





Abstract
Nucleotide substitutions, insertions, and deletions constitute the principal molecular mechanisms generating genetic variation on small length scales. In contrast to substitutions, the nature of short DNA insertions and deletions (indels) is far less understood. With the recent availability of whole-genome multiple alignments between human and other primates, detailed investigations on indel characteristics and origin have come within reach. Here, we show that the majority of short (1–100 bp) DNA insertions in the human lineage are tandem duplications of directly adjacent sequence segments with conserved polarity. Indels in microsatellites comprise only a small fraction. The underlying molecular processes generating indels do not necessarily rely on the presence of preexisting duplicates, as would be expected for unequal crossing over, as well as replication slippage. Instead, our findings point toward a mechanism that preferentially occurs in the male germline and is not recombination-mediated. Surprisingly, nonframeshifting tandem duplications and deletions in coding regions still occur at approximately 50% of their genomic background rates. As is already well established in the context of gene and segmental duplications, our results demonstrate that duplications are also likely to constitute the predominant process for rapid generation of new genetic material and function on smaller scales.
Introduction
The identification and precise characterization of the fundamental molecular processes that induce genomic variation will shed light on evolution's key mechanisms underlying the emergence of genetic innovation and adaptive evolution. In this context, DNA insertions and deletions are likely to play a pivotal role. Ubiquitous throughout evolution they occur on all scales ranging from single nucleotides up to whole-genome duplications (International Human Genome Sequencing Consortium 2001; Britten et al. 2003; Kent et al. 2003; Thomas et al. 2003; Zhang and Gerstein 2003; Cheng et al. 2005; Chimpanzee Sequencing and Analysis Consortium 2005; Gregory 2005; Bailey and Eichler 2006; Chen et al. 2007; Redon et al. 2006).
DNA insertions of larger segments in eukaryotic genomes, irrespective of the various molecular causes, typically involve duplications of parts of the genome. Examples include insertions of transposable elements, gene duplications, or large-scale segmental duplications. From a mechanistic point of view, the ubiquity of duplications reflects intrinsic features of the prevalent molecular processes generating insertions of DNA segments, such as replication slippage, retrotransposition, or unequal crossing over (UCO).
An evolutionary approach focuses on a possibly beneficial role of duplication events. For example, following the duplication of a selectively constrained gene, one copy is allowed to evolve freely and can possibly acquire new functions, whereas the remaining copy will continue to perform the original task. Initially established by Ohno in his seminal work on proteome evolution by gene duplication (Ohno et al. 1968), the concept of duplication-driven evolution has nowadays been extended from genes to also larger segmental duplications (Bailey and Eichler 2006). Consequently, this generalization to longer chromosomal segments raises the question whether in a similar fashion duplications might also play an important evolutionary role on smaller length scales ranging down to single nucleotides.
Although such small DNA insertions comprise by far the largest number of all insertion events, for example, throughout recent human evolution (Britten et al. 2003; Kent et al. 2003; Thomas et al. 2003; Chimpanzee Sequencing and Analysis Consortium 2005), profound knowledge about their characteristics, underlying molecular processes, and evolutionary role is sparse. Yet, it is commonly believed that short indels are primarily generated by replication slippage or UCO (Levinson and Gutman 1987), and both processes indeed generate tandem duplication insertions. Additional indication of a duplication mechanism on small length scales is provided by the overrepresentation of short paired duplicates in mammalian genomes (Achaz et al. 2001; Thomas et al. 2004).
Here, we present a genome-wide analysis of short (1–100 bp) indels in the human genome since its split from the common ancestor with chimpanzee. Insertions are explicitly distinguished from deletions by comparison with an out-group species. We use rhesus for that purpose, which allows us to retain a good coverage of the human genome. We show that tandem duplication is indeed the predominant mechanism for the generation of recent short DNA insertions in the human lineage. However, many indels do not show replication slippage or UCO-compatible characteristics. We instead propose that a large fraction of indels may result from imperfect repair of double-strand breaks by nonhomologous end joining (NHEJ).
In protein-coding regions, we find nonframeshifting indels to be less deleterious compared to nonsynonymous substitutions. Together with the fact that the majority of coding insertions are tandem duplications, this hints toward a possibly important evolutionary role of duplications also among small insertions. Our results therefore suggest that the concept of duplication-driven evolution is likely to span the entire range of genomic length scales.
