





Abstract
Over a hundred families of non-long terminal repeat retrotransposons (non-LTRs) were found in the newly released Anopheles gambiae genome assembly during a reiterative and comprehensive search using the conserved reverse transcriptase (RT) domains of known non-LTRs as the starting queries. These families, which are defined by at least 20% amino acid sequence divergence in their RT domains, range from a few to approximately 2,000 copies and occupy at least 3% of the genome. In addition to having an unprecedented number of diverse families, A. gambiae non-LTRs represent 8 of the 15 previously defined clades plus two novel clades, Loner and Outcast, more than what has been reported for any genome. Five families were found belonging to the L1 clade, which had no invertebrate representatives to date. One unique family named Sponge contains only a complete open reading frame (ORF) for the Gag-like protein and appears to have been mobilized by a family of the CR1 clade. Although most families appear to be inactive as expected, all clades except R4 have families with characteristics suggesting recent activity. At least 21 families have multiple full-length copies with over 99% nucleotide identity and some or all of the following characteristics: target site duplications (TSDs), intact ORFs, and corresponding expressed sequence tags (ESTs). The incredible diversity and the maintenance of multiple recently active lineages within different clades indicate a complex evolutionary scenario. A. gambiae non-LTRs have the potential to be developed as tools for population genetic studies and genetic manipulations of this primary vector of the devastating disease malaria. The semi-automated reiterative search approach described here may be used with any genome assembly to systematically survey and characterize non-LTRs as well as other transposable elements that encode a conserved protein.
Introduction
Transposable elements (TEs), or mobile genetic elements, are integral components of the eukaryotic genomes. Because they have the ability to replicate and spread in the genome as primarily “selfish” genetic units (Doolittle and Sapienza 1980), TEs tend to occupy significant portions of the genome. For example, at least 45% of the human genome (Lander et al. 2001) and 16% of the newly reported Anopheles gambiae genome (Holt et al. 2002) are TE-derived sequences. Recent evidence suggests that the selfish property may have enabled TEs to provide the genome with potent agents to generate tremendous genetic and genomic plasticity (Kidwell and Lisch 2001). These elements transpose through either RNA-mediated or DNA-mediated mechanisms (Finnegan 1992). DNA-mediated TEs generally transpose by a cut-and-paste process, directly from DNA to DNA. RNA-mediated TEs transpose by a replicative process that involves transcription, reverse transcription, and integration of cDNA molecules. Transposable elements in this category include the long terminal repeat (LTR) retrotransposons, non-LTR retrotransposons (non-LTRs) or long interspersed nuclear elements (LINEs), and short interspersed nuclear elements (SINEs).
Non-LTRs are generally 3–8 kb long and have been found in virtually all eukaryotes studied. Most non-LTRs have a reverse transcriptase (RT) domain that is essential for their retrotransposition. The RT domain has been used for phylogenetic classification of non-LTR retrotransposons into 11 clades, all of which date back to the Precambrian era, approximately 600 MYA (Malik, Burke, and Eickbush 1999). The total number of clades has been recently increased to 15, with the addition of NeSL-1 (Malik and Eickbush 2000), Ingi, Rex1 (Eickbush and Malik 2002), and L2 clades (Lovsin, Gubensek, and Kordi 2001). In addition to the RT domain, many non-LTRs encode an additional protein related to the retroviral Gag genes that is a nucleic acid binding–protein or nucleocapsid. Studies of this Gag-like protein from L1 in mice show that it acts as a nucleic acid chaperone (Martin and Bushman 2001). Some elements also have a ribonuclease H (RNase H) and/or AP endonuclease (APE) domain encoded. Other typical structural characteristics found in various non-LTR families are internal pol II promoters and 3′ ends containing canonical AATAAA polyadenylation signals, poly (dA) tails, or adenosine-rich tandem repeats. Target Primed Reverse Transcription has been proposed as the mechanism of retrotransposition for R2 of Bombyx mori and may be generally true for all non-LTR elements (Luan et al. 1993). Target site duplications (TSDs) are generated by most non-LTRs as a result of insertion of a new copy.
