genscript into pMAL-c5E (supplementary table 2, Supplementary Material online).
The codon-optimized PCV1 RdRp was inserted by Genscript into pUC57. Ligation of PCV1-RdRp into the modified pET28b+ expression plasmid (pSL0220) was mediated by NcoI and XhoI restriction sites at the 5′ and 3′ ends, respectively. Plasmid pUC57-PCV1-RdRp was digested by NcoI and XhoI in the appropriate buffer. After digestion, DNA was purified and cloned into expression plasmid pSL0220 with N-terminal glutathione-S-transferase (GST)-tag and C-terminal 6xHis-tag, which was generously provided by Dr Scott Lindner, using T4 DNA ligase (New England Biolabs) according to the manufacturer’s instruction. The final construct (pSL0220-PCV1-RdRp) was confirmed by sequencing at the Pennsylvania State University Core Facility.
The PCV1 RdRp was inserted into plasmid pET28 with N-terminal maltose-binding protein (MBP)-tag and C-terminal 6×His-tag using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs). The plasmid and RdRp gene were amplified with primers containing overlapping regions (supplementary table 1, Supplementary Material online) using Q5 master mix (New England Biolabs), according to the manufacturer’s instruction, to make linearized amplicons with sequences complementary to each other. The construct was confirmed by DNA sequencing.
In Vitro Translation Assay of PCV1 RdRp
The pSL0220-PCV1-RdRp construct with N-terminal GST-tag was used as DNA template. The T7 promotor, the ribosome‐binding site, and the T7 terminator, all the elements required for protein production, are included in the construct. The DNA template was mixed with the PURExpress® in vitro Protein Synthesis kit containing E. coli ribosomes (New England Biolabs), according to the manufacturer's instructions. Reaction of a final volume of 25 μl was mixed as recommended, including 150 ng of template DNA and 20 units RNase inhibitor (Sigma). The reaction was incubated at 37°C for 2 h and subsequently analyzed by SDS-PAGE.
RT Activity of In Vitro Translated PCV1 RdRp
The ability of in vitro translated PCV1 RdRp to synthesize cDNA from RNA template was analyzed in a 20 μl RT reaction consisting of 5 μl (∼2 μg) of PCV1 dsRNA as a template. The RT mixtures were prepared as described above including boiling dsRNA templates to denature, and 2-3 μl of RdRp protein was added to mixtures. MMuLV-RT was used in positive control reaction, and two negative control reactions were prepared without RT enzyme and without primer. The MMuLV reaction was incubated at 42°C for 2 h, while the PCV1 reaction was held at room temperature for the same amount of time. The reactions were treated with boiled RNase A (Sigma, USA), and synthesized cDNAs were cleaned and amplified in a PCR reaction as above. The RT-PCR products were separated on a 1.2% agarose gel.
Recombinant Protein Expression
Recombinant RdRp proteins of PCV1 and eight partitiviruses were expressed as fusion proteins with an N-terminal MBP-tag using the pET28 and pMAL-c5E vectors, respectively. Expression of recombinant fusion proteins was performed in the Shuffle B strain of E. coli cells (New England Biolabs). The cells were grown at 30°C to OD600 of 0.5–1 by shaking at 250 rpm. Then the cells were induced by the addition of isopropyl β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.4 mM and allowed to grow at 16°C overnight. The cells were harvested by centrifugation and stored at −80°C until used. The frozen cells were thawed and lysed by cell disruption in buffer A containing 20 mM Tris-HCl, pH 7.5, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, and 0.1% Triton X-100. The disrupted cells were centrifuged, and the supernatants were used to purify the fusion proteins by AKTA-FPLC (GE Healthcare) via affinity chromatography using an amylose resin (New England Biolabs) column equilibrated with buffer A. The columns were washed for an additional 20 column volumes with buffer A. The fusion RdRp proteins were eluted from the column with elution buffer (buffer A + 10 mM maltose). All extraction and purification procedures were performed at 4°C unless otherwise stated. Successful purification of the proteins was validated by SDS-PAGE. To establish the identity of the recombinant protein, the MBP-PCV1-RdRp was sent for mass spectrophotometry.