Methods
Identifying Insertions and Deletions
Our comparative genomics analysis utilizes the recently available University of California-Santa Cruz (UCSC) whole-genome multiple alignments of 16 vertebrates including human. From these multiple alignments we extracted the human (hg18, Mar 2006), chimp (panTro1, Nov 2003), and rhesus (rheMac2, Jan 2006) tracks. The resulting 3 species alignments cover 85% of the human genome and feature gap lengths of up to 100 bp in each species. If not a result of erroneous alignment, these gaps correspond to insertion or deletion events along branches of the phylogenetic tree ((human, chimp), rhesus).
The ranking of different gap motifs with more than 105 hits is shown in table 1. Motifs 3–6 represent elementary events that can unambiguously be explained by a single insertion or deletion event in one branch of the phylogenetic tree (Sinha and Siggia 2005; Chen et al. 2007). In particular, motifs 3 and 4 are insertions and deletions in chimp, 5 and 6 are deletions and insertions in human. Motifs 1 and 2 can also be explained by a single indel event, but due to the unknown status of the root we cannot distinguish whether the event occurred in the rhesus lineage, or on the branch from the root to the common ancestor of human and chimp. Overall, motifs 1–6 comprise 93.75% of the number of all events and account for 84.75% of base pairs comprised in gap motifs. In contrast, gap motifs with not exactly overlapping gaps require at least 2 indel events for their explanation. For instance, motif 7 can represent 2 deletion events, 1 in human and the other in chimp. However, the motif can also be explained by 2 insertion events in chimp and rhesus. For our analysis, we focused on unambiguous insertions and deletions in the human branch after its split from the common ancestor with chimp (motifs 6 and 5, respectively, in table 1). If a deletion occurred in human, the corresponding chimp sequence was taken as an approximation of the ancestral sequence.
Ranking of Gap Motifs
| Motif | Hits | Kilobase Pairs | |
| 1 | ––– | 6,124,068 | 27,948 |
| ––– | |||
| ––– | |||
| 2 | ––– | 5,457,464 | 32,481 |
| ––– | |||
| ––– | |||
| 3 | ––– | 1,171,399 | 4,915 |
| ––– | |||
| ––– | |||
| 4 | ––– | 1,129,247 | 3,935 |
| ––– | |||
| ––– | |||
| 5 | ––– | 749,968 | 2,416 |
| ––– | |||
| ––– | |||
| 6 | ––– | 450,737 | 1,773 |
| ––– | |||
| ––– | |||
| 7 | –—– | 116,221 | 1,099 |
| ––— | |||
| –—– | |||
| 8 | ––— | 114,681 | 1,122 |
| –—– | |||
| ––— | |||
| 9 | —–– | 113,403 | 1,183 |
| ––— | |||
| –—– | |||
| 10 | ––— | 105,025 | 1,120 |
| —–– | |||
| –—– | |||
| Others | 555,805 | 8,664 |
| Motif | Hits | Kilobase Pairs | |
| 1 | ––– | 6,124,068 | 27,948 |
| ––– | |||
| ––– | |||
| 2 | ––– | 5,457,464 | 32,481 |
| ––– | |||
| ––– | |||
| 3 | ––– | 1,171,399 | 4,915 |
| ––– | |||
| ––– | |||
| 4 | ––– | 1,129,247 | 3,935 |
| ––– | |||
| ––– | |||
| 5 | ––– | 749,968 | 2,416 |
| ––– | |||
| ––– | |||
| 6 | ––– | 450,737 | 1,773 |
| ––– | |||
| ––– | |||
| 7 | –—– | 116,221 | 1,099 |
| ––— | |||
| –—– | |||
| 8 | ––— | 114,681 | 1,122 |
| –—– | |||
| ––— | |||
| 9 | —–– | 113,403 | 1,183 |
| ––— | |||
| –—– | |||
| 10 | ––— | 105,025 | 1,120 |
| —–– | |||
| –—– | |||
| Others | 555,805 | 8,664 |
NOTE.—The 3 rows of each gap motif correspond to the 3 species tracks and indicate presence (bars) and absence (empty spaces) of sequence segments of variable length in the 3 species multiple alignments. Species order is human (top row), chimp (middle row), and rhesus (bottom row). The overall number of base pairs for each motif was calculated by summing the distances between the 5′ and 3′ ungapped flanks for all gap-containing regions in our multiple alignments, which feature the given gap motif.