Non-LTRs may have had significant influences on eukaryotic genome evolution (Brosius 1999; Makalowski 2000; Kidwell and Lisch 2001). They have been shown to occupy a large portion of some genomes, contain sequences that affect the expression of nearby genes, serve as homologous sites for recombination, and contribute to novel exons. For example, L1 from humans and a SINE named Alu, which is thought to be retrotransposed by L1, make up more than 27% of the human genome (Lander et al. 2001). In addition, L1 has been shown to be capable of 3′ transduction (Moran, DeBerardinis, and Kazazian 1999), and is believed to be responsible for the duplication of up to 1% of the human genome (Goodier, Ostertag, and Kazazian 2000; Pickeral et al. 2000). L1 is also believed to be responsible for the production of processed pseudogenes because they have the characteristics of retrotransposed sequences. Although non-LTRs occupy a significant bulk of the human genome, they represent only two of the 15 described clades (Malik, Burke, and Eickbush 1999; Lander et al. 2001). Large-scale surveys of Arabidopsis thaliana, Caenorhabditis elegans, and a few other smaller eukaryotic genomes showed similar lack of diversity. However, in Drosophila melanogaster (Berezikov, Bucheton, and Busseau 2000), more than 10 families of non-LTRs were discovered which represent six of the 15 clades. The recently reported Fugu rubripes genome (Aparicio et al. 2002) also showed significant diversity, containing non-LTRs from five clades. Here we report the discovery and characterization of a large number of non-LTRs in the newly released A. gambiae genome assembly.
Materials and Methods
Search Strategy
A systematic strategy (fig. 1), which incorporates multi-query Blast (Altschul et al. 1997) and a few computer programs described below, was used to search for non-LTRs in the A. gambiae genome assembly (Holt et al. 2002). The assembly consisted of 8987 scaffolds comprising 278 Mbp of the A. gambiae genome. All search programs were executed on a Dell 530 Linux workstation with twin 2.0 GHz processors, 1.5 Gb RAM, and an 80 Gb hard drive. Amino acid sequences containing the conserved 0–5 regions of the RT domain (Malik, Burke, and Eickbush 1999) of one representative non-LTR element in each of the 12 established clades (Malik and Eickbush 2000) were used as starting queries in a tBlastN search (E-value cut-off of 1e-5). Starting query sequences included the conserved RT regions of known mosquito retrotransposons JAM1, JuanA, Lian, and MosqI from Aedes aegypti (Mouches, Bensaadi, and Salvado 1992; Warren, Hughes, and Crampton 1997; Tu, Isoe, and Guzova 1998; Tu and Hill 1999). To explain our strategy, we use the example of a representative non-LTR as shown in figure 1A. The initial step is a tBlastN search with this query, the result of which is then organized using TEpost. Because low stringency parameters were used in tBlastN to ensure full coverage, multiple families of non-LTRs are expected in the Blast output. To organize the hits into different families on the basis of sequence similarity, a reiterative approach is employed. Starting with the nucleotide sequence of the most significant hit Xi as the query, BlastN searches were performed on the A. gambiae genome assembly to identify all related sequences and their coordinates (see below for parameters of BlastN and subsequent TEpost). All the BlastN hits that had more than 80% nucleotide sequence identity as the query Xi were considered members of the Xi family (see details below), which were subsequently masked from the genome assembly using TEmask. (In later phylogenetic analysis, for the purpose of reducing crowding and redundancy in the tree, a family was defined as a group of sequences having ≥80% amino acid identity in the same region that was used as queries described above. This resulted in the merging of only a few families that had similar structural characteristics.) After database masking, the cycle was repeated starting with tBlastN using the same query described above. However, the cumulatively masked database was used during each cycle to ensure that only new families produced hits. This reiterative search was continued exhaustively until virtually all families were defined. The use of a large number of representative non-LTRs as queries in tBlastN was designed to ensure discovery of non-LTRs from all clades and, possibly, novel divergent elements (not shown in figure 1A). However, we later discovered that over 95% of RT families can be identified using the RT sequence from only one known non-LTR during the systematic reiterative approach. In cases where we used multiple RT queries in one tBlastN search, TEcombine was used to produce a non-redundant list of hits. Nucleotide sequences for known A. gambiae non-LTRs Q, T1, RT1, RT2 (Besansky 1990; Besansky et al. 1992; Besansky, Bedell, and Mukabayire 1994), and the fragments Vash, Guildstern, and AgJuan (Hill et al. 2001) were also used in BlastN searches. At this point only 70 sequences at least 100 bp long resulting from all of the tBlastN searches remain uncharacterized; all of them appear to be copies of old, degenerate non-LTRs. It should be noted that the A. gambiae genome assembly was chopped up into 100 Kbp fragments before use to make searching and data processing more efficient. The potential problem arising from chopping a RT sequence in the middle is insignificant.