RT Activity of RdRp Recombinant Proteins
Reverse Transcription Polymerase Chain Reaction
To analyze the RT activity of RdRp recombinant proteins of partitiviruses, cDNA synthesis was performed using 100 ng RNA (transcript of PCV2 RdRp), 1 mM dNTPs, 1 μM reverse primer (PCV2-RdRp-T7-R, supplementary table 1, Supplementary Material online), 1x ProtoScript II Reaction buffer (New England Biolabs), and 2 μM of recombinant proteins (as RT enzyme) in 10 μl total reaction volume. In the positive control reaction, 0.5 μl of ProtoScript II was added to the mixture as reverse transcriptase. The reactions without a template or RT enzyme are considered as two negative controls. The reactions with recombinant proteins and ProtoScript II were incubated for 3 h at 27°C and 42°C, respectively. After cDNA synthesis, 1 μl (10 mg/ml) of boiled ribonuclease A (Sigma, USA) was added to the reactions and incubated for 15 min at room temperature. The samples then were heated to 85°C for 2 min, and single-stranded cDNA was purified using a Monarch PCR & DNA clean-up kit (New England Biolabs) according to the manufacturer’s protocol. The cleaned cDNA was amplified by PCR using Q5 High-Fidelity 2X Master Mix (New England Biolabs) according to the manufacturer’s instructions. The activity and fidelity of RdRp protein (ACDMV RdRp) as an RT enzyme was confirmed by direct DNA sequencing of full-length PCR product of enzyme reaction.
Quantitative Reverse Transcription Polymerase Chain Reaction
Since RT-qPCR is a more sensitive detection method, two housekeeping genes, ACTB and GAPDH, were selected to evaluate the RT activity of RdRp recombinant proteins of ACDMV and HubeiPV53 by doing RT-qPCR using 50 ng human T-cell leukemia total RNA. The cDNA syntheses were performed as described above. Three negative controls, no template, no primer, and no RT enzyme, and two positive controls, ProtoScript II and LunaScript Super mix kit (New England Biolabs), were assayed for each primer. The qPCR was performed using Luna Universal qPCR Master Mix (New England Biolabs) and a CFX96 real-time PCR system (Bio-Rad, USA), and analyzed using CFX Manager software. Each reaction mixture contained 1 μl of cDNA, 0.25 μM of each gene-specific forward and reverse primer (supplementary table 1, Supplementary Material online), 10 μl of 2× Luna Universal qPCR Master Mix, and nuclease-free water in a total volume of 20 μl. The thermal cycling conditions were as follows: 60 s at 95°C, followed by 44 cycles of 15 s at 95°C and 30 s at 60°C, and a final melt-curve step at 60-95°C to identify primer dimers and nonspecific amplification. At least three technical replicates were performed for each biological sample. The products were confirmed by running on a 2.5% agarose gel followed by SYBER gold staining.
RT Activity of 3Dpol RdRp
To investigate the RT activity of other RdRps, the ability of RdRp of Poliovirus (3Dpol) to use dNTPs versus NTPs was monitored by extension of a fluorescein amidite (FAM) labeled RNA or DNA primers as follows. 3Dpol (1 μM) was mixed with 1 μM 5′-FAM-RNA primer (supplementary table 1, Supplementary Material online), 2 μM synthetic RNA substrate (supplementary table 1, Supplementary Material online), 10 mM dNTPs/25 mM NTPs, RdRp buffer (50 mM HEPES, 5 mM MgCl2, 60 μM ZnCl2, and 10 mM 2-mercaptoethanol) (Arnold and Cameron 2000). For DNA primer, 3Dpol (1 μM) was mixed with 1 μM 5′-FAM-DNA primer (supplementary table 1, Supplementary Material online), 2 μM synthetic RNA substrate (supplementary table 1, Supplementary Material online), 10 mM dNTPs/25 mM NTPs, and 1x ProtoScript II Reaction buffer (New England Biolabs). After incubation at 30°C for 1 h, reactions were terminated by heating at 70°C for 5 min. As a control, a reaction was incubated without 3Dpol. Reactions were analyzed by Capillary gel electrophoresis (CE), and the results were analyzed using Peak Scanner software version 1.0 (Applied Biosystems).