Ranking of Gap Motifs
| Motif | Hits | Kilobase Pairs | |
| 1 | ––– | 6,124,068 | 27,948 |
| ––– | |||
| ––– | |||
| 2 | ––– | 5,457,464 | 32,481 |
| ––– | |||
| ––– | |||
| 3 | ––– | 1,171,399 | 4,915 |
| ––– | |||
| ––– | |||
| 4 | ––– | 1,129,247 | 3,935 |
| ––– | |||
| ––– | |||
| 5 | ––– | 749,968 | 2,416 |
| ––– | |||
| ––– | |||
| 6 | ––– | 450,737 | 1,773 |
| ––– | |||
| ––– | |||
| 7 | –—– | 116,221 | 1,099 |
| ––— | |||
| –—– | |||
| 8 | ––— | 114,681 | 1,122 |
| –—– | |||
| ––— | |||
| 9 | —–– | 113,403 | 1,183 |
| ––— | |||
| –—– | |||
| 10 | ––— | 105,025 | 1,120 |
| —–– | |||
| –—– | |||
| Others | 555,805 | 8,664 |
| Motif | Hits | Kilobase Pairs | |
| 1 | ––– | 6,124,068 | 27,948 |
| ––– | |||
| ––– | |||
| 2 | ––– | 5,457,464 | 32,481 |
| ––– | |||
| ––– | |||
| 3 | ––– | 1,171,399 | 4,915 |
| ––– | |||
| ––– | |||
| 4 | ––– | 1,129,247 | 3,935 |
| ––– | |||
| ––– | |||
| 5 | ––– | 749,968 | 2,416 |
| ––– | |||
| ––– | |||
| 6 | ––– | 450,737 | 1,773 |
| ––– | |||
| ––– | |||
| 7 | –—– | 116,221 | 1,099 |
| ––— | |||
| –—– | |||
| 8 | ––— | 114,681 | 1,122 |
| –—– | |||
| ––— | |||
| 9 | —–– | 113,403 | 1,183 |
| ––— | |||
| –—– | |||
| 10 | ––— | 105,025 | 1,120 |
| —–– | |||
| –—– | |||
| Others | 555,805 | 8,664 |
NOTE.—The 3 rows of each gap motif correspond to the 3 species tracks and indicate presence (bars) and absence (empty spaces) of sequence segments of variable length in the 3 species multiple alignments. Species order is human (top row), chimp (middle row), and rhesus (bottom row). The overall number of base pairs for each motif was calculated by summing the distances between the 5′ and 3′ ungapped flanks for all gap-containing regions in our multiple alignments, which feature the given gap motif.
To further increase the quality of our set, we performed a second filtering step excluding those indels from our analysis, which have more than one mismatch or gap in the 3 species alignments of their 10 bp upstream or downstream flanks. This second step additionally filtered out approximately 50% of events along the human branch for the sake of the resulting set now being highly unlikely to result from alignment errors.
Quality Assessment and Indel Annotation
For each contig in the human assembly (Ensembl version 38, Apr 2006) “Base Quality tracks,” if available, were retrieved from GenBank using the asn2fsa tool of the National Center for Biotechnology Information (NCBI) toolbox. Indels were annotated as simple sequence repeat (SSR) if at least half of their sequence was identified as SSR by the DUST module of Blast (Altschul et al. 1997). We classified indels as coding if they fall into (or have overlap with) a protein-coding region of an exon according to the NCBI36 annotation of the human genome.
Indel Trace Extension
In our analysis, we want to judge whether an insertion of a sequence segment is in fact a duplication of an adjacent sequence segment, rather than just a random piece of DNA. If so, we would further like to know whether duplicates were already present at the insertion site before the duplication event occurred. Likewise, we want to detect deletion events which resulted from removing 1 of the 2 copies of a preexisting duplicate. Measuring the trace extension of an indel allows us to address these questions in a quantitative way and will be described in the following.
The trace extension of an indel is a quantity derived from the alignment dot-matrix in the vicinity of the indel event. In our case, dot-matrices are constructed from the homologous sequence segments of human and chimp, which we extracted from the 3 species multiple alignments. A pairwise alignment of 2 sequences corresponds to a specific path in the dot-matrix of the 2 sequences. An exemplary dot-matrix is shown in the top part of figure 1. The given alignment between the 2 sequences (solid line connecting points 1, 2, 3, 3′, 4′, and 5′) describes a situation where either a sequence segment was inserted in species 1 or deleted in species 2. If by comparison with the out-group species (rhesus), the indel is identified as an insertion in human, species 1 is assigned to human and species 2 to chimp, and vice versa in case of a deletion. The inserted or deleted sequence is identified as the corresponding segment in species 1 between points 3 and 3′. However, for the given scenario the 2 paths (1, 2, 2′, 3′, 4′, 5′) and (1, 2, 3, 4, 4′, 5′) describe alternative alignments that do not involve a different number of gaps and mismatches and will therefore get assigned equal alignment scores. Hence, it is not possible to unambiguously identify the precise sequence of the inserted or deleted segment. This type of ambiguity is generic among indel events involving tandem duplicates. Which specific alignment is actually chosen by the alignment algorithm depends on the particular implementation. The length l of an indel can be defined as the horizontal distance between the starting and end points 3 and 3′ of the indel in the original pairwise alignment. It is independent of the particular alignment chosen.