To test the robustness of family structure we examined the Ag-Loner-1 family that is closely related to the Ag-Loner-2 family. Using a nucleotide sequence of an Ag-Loner-1 copy corresponding to the RT domain (the same as that used for phylogenetic analysis), we performed a BlastN search against the genome. The resulting hits were filtered using our described programs, retaining only sequences with 80% or more identity and a length of at least 500 nucleotides (the query was 687 nt). After alignment of these sequences and the generation of a pairwise distance matrix, the most divergent sequence to the query was chosen to repeat the above procedure. The resulting “family structure” was identical to that of the first search and did not include any copies of the Ag-Loner-2 family. The above procedure was repeated with Ag-Jen-3 and the result was the same, the family structure was unchanged. The robustness of the family structure might be explained by the relatively inclusive criteria that we used to define a family.
Identification of Full-Length Elements
Alignment of the nucleotide sequences plus flanking genomic sequence of each family was performed with ClustalW to determine transposon boundaries and full-length elements (fig. 1B). The alignment was also used to identify TSDs. The alignment was done using ClustalW version 1.81 for the Linux operating system (Thompson, Higgins, and Gibson 1994) and viewed with ClustalX versison 1.81 for Windows (Thompson et al. 1997). The following parameters were used: pairwise gap penalty (open = 10, extension = 0.1), multiple gap penalty (open = 10, extension = 0.2). The alignment process was facilitated by the use of FromTEpost to retrieve all qualified hits into a fasta file. In practice, the search for non-LTR families and the identification of full-length elements were performed concurrently. Identified full-length elements were used in TEmask whenever available.
Software Description
Software modules designed for this study include four C programs TEpost, TEcombine, FromTEpost, and TEmask, all of which are available for download on our Web site (www.biochem.vt.edu/aedes or http://128.173.80.165). Details of the programs can be found in the corresponding readme files on our Web site. TEpost uses a Blast output file (produced by single or multiple queries) as an input file and produces an output file listing each Blast hit in a row along with several characteristics associated with that hit. The parameters for TEpost are these: maximum transposon length, maximum transposon gap, and minimal overlap. Maximum transposon length restricts the hits reported to those with a length less than the specified value. Because of the nature of Blast and the presence of indels or other chromosomal rearrangements, Blast hits corresponding to one transposon copy can be reported as multiple hits and can result in an overestimation of copy number. The gap length parameter was added to reduce this occurrence by grouping fragmented hits associated with one transposon copy as a single match. The formula for gap length is |(Q2–Q1)–(S2–S1)| where Q1 and Q2 are the start and end of the query aligning with the Blast hits, and S1 and S2 are the start and end of the subject in the Blast hits. Please note that the start and end positions are from neighboring hits being considered as potentially one “continuous” match. Situations where query and subjects are on opposite strands are treated separately. A maximum cut-off value is used so that hits having gaps exceeding this value are recorded as separate and are assumed to originate from different transposon copies. Additional optional parameters that filter the TEpost output are E-value, hit length, and percent identity that are set as cut-off limits. When multiple queries from the same region of a transposon are used for Blast searches, it is expected that the same hit will be reported for more than one query. This creates a problem when TEpost files generated from Blast searches using different queries are combined. TEcombine was designed to eliminate these redundant hits by retaining only the hit listing with the most significant E-value. Minimal overlap is a parameter used here in the same way as that described for TEpost. Both TEpost and TEcombine output files can be copied/pasted into Microsoft Excel for sorting and viewing. FromTEpost uses both TEpost and TEcombine files as input to produce fasta sequence files of the recorded hits. Flanking sequences can be included by choice of the user. Sequences on plus and minus strands are reported separately. TEmask uses information from either a TEpost or TEcombine file to mask a database for all of the recorded hits. As with the previously described program RepeatMasker (Smit and Green, http://ftp.genome.washington.edu/RM/RepeatMasker.html), TEmask ensures that discovered families are not hit again in subsequent Blast searches.