In addition, the different concentrations (1, 2, and 3 μM) of 3Dpol were added to mixtures with 100 ng RNA (Transcript of PCV2 RdRp), 1 mM dNTPs, 1 μM reverse primer (PCV2-RdRp-T7-R, supplementary table 1, Supplementary Material online), and 1× ProtoScript II Reaction buffer (New England Biolabs) in 10 μl total reaction volume to study the ability of enzyme to make cDNA. As negative controls, the reactions were incubated without a template or enzyme. And a mixture with ProtoScript II was considered as positive control. Also, two different concentrations (1 and 2 μM) of RdRp recombinant protein of FpPV2 were used in RT mixtures for the comparison of RT activity. The reactions with FpPV2-RdRp/3Dpol and ProtoScript II were incubated for 3 h at 27°C and 42°C, respectively. After cDNA cleaning, PCR was done as described above for RdRp recombinant proteins.
DNA Stage of PCV1
The presence of a DNA stage of PCV1 in Jalapeño pepper cytosol was investigated using qPCR. Cytosol was prepared from 2 g of pepper leaf tissue, using liquid nitrogen to freeze the cells followed by mortar and pestle disruption. The broken cells were resuspended in 6 ml of 100 mM Tris-HCl pH 7.5 and kept on ice for 20 min. The resuspended cells were filtered through two layers of Miracloth presoaked with 100 mM Tris-HCl pH 7.5. Cell fractionation was done using differential centrifugation. The broken cells were centrifuged 2 times at 10,000 g for 10 min to pellet unbroken cells and nuclei, and then the supernatant was transferred to a new tube and centrifuged at 20,000 g for 25 min to remove organelles and membrane. The supernatant was the cytosol component that is free of contaminating nuclear material. DNA was extracted using phenol-chloroform followed by ethanol precipitation.
Amplifications were performed using a Roche LightCycler 480 Instrument II (Roche, Mannheim, Germany). The primer sequences are given in supplementary table 1, Supplementary Material online. Serial dilutions (from 10 to 10−16) of a plasmid DNA (pGEMT-9-2) were used to determine the detection limit of the qPCR assay. The plasmid DNA concentration was determined with NanoDrop. Each reaction mixture was prepared in a 20 μl final volume including 10 μl of 2× Luna Universal qPCR Master Mix (New England Biolabs), 0.25 μM of each specific plasmid and PCV1-RdRp forward and reverse primers, and 1 μl of template DNA (cytosol component, or plasmid standard). The qPCR was carried out with an initial denaturation step at 95°C for 60 s followed by 44 cycles of denaturation at 95°C for 15 s and annealing/extension at 60°C for 30 s, and a final melt-curve step at 60–95°C. All samples were tested in triplicate and a “no template DNA” reaction was used as negative control. Cp data were processed and analyzed using LightCycler 480 software (Roche, Mannheim, Germany).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Acknowledgments
This work was supported by the Pennsylvania State University College of Agricultural Science, and the Huck Institutes of Life Sciences, and by New England Biolabs. We express our gratitude to Drs. Craig Cameron and Jamie Arnold for helpful discussions, and for providing 3Dpol, Dr Scott Lindner, Dr Juan José López-Moya, Jacob Perryman, and Allen Minns for their assistance in designing constructs of PCV1 RdRp and expressing the protein for preliminary observation of solubility. We thank New England Biolabs colleagues Dr Minyong Chen, Dr Saulius Vainauskas, Dr Jennifer Ong, and Dr Thomas Evans for helpful discussions and critical feedback.
Author Contributions
M.P. conducted the analyses and wrote the first draft of the manuscript; S.G. and M.R. supervised the project, M.R. revised and wrote the final draft of the manuscript.
Data Availability
All data is available in the main text or the Supplementary Material online.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}