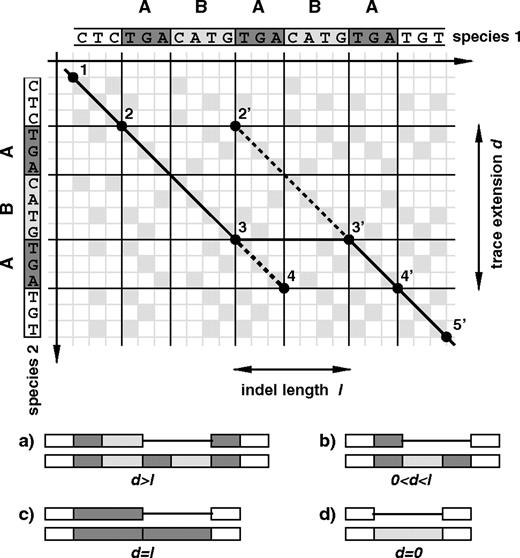
Identifying indel duplication signatures by the trace extension method. In the top part of the figure the dot-matrix of an exemplary indel event is shown. Different relations between the indel length l and its trace extension d corresponds to different classes of events. The 4 major classes are sketched in the bottom part of the plot (see Methods).
To define the trace extension of an indel, we locate 2 specific points (4 and 2′) in the dot-matrix according to the following procedure: From the starting point 3 of the indel in the original pairwise alignment, we measure how far we can extend an alternative “forward” alignment path (dashed line) that has to obey positive score along the entire path and is not allowed to rejoin the original alignment path (solid line). We use a gapped Smith–Waterman alignment algorithm with a scoring function that assigns a score of 1 to matching nucleotides, whereas mismatches and gaps are penalized equally with a score of −9. This results in positive alignment scores between the 2 aligned sequences if their similarity according to edit distance (number of gaps + mismatches) is larger than 90%. As mentioned in the text, in one particular analysis we only require 80% sequence similarity, which is achieved by a higher match score of 2. Point 4 is then defined as the point of maximal score of such an alternative forward alignment. Likewise, 2′ is derived by an analogous “backward” alignment starting at 3′. We now define the trace extension d as the vertical distance between points 4 and 2′. Alike the indel length l, the trace extension d is also independent of the particular original alignment path chosen if there are ambiguous paths. Our software used to calculate indel trace extensions is implemented in C++ and is available upon request from the corresponding author.
Calculating the trace extension d of an insertion or deletion event allows to identify its duplication characteristics because different relations between d and l correspond to different classes of events. The 4 major classes are sketched in the bottom part of figure 1: a) The case d > l indicates tandem duplication insertions of the form ABA → ABABA, or deletion events ABABA → ABA, and corresponds to the sketched scenario in the upper dot-matrix of figure 1. The length of the preexisting duplicate A is d − l. b) Partially duplicated insertions A → ABA, or deletion events of the form ABA → A, have 0 < d < l. In this case, the length of A is specified by d. c) Events with d = l are tandem duplication insertions A → AA lacking preexisting duplicates or complete deletions of one copy of a preexisting tandem duplicate AA → A. d) Nonduplication insertions, or deletions which did not result from taking out one copy of a preexisting duplicate, have d = 0. Notice that insertions with d ≥ l are always tandem duplications, irrespective of the length of a possibly present preexisting duplicate. This corresponds to cases a) and c). In contrast, b) is not considered as a tandem duplication since the fragment B was not present in the ancestral sequence.
Results and Discussion
Insertion and Deletion Statistics
We utilized human–chimp–rhesus multiple alignments to identify indels in the human branch since its split from the common ancestor with chimp. For our purposes, the considered data set represent the most suitable set available at the moment; major advantages are small divergence among all 3 species (pairwise similarities with respect to single-nucleotide mutations are 98.8% between human and chimp, 93.5% between human and rhesus, and 93.3% between chimp and rhesus), good sequence quality, and high coverage (85% of the human genome). Using rhesus as out-group, we can explicitly partition indels into insertions and deletions in the human branch by means of maximum parsimony (Sinha and Siggia 2005; Chen et al. 2007). Because we aim at the identification of reliable characteristics of insertions and deletions, we performed rigorous filtering keeping only unambiguous events located in high-quality alignments.