Copy Number Estimation
Most non-LTR retrotranspsoson copies are truncated at the 5′ end as a result of incomplete reverse transcription during retrotransposition. Copy numbers are therefore expected to be different for estimations based on the 5′ and 3′ ends. Full-length elements were identified by multiple sequence alignment for use as queries in copy number determination. When full-length copies were not identified, we used the longest obtainable sequence in a family (including the 3′ terminus when possible). A multiple query fasta file (one sequence per family) was used in a BlastN search with an E-value cut-off of 1e-10. The Blast output was then processed using TEpost and TEcombine, during which only sequences that showed more than 80% nucleotide identity were included for copy number estimation. Parameters used were these: minimum hit length, 50; maximum transposon length, 10,000; maximum transposon gap, 3,000; minimal overlap, 50. To determine the minimum percentage of the genome occupied by non-LTRs, we summed the total number of bases resulting from Blast hits. Manual inspection of the output files used for copy number determination showed that a few copies were reported twice because of hits resulting from different parts of the query that exceeded the allowed gap. These occurrences were insignificant, however, and the numbers were adjusted when any were identified. Some queries contained other repetitive elements that resulted in increased copy numbers, but these hits were excluded from the reported copy numbers. It should be noted that we have used a working definition of a non-LTR family as a group of sequences having ≥80% amino acid identity in the conserved RT regions. This definition is more inclusive than the criterion used for copy number estimation, which is 80% nucleotide identity in the conserved RT region. Therefore, our current copy number estimations may be slight underestimates in some cases.
Phylogenetic Analysis
Phylogenetic analyses were performed using multiple sequence alignments of approximately 260 amino acid characters in the conserved regions 0–5 of the RT domains. These alignments were obtained using ClustalW version 1.81 for the Linux operating system (Thompson, Higgins, and Gibson 1994) and viewed with ClustalX version 1.81 for Windows (Thompson et al. 1997). Parameters used for these alignments were these: pairwise gap penalty (open = 30, extension = 0.8), multiple gap penalty (open = 10, extension = 0.25). Only one representative was used from each family found in the A. gambiae genome. In the case of the alignment used in the overall phylogenetic analysis shown in figure 2, minor adjustments were made in the alignment at conserved region 5 (Malik, Burke, and Eickbush 1999) for two familes of the R4 clade, Dong of Bombyx mori and Ag-R4-1. All phylogenetic analyses were performed with PAUP* version 4.0b10 (Swofford 2002). Both Neighbor-Joining and maximum parsimony trees were constructed with bootstrap support of 500 replicates. In cases where it was not possible to obtain amino acid sequence including all regions 0–5, the longest obtainable amino acid sequence was used. Outgroup selections and specific parameters for the phylogenetic analyses are described in the legends of figures 2 and 3, respectively.
Results
The A. gambiae Genome Harbors a Rich Diversity of Non-LTRs
We have recently reported the presence of several non-LTR groups in the A. gambiae genome (Holt et al. 2002). At the time of data submission for the previous report our survey was incomplete and no characterization or phylogenetic analysis of the limited number of non-LTRs was presented. As shown in table 1 and Appendix A, here we present the characterization and classification of 104 families of non-LTRs in A. gambiae, including four previously characterized families (Besansky 1990, 1999; Besansky, Bedell, and Mukabayire 1994). A family is defined here as having at least 80% amino acid identity in the part of the RT domain used for phylogenetic analysis (see Materials and Methods). As shown in figure 2, the phylogeny of A. gambiae non-LTRs is analyzed in the context of known non-LTRs of other organisms from established clades (Eickbush and Malik 2002). The 104 A. gambiae non-LTR families represent divergent lineages in eight previously established non-LTR clades (R4, L1, RTE, R1, L2, CR1, Jockey, and I) and two new clades named Loner and Outcast, which are described later. Most of these clades were well supported by bootstrap analysis using either the Neighbor-Joining or the maximum parsimony methods (fig. 2). No families were found corresponding to the CRE, NeSL-1, R2, Tad1, LOA, Ingi, or Rex1 clades. Representation appears to be biased toward the most derived clades in non-LTR evolution, especially the CR1 and Jockey clades, with 31 and 25 families, respectively. The R1 clade also contains significant diversity, having 16 families. The rich diversity of non-LTRs within A. gambiae, both in terms of the number of clades represented and the number of divergent families within a clade, is unprecedented (table 1 and Appendix A). To provide an evolutionary context for the large number of A. gambiae non-LTRs in the CR1 and Jockey clades, a phylogenetic tree was constructed with these non-LTRs and CR1 and Jockey elements from a diverse group of organisms including non-Anopheles mosquitoes and other Dipteran insects (fig. 3). All of the 31 A. gambiae CR1 elements are grouped together, with the Drosophila CR1 being a sister branch. At the same time, there appear to be three groups (I, II, and III) of the A. gambiae Jockey elements, which may have different sister elements either from other mosquitoes or from other Dipteran insects. It should be noted that the bootstrap supports for these groupings are weak, and only few relevant sequences in closely related species are available for the analysis. The evolutionary dynamics that may be responsible for the inter- and intracladal diversity of non-LTRs in A. gambiae are discussed later.