Our resulting data set contains 225,744 insertions and 429,048 deletions of high quality in the human branch. A table containing chromosomal position, length, and inserted/deleted sequence of all identified insertions and deletions is provided online (http://evogen.molgen.mpg.de/data/indels.txt.gz). Insertions overall comprise 0.76 Mbp of sequence, deletions 1.36 Mbp. Their length distribution is shown in figure 2. In the 2,384 Mbp of human genomic sequence covered by our multiple alignments this accounts on average for 1 inserted base per 3.1 kbp, and 1 deleted base per 1.8 kbp. However, these rates should be regarded as conservative lower bounds of actual insertion and deletion rates in the human genome. Rather than to derive true indel rates, our study is designed to investigate detailed characteristics and possible origin of inserted and deleted segments. For that purpose, we require a reliable set of high-quality indels and applied a strictly conservative filtering procedure. Our numbers are therefore much smaller compared with previous studies. For instance, in 2 Mbp of pairwise human–chimp alignments Britten et al. (2003) have measured a cumulative total of 20 kbp located in gaps of length 1–100 bp. Assuming all of these gaps to reflect indel events (which is likely to overestimate the number of actual events due to the known low quality of the chimp sequence) and equal indel rates in the human and chimp lineage, this would indicate an approximately 5 times higher actual rate of inserted or deleted bases resulting from 1–100 bp long indel events, compared with our lower bounds.
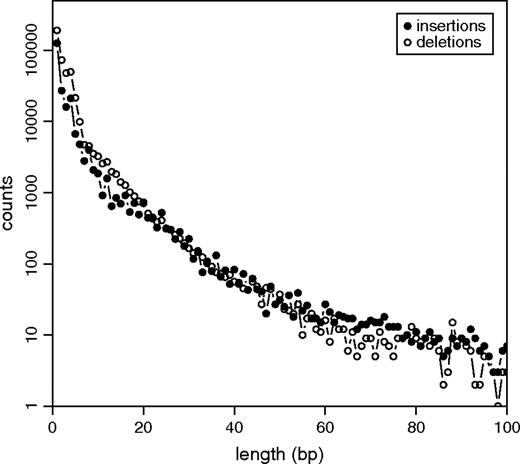
Length distribution of the identified 225,744 insertions and 429,048 deletions in our data set. Short indels comprise by far the largest number of all indels. Single-nucleotide insertions (deletions) already account for 56% (44%) of all insertions (deletions) on the investigated scales. Note that these numbers are only lower bounds on true indel numbers due to our conservative filtering for high-quality events.
We tested whether our set of identified insertions and deletions might be likely to have originated from sequencing or assembly errors by analyzing sequence quality values, which could be obtained for 420 Mbp, that is, more than 10% of the human genomic sequence. About 325 Mbp (77%) of these bases are of high quality with quality values of 90 or more. 95 Mbp (23%) are of low quality. For 96 kbp of inserted sequence segments, base-quality information is available. In this set, 74 kbp (77%) have high-quality values of 90 or more. Similarly, we tested the 2 bases 5′ and 3′ to deleted sequence segments. In total there are 106 kbp of flanking bases with base-quality information, 83 kbp (78%) of which have quality values of 90 and more. Sequence quality in inserted sequence segments and around deleted segments hence reflects that of the genomic background, disproving that indels are preferentially identified in low quality sequence regions.
In addition, assuming that a considerable amount of indels in our set (which overall comprises more than 2 Mbp, i.e., 0.06% of the entire genome) reflects sequencing or assembly errors would also imply much lower sequence accuracy than the claimed 99.99% of the human genome sequence (Schmutz et al. 2004). Sequencing or assembly errors in chimp and rhesus are much less likely to give rise to wrongly identified insertions or deletions because this would require equal errors in both species' sequences. We conclude that sequencing errors are unlikely to play a major role in our analysis.
Indels in Microsatellites
Elongation and contraction of microsatellites—tracts of short SSRs—pose an established mechanism for the generation of short indels (Toth et al. 2000). Microsatellites comprise about 3% of the human genome and show a high degree of copy number variation between species and polymorphism within the human population (International Human Genome Sequencing Consortium 2001).
We find SSR insertions to account for 15% (112 kbp) of the number of all insertions and 5% (70 kbp) of all deletions in our data set (supplementary fig. 1, Supplementary Material online). Although SSR indels in microsatellites occur at a higher rate compared with non-SSR indels in the genomic background, they make up only a small fraction of all indels in our data set.
The prevalence of SSR insertions over deletions strongly supports the hypothesis of an overall microsatellite expansion in the human lineage (Amos et al. 1996; Webster et al. 2002).
Tandem Duplications and Molecular Mechanisms
In the following, we focus on the characteristics and possible origin of non-SSR–related indels, which constitute 85% of all insertions and 95% of all deletions. Two mechanisms are commonly regarded as the primary processes capable of inserting and deleting short DNA segments, replication slippage and UCO (they are also assumed responsible for SSR length variation). Both processes feature distinct intrinsic characteristics because they require the original presence of 2 close copies of a DNA segment, UCO for the ectopic recombination between the 2 copies, replication slippage for the slipped strand misalignment (Levinson and Gutman 1987). Signatures of UCO and replication slippage on sequence level are both of the form ABA → ABABA (insertions) and ABA → A (deletions), where A's denote copies of a DNA segment, which might be separated by a spacer segment B. Consequently, insertions resulting from either of the 2 processes are tandem duplications of juxtapositional sequence, and deletions result in loss of the spacer B and one copy of the preexisting duplicate A.
Events with these signatures show specific local particularities in the dot-matrices of the aligned sequences, which can be analyzed by measurement of the trace extension (see Methods). This method provides a powerful tool to identify tandem duplication insertions and can also determine the length of preexisting duplicates.
To test whether generation of non-SSR indels is compatible with UCO or replication slippage, we computed the trace extension d for all indels in our data set. As an example, the distribution of d for indels of length l = 8 bp is shown in figure 3 (additional plots for different indel lengths are shown in supplementary fig. 2, Supplementary Material online). The trace extension analysis revealed that indeed 84% of all non-SSR insertions are tandem duplications, indicated by d ≥ l. As shown in figure 4, the proportion of duplications is generally higher for short insertions compared with longer insertions. For instance, 91% of all single-nucleotide insertions are tandem duplications in contrast to only 42% of all 30 bp long insertions. We further checked whether the remaining 16% of insertions, which are not identified as tandem duplications (d < l), are inverted copies, complementary duplications, or complementary inversions of adjacent sequence and found none of these classes to yield significant contributions. However, the fraction of insertions with d < l could be substantially reduced by relaxing the required similarity between duplicates from 90% to 80% (fig. 4). We hence suggest that many insertions with d < l might also have originated from tandem duplication events, but divergence between the 2 duplicates is too high, or multiple indel events might have occurred at the same locus. At first glance, sequence divergence of more than 10% during the investigated rather short evolutionary timescale seems exceptionally large. Yet, this could be due to a possibly much higher rate of insertions and deletions in generically unstable regions of the genome. In fact, the generation of tandem duplication insertions will make these regions even more prone for further insertion and deletion events to occur since the newly generated tandem copies can promote further UCO and replication slippage events.
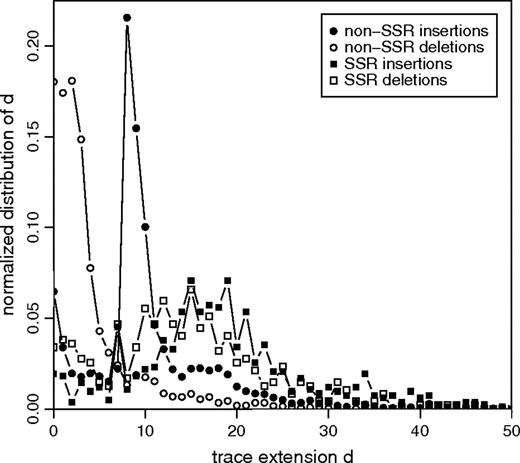
Normalized distributions of the trace extension d for indels of length l = 8 bp. The distinct peak at d = l for non-SSR insertions represents tandem duplications of the form A → AA. In contrast to non-SSR indels, SSR insertions and deletions have broad distributions, and d is significantly larger than l for most indels, as expected for UCO or replication slippage.
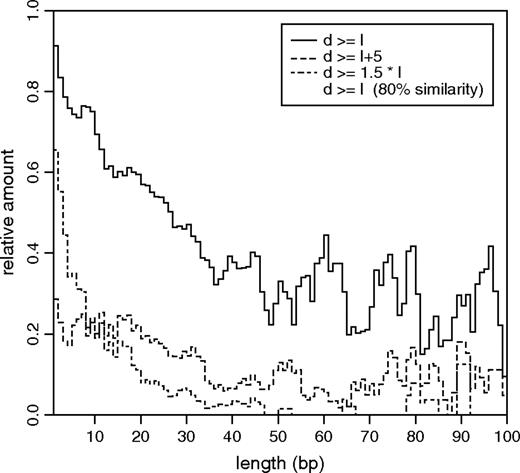
Duplication signatures of non-SSR insertions. The solid line is the ratio of tandem duplications (d ≥ l) among all non-SSR insertions per insertion length l. The dashed line is the proportion of non-SSR insertions with d ≥ l + 5, that is, tandem duplications of the form ABA → ABABA featuring preexisting duplicates A longer than 4 bp. In comparison to insertions with a fixed preexisting duplicate length, the relative amount of insertions with preexisting duplicates at least half as long as the indel decreases rapidly with increasing indel length (dot-dashed line). The dotted line demonstrates how the proportion of tandem duplications among all non-SSR insertions can be increased by reducing the required sequence similarity for our trace extension analysis from 90% to 80%. All curves have been smoothed using running averages over 3 bp.