Several Families Have Characteristics Suggesting Recent Activity
Sequence analysis can give clues to the history of transpositional activity of an element. These characteristics include the presence of full-length elements, intact open reading frames (ORFs), multiple copies with high nucleotide identity, and TSDs. High nucleotide identity indicates recent amplification from a source element, without enough time for divergence caused by nucleotide substitution and other mutations. Most families identified appear to be inactive, as expected; however, all clades except R4 have families with multiple copies having nucleotide identities over 99% (table 1). Sequence identities of families in the R1 clade are excluded from table 1 because it was difficult to obtain full-length copies. Twenty-one families from eight clades were found to have at least two copies with nucleotide identity of 99% or more. Thirteen of these families from seven clades have copies with 99.9% or more identity. The presence of perfect TSDs can also serve as an indicator of recent transposition. Fifteen families in six clades have multiple full-length copies flanked by TSDs. Although most elements create unique TSDs upon insertion, TSDs could not be found for families of the CR1 and the closely related L2 clades. The inability to find TSDs had been previously reported for Q and T1 of the CR1 clade (Besansky 1990; Besansky, Bedell, and Mukabayire 1994). In addition, seven families listed in table 1 had significant hits when Blast searches were carried out against over 94,000 A. gambiae expressed sequence tags (ESTs) downloaded from National Center for Biotechnology Information (NCBI). The above hits had E-values of less than 9e-97 and identities from 93%–100% to the query sequences. Another 14 families in Appendix A have significant hits to ESTs as well. One caveat is that ESTs could arise from spurious transcription, for example if a copy resides within a host gene. This occurrence is expected to be more likely for high copy number elements. Consistent with the EST analysis, our initial reverse transcription polymerase chain reaction (RT-PCR) analysis using mRNA isolated from an A. gambiae cell line (Mos.55) demonstrated the presence of transcripts of a Jockey element (data not shown). In summary, the high level of intra-family sequence identity, the existence of full-length copies flanked by TSDs, and the detection of possible transcripts all are consistent with the hypothesis that many of the non-LTRs are either currently or recently active in A. gambiae.
Loner and Outcast are New Clades, Each Comprised of Divergent Families
Phylogenetic analysis (fig. 2) resulted in two novel deep branching groups, new clades named Loner and Outcast. Both are well supported by bootstrap analyses (100%) using Neighbor-Joining and maximum parsimony methods. Domains found present in ORF1 and ORF2 of these new clades are compared in figure 4. ORF1 from elements of both clades contains three repeated cystine-histidine (Cys/His) motifs (CCHC) characteristic of nucleic acid–binding domains. A single Cys/His motif was also found in the 3′ end of ORF2 in families of these clades, similar to what has been described for elements in other clades (Malik, Burke, and Eickbush 1999). ORF2 from elements of these clades contains domains in the order of APE, RT, and RNase H. Whereas ORF2 of Ag-Loner-1 appears to require a frameshift for translation, the ORF2 of Ag-Outcast-6 appears to start from an ATG 95 nucleotides downstream of the ORF1 stop. Three criteria have been proposed to define a clade, including shared structural features, ample phylogenetic support for the group, and an origin dating back to the Precambrian era (Malik, Burke, and Eickbush 1999). The first two of the three proposed criteria are met for both the Loner and Outcast clades. The last criterion can be assumed based on the comparison of the branch origins of Loner and Outcast to those of other established clades. Multiple divergent families were also found within each clade. Copy numbers are relatively low for most of the families in these clades, ranging from 4 to 86 (table 1 and appendix A). No representatives were found in other organisms on the basis of tBlastN searches against the NCBI nonredundant nucleotide database using amino acid query sequences including the RT domains of the Loner and Outcast families. Loner and Outcast clades both contain families that appear to have been recently active (table 1). The Ag-Loner-1 family has two copies with 99.3% nucleotide identity and a significant hit to an EST. Four copies of Ag-Outcast-6 are virtually identical, having nucleotide identities of 99.9%–100% when compared to the consensus sequence of three full-length copies. Two other families Ag-Outcast-2 and Ag-Outcast-5 also have copies with a high degree of nucleotide identity. Both clades have families with TSDs (table 1).