A striking feature of the measured trace extension distributions is the distinct peak at d = l for non-SSR insertions. This peak indicates tandem duplications of the form A → AA and is common among insertions in all investigated length classes (supplementary fig. 2, Supplementary Material online). Insertions with d = l are unlikely to have originated by UCO or replication slippage because no preexisting duplicates were present prior to the insertion event. The steep right flank of the peak at d = l for non-SSR insertions and the rapid decay of the distribution for d > 0 among non-SSR deletions indicates a general lack of preexisting duplicates longer than a few base pairs (the length of a preexisting duplicate is d − l for insertions and d for deletions). Only 25% of all non-SSR insertions and 17% of deletions show signatures of preexisting duplicates longer than 4 bp. However, the conclusions that can be drawn from the length of a preexisiting duplicate about a likely evolutionary mechanism of indel generation depend on the length of the indel. Most 1 bp long non-SSR indels, for instance, originate from preexisting duplicates of 1–4 bp. This indicates the presence of a mononucleotide stretch prior to the indel event, short enough not to be annotated as SSR. A 1-nucleotide slippage of DNA polymerase within this stretch poses a likely scenario for the origin of most such indels. On the other hand, for indels considerably longer than 4 bp, the presence of 1–4 bp long preexisting duplicates implies that both copies of the preexisting duplicates are separated by a distance much larger than their lengths (see fig. 1). It is rather unlikely that such far-spaced and short duplicates can trigger ectopic recombination between the 2 copies in case of UCO or lead to a far backward or forward slippage of DNA polymerase during replication. Thus, in order to determine whether an indel is likely to have originated by UCO or replication slippage, it is more adequate to investigate the ratio (d − l)/l of preexisting duplicate length to insertion length (for deletions, the ratio is d/l). For a reasonable ratio of 1.5 which means that the length of the preexisting duplicate is at least as long as the spacer between the 2 copies, it is shown in figure 4 that the ratio of non-SSR insertions with (d − l)/l ≥ 1.5 decreases rapidly from approximately 65% of all 1 bp long non-SSR insertions to less than 10% of all 20 bp long non-SSR insertions. Similar behavior is observed for deletions (data not shown). We conclude from this data that the majority of short indels are compatible with UCO or replication slippage, whereas many longer indels (l > 5) are unlikely to have originated by these processes.
Instead of UCO or replication slippage, we propose that a considerable fraction of longer indels might be generated by a different mechanism, based on the imperfect repair of DNA double-strand breaks by NHEJ (van Gent et al. 2001; Lieber et al. 2003). NHEJ is the most common double-strand break repair pathway in many organisms and is evolutionary conserved throughout all kingdoms of life. After a DNA break with single-stranded overhangs both ends rejoin by base pairing between opposite single strands. This process is known to often result in gain or loss of DNA, especially if overhangs are damaged (Roth et al. 1985; Pfeiffer et al. 1994). If base pairing erroneously occurs between microhomologies at the tips of the overhangs, the succeeding filling in of the remaining single-stranded intervals generates tandem duplication insertions. Ligation following the excision of nucleotides at the overhangs can result in deletions. NHEJ requires only short microhomologies of 1–4 bp and can even ligate overhangs without homologies at all (Roth et al. 1985). As double-strand breaks are especially deleterious, it is not surprising that the repair mechanism accepts changes in the nucleotide sequence for the sake of preserved chromosomal integrity. In accordance with our findings long preexisting duplicates are not crucial for indels to be generated by this mechanism.
Further insight into the partial contributions of UCO, replication slippage, and NHEJ to indel generation can be obtained by a separate measurement of indel rates in autosomes and the 2 sex chromosomes. We find that compared with autosomes, the rates of insertions and deletions in the X chromosome are decelerated (IX/IA = 0.85, DX/DA = 0.83), whereas higher rates are observed in the Y chromosome (IY/IA = 1.15, DY/DA = 1.35) (supplementary table 1, Supplementary Material online). Different indel length classes show qualitatively similar behavior (supplementary fig. 3, Supplementary Material online). These findings point toward 2 general characteristics of the underlying molecular processes: indels are preferentially generated in the male germ line, and indel generation is not recombination mediated. Both results do not speak in favor of UCO as predominant mechanism. In contrast, replication slippage and NHEJ do not require recombination events. The suggested preferential occurrence of indels in the male germ line might be related to the higher number of germ cell divisions in males making them more prone to double-strand breaks as well as replication errors.