A. gambiae Has the First Reported Invertebrate Representatives of the L1 Clade
Elements of the L1 clade have been found in diverse organisms such as mammals, plants, fish, and yeast, but none have been found in invertebrates until now. Five divergent families were discovered in A. gambiae, two of which have multiple full-length copies with high nucleotide identities (table 1) and therefore appear to have been active recently. Four copies of Ag-L1-5 have identities from 99.7% to 99.9% and two of these copies have only two bases different over the full 4.5-kb length of the element. Target site duplications could not be determined for one of the four elements. However, all copies are the same length with two ORFs and unique flanking sequences, and therefore this copy is also presumably full-length. Ag-L1-2 has two full-length copies with 99.7% identity, TSDs, and unique flanking sequences. These families have an APE upstream of the RT in ORF2, like other elements in the L1 clade (fig. 4; Malik, Burke, and Eickbush 1999). Interestingly, the ORF1 of Ag-L1-2 and Ag-L1-5 families contain a single Cys/His motif (C-X2-C-X4-H2-X3-C) which is in contrast to the leucine zipper motif found in ORF1 of a human L1 (Holmes, Swinger, and Swergold 1992). Most of the Ag-L1 families have relatively low copy numbers that range from 7 to 60 (table 1 and Appendix A).
A. gambiae Has Representatives of the Recently Described L2 Clade
Recently L2 was proposed as a new clade (Lovsin, Gubensek, and Kordi 2001), having diverse representatives from insect, sea urchin, snake, and fish. This clade was once considered part of the CR1 clade, but later was established as distinct from CR1. In addition to three families in A. gambiae (Ag-L2-1, 2, and 3) and the Takifugu rubripes representative (Lovsin, Gubensek, and Kordi 2001), two diverse representatives were found from the Zebrafish in support of this clade. ORF2 of Ag-L2 families contain an APE upstream of the RT, like families in the CR1 clade. Representatives from diverse organisms, phylogenetic support, and the deep branching of this group support this as a distinct clade from CR1. The Ag-L2-1 family has two 5.2 kb copies with nucleotide identities of 99.9% but their ORF2 proteins contain frameshifts. Ag-L2-2 and Ag-L2-3 families have multiple copies with identities of 96% or less spanning roughly 300–500 nucleotides.
Sponge, a Unique Gag-Only Non-LTR Retrotransposon
While analyzing sequences of the Ag-CR1-3 family, a group of sequences of identical length were found that were much shorter than Ag-CR1-3. Further analysis revealed these sequences to be a distinct family that contained a 1.9-kb deletion in ORF2 that eliminates the RT domain (fig. 4). This family was named Ag-Sponge for its dependency on another element for retrotransposition. It has at least 13 full-length copies that are approximately 2.4 kb long (not shown). These full-length copies have the same 5′ and 3′ termini, unique flanking genomic sequences, and a high degree of nucleotide identity, suggesting that they were once an intact unit of retrotransposition. Ag-Sponge and Ag-CR1-3 have 98% nucleotide identity covering 160 bases in their 3′ ends, including their tandem repeats. The remainder of Ag-Sponge has approximately 95% identity to Ag-CR1-3 and contains another 400 bp deletion in the APE region of ORF2. Sponge probably relied on the reverse transcriptase of an autonomous non-LTR, likely that of Ag-CR1-3. It should be noted that the discovery of a family of internally deleted non-autonomous non-LTRs is interesting because most nonautonomous non-LTRs are 5′ truncations that may no longer be able to form a unit for retrotransposition.
3′ Sequence Characteristics
The 3′ ends of non-LTRs contain important sequence characteristics such as polyadenylation signals, poly (dA) tails or unique tandem repeats, and in some cases conserved secondary structure (Finnegan 1997; Mathews et al. 1997). The first three of these characteristics are listed in table 1 and Appendix A for A. gambiae non-LTRs. The presence of these characteristics is variable for any given family. Considering all clades, the majority of families have polyadenylation signals but do not have poly (dA) tails in the 3′ end. For most elements with poly (dA) tails in the genomic sequence, the canonical AATAAA polyadenylation signal can be found upstream. Interestingly, there are some families in the R1 clade that have poly (dA) tails, but no canonical AATAAA polyadenylation signal. Other families have neither poly (dA) signals nor poly (dA) tails. Most A. gambiae elements have a 3′ tandem repeat, with exceptions in the L1 and R1 clades. The repeat unit varies in length for different families, from the two base pair repeat AT for Ag-Jock-13 of the Jockey clade to the variety of 8 bp repeats for families of the CR1 clade. In some cases, the sequence of the tandem repeat units reported in the order 1, 2, 3 may actually be in the order 2, 3, 1 because it was difficult to determine the start and finish of the repeat unit (e.g., Ag-Outcast-2 and Ag-Outcast-5 have GAA and AAG repeats). A notable feature is that the presence of a tandem repeat and a polyA tail are apparently mutually exclusive. For most families, the total length of the 3′ tandem repeat is restricted to approximately 30 bp or less and starts close to or immediately downstream of the AATAAA signal. Another notable feature is that there appears to be a general correlation between the 3′ repeat sequence and RT phylogeny with a few exceptions.