Evolutionary Role of Small Indels
Our findings provide direct evidence that the generation of tandem duplications is the predominant process of DNA insertion on small length scales. This observation can also explain the ubiquity of short paired duplicates in mammalian genomes. For instance, copies of 25–100 bp long segments (which do not include known repetitive elements) have been found to be highly overrepresented in vertebrate genomes, yet the 2 copies are usually separated by spacers ranging from a few base pairs up to several kilobase pairs (Thomas et al. 2004). It has been proposed by Achaz et al. (2001) that spaced duplets arise by direct tandem duplications and separation evolves by subsequent insertion or rearrangement events. However, the nature of the separation mechanism is still controversial (Thomas et al. 2004). Our analysis shows that spaced duplicates are indeed likely to have originated from juxtapositional copies.
Short tandem duplications also constitute the vast majority (81%) of all insertions in coding regions, raising the question of their evolutionary role regarding the emergence of genetic innovation and adaptive evolution. To quantify the amount of purifying selection associated with these mutational processes, we calculated the ratio of indel rates in coding regions and those measured in the noncoding background (fig. 5). As expected, frameshifting indels are highly suppressed in coding regions. Nonframeshifting indels, on the other hand, have Ic/Inc ∼ Dc/Dnc ∼ 0.5.
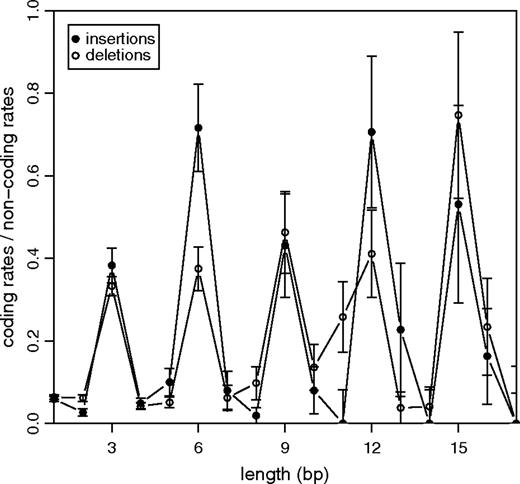
Comparison of insertion and deletion rates between coding and noncoding regions. Rates were calculated by dividing the numbers of insertions and deletions for the 2 classes by the length of sequence each class comprises in our multiple alignments. Numbers of coding and noncoding indels and the calculation of error bars are presented in supplementary table 2 (Supplementary Material online).
This ratio is surprisingly high compared to the ratio of the nonsynonymous single-nucleotide mutation rate in coding regions and the mutation rate in noncoding regions, which is only KA/KI ∼ 0.23 between human and chimp (Chimpanzee Sequencing and Analysis Consortium 2005). Assuming the majority of indels in noncoding regions to be selectively neutral, the observed ratio implies that every second nonframeshifting indel in a coding region is not sufficiently deleterious to be removed by natural selection, in contrast to only 1 out of 4 nonsynonymous substitutions (Sabeti et al. 2006). Hence, in most of the cases amino acid insertions or deletions seem to have a considerably smaller impact on protein structure and function than substituting one amino acid by another.
Gene duplications, large segmental duplications, and entire genome duplications have been widely accepted to promote adaptive evolution and the generation of new genetic functions on large scales (Ohno et al. 1968; Lynch and Conery 2000; Taylor and Raes 2004; Bailey and Eichler 2006). This raises the question to what extent also smaller duplications can contribute to adaptive evolution by generating selectively beneficial variants of proteins or regulatory regions. Several qualitative considerations already point toward a possibly beneficial role of duplications also on intermediate length scales. For instance, duplications of small genomic segments have been suggested to accelerate evolution, for example, by copy number variations of cis-regulatory motifs (Chuzhanova et al. 2000), or duplication-driven generation of protein domains. It is shown by our analysis that tandem duplication events indeed account for the majority of recently inserted genetic material into the human genome on length scales ranging from short DNA motifs down to single nucleotides. Moreover, we found that nonframeshifting insertions and deletions in protein-coding regions are on average less deleterious compared with nonsynonymous substitutions. Whether particular duplications of short DNA motifs were actually fixed due to positive selection poses an interesting question for future research that could be addressed by analyzing the degree of polymorphism among small indels.
We thank Richard Durbin, Evan Eichler, Dmitri Petrov, and Martin Vingron for valuable discussions and comments.
References
Author notes
Arndt von Haeseler, Associate Editor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}