Non-LTR Families Vary Widely in Copy Number
To determine copy number, the longest obtainable sequence of a family was used as queries for a Blast search. These queries included the 3′ terminus when possible. The majority of hits were not full-length, either because of incomplete reverse transcription or because they did not fall within the specified parameters (see Materials and Methods for parameters used). Copy numbers are highly variable and range from just a few to about 2,000 for A. gambiae non-LTRs (table 1 and Appendix A). Most families are of low copy number with exceptions in the RTE and CR1 clades. Ag-JAMMIN-1 and Ag-JAMMIN-2 families of the RTE clade both have copy numbers dramatically higher than any other A. gambiae non-LTR family. Together they contribute to about 25% of the non-LTR portion of the genome. The T1, Ag-CR1-13, and Ag-CR1-3 families of the CR1 clade also have attained significantly higher copy numbers than most families. We have determined that non-LTRs contribute to at least 3% of the A. gambiae genome. Reported TE copy numbers will often be an underestimate because older elements will be mutated beyond recognition. This will also result in a conservative estimate when determining the percentage of a genome comprised by non-LTRs.
Discussion
A Systematic Computational Approach to Identify, Classify, and Characterize Non-LTRs
The semi-automated reiterative strategy presented here (figure 1 and Materials and Methods) has proven useful for systematic and rapid identification of non-LTRs on a whole genome scale. The incorporation of a few computer programs described here with multi-query Blast (Altschul et al. 1997) greatly facilitated the discovery and characterization process. The use of the highly conserved RT domain and the inclusion of representatives from different known clades as queries helped toward a comprehensive coverage. The potentially comprehensive nature of our approach was demonstrated as two new clades were identified. The inclusive nature of our approach was further indicated when we observed that non-LTRs across all known clades were identified using the reiterative approach with a single Drosophila representative in the Jockey clade as the starting query (data not shown). In addition, the reiterative feature of the strategy described in this study provides an opportunity for further automation by linking the modules described in figure 1A. As a control, we tested an early version of such a fully automated program to identify all non-LTR families in the A. gambiae genome. Preliminary results showed that 102 of the 104 non-LTR families were identified in one run of the program that takes approximately 15 hours on the computer system described in Materials and Methods. A modification further reduced the run time to less than two hours, in which only sequences that significantly match the queries were used in subsequent searches. Only non-mosquito non-LTR sequences were used as queries in the automated survey, suggesting that as long as input files contain diverse representatives of a superfamily, this method should work for any TE with coding capacity in any genome. Two other methods have previously been employed to find repetitive elements in genomes. Recon (Bao and Eddy 2002), an automated comprehensive method, works to identify all repetitive sequences using a multiple sequence alignment algorithm. Recon requires no previous information about the TE sequences yet it is computationally intensive. Berezikov, Bucheton, and Busseau (2000) used software based on profile hidden Markov models to find all sequences matching the full-length reverse transcriptase with the conserved FYXDD motif common to all RTs. This method is potentially comprehensive for all RT–containing sequences, but it requires further classification of the sequences into retroviral, LTR retrotransposon, or non-LTR families. The strategy described in the current study and its recent modifications offer an efficient alternative for simultaneous identification and classification of TEs in any given genome.
Unprecedented Diversity, Recent Activity in Multiple Lineages, and Evolutionary Implications
Over a hundred families of non-LTRs were found in the A. gambiae genome, and they occupy at least 3% of the genome. These families, defined by at least 20% amino acid sequence divergence in their RT domains, represent 10 different clades including eight of the 15 previously described clades plus two novel clades, Loner and Outcast. To our knowledge, such a level of inter- and intracladal diversity of non-LTRs has not been reported in any genome. Considering that there are interscaffold and intrascaffold gaps (Holt et al. 2002), there still may be unidentified families in the genome. In this regard, A. gambiae is in great contrast to the human genome, where only three families in the L1 and L2 clades were found to occupy over 20% of the genome (Lander et al. 2001). Large-scale surveys of a few smaller eukaryotic genomes showed a similar lack of diversity (table 2). However, the recently reported Fugu rubripes genome (Aparicio et al. 2002) showed a significant level of diversity, containing non-LTRs from five clades. In D. melanogaster, more than 10 families of non-LTRs were reported (Berezikov, Bucheton, and Busseau 2000), which represent six of the 15 clades, four of which are found in A. gambiae (I, Jockey, CR1, and R1; table 2). Because their definition of a family was perhaps more inclusive than the one used in this study and because significant portions of the heterochromatic regions were not analyzed (Berezikov, Bucheton, and Busseau 2000), there may be even greater diversity waiting to be discovered in D. melanogaster. The number of described non-LTR clades in all eukaryotes has increased dramatically from 11 (Malik, Burke, and Eickbush 1999) to 17 over the past few years. The availability of increasing numbers of genomic sequences and the development of computational approaches for database searches will further facilitate our understanding of this divergent group of long-time residents in a broad range of genomes. As genomic data accumulate, it will be interesting to determine whether non-LTRs in the Loner and Outcast clades will be found in other species, and whether non-LTRs in the L1 clade will be found in other invertebrates.
Families in the novel clades Loner and Outcast have domains that are in accordance with their phylogenetic placement. Malik, Burke, and Eickbush (1999) proposed that in non-LTR evolution, domains were acquired in the order RT, APE, and RNase H. Although the branch length is short, the lineage leading to the Loner and Outcast clades was derived prior to the I clade and after that of the lineages leading to the LOA, R1, and Tad1 clades (fig. 4). All of these clades have the RNase H domain in at least some representatives (Malik, Burke, and Eickbush 1999). Therefore, the presence of the RNase H domain in families of the Loner and Outcast clades is not surprising.
At least 21 of the highly diverse non-LTR families show evidence of recent activity. All clades except R4 have one or more families with some or all of the sequence characteristics, suggesting recent transposition activity such as multiple full-length copies with over 99% nucleotide identity, target site duplications (TSDs), intact ORFs, and corresponding ESTs. Therefore it is clear that these divergent clades for the most part are not ancient “fossils.” Rather, many families in these clades are or have recently been active components of the A. gambiae genome. It is remarkable that a large number of diverse non-LTR lineages have been maintained in the genome, and many show evidence of recent activity (table 1 and Appendix A). In A. gambiae, both intra- and inter-cladal diversity are biased toward expansion in lineages of the non-site–specific type, with the exception of some non-LTRs in the R1 clade that showed specific target sites. Elements in CRE, NeSL-1, R2, and R4 clades encode restriction enzyme–like endonucleases that confer site-specificity and represent the primordial non-LTRs. Of these clades, only degenererate copies of R4-like elements were found in the A. gambiae genome. In contrast, the CR1 and Jockey clades have flourished, having more families than any other clade (fig. 1). At least six families in the Jockey clade and five families in the CR1 clade appear to have been recently active based on sequence analysis. Some of these families have significant hits to A. gambiae ESTs. These estimates are conservative because in the CR1 clade, frequent 5′ truncations and the lack of TSDs made it difficult to identify full-length elements for many families.
High levels of diversity and the maintenance of multiple recently active lineages within different clades in the A. gambiae genome indicate a complex evolutionary scenario. Unlike DNA-mediated TEs, for which horizontal transfer is well documented, analysis by Eickbush and Malik (2002) suggests that there is no reason to believe that non-LTRs have been involved in horizontal transfer. They showed that non-LTRs previously implicated in horizontal transfers on the basis of phylogenetic incongruence actually obey expectations for vertical transmission. The high degree of intracladal diversity described in our current study suggests that great caution should be applied when inferring horizontal transfer on the basis of phylogenetic analysis, because the presence of a large number of paralogous families can easily confound the analysis. If we accept the assumption of vertical transmission, A. gambiae non-LTRs of the CR1 and Jockey clades must have gone through a tremendous diversification (fig. 3). Without extensive CR1 and Jockey sequences from other mosquitoes, it is difficult to determine whether the diversification has occurred prior to the common ancestor of mosquitoes or within the lineage leading to A. gambiae during mosquito evolution. However, most of the diversification events may have occurred after the divergence that led to D. melanogaster and A. gambiae lineages. Given the presence of multiple recently active lineages within the CR1 and Jockey clades, it is tempting to speculate that the observed diversity is driven by positive selection generated by competition among different non-LTR families or by attempts to escape suppressive mechanisms by the host. Nucleotide sequences are too divergent between these families for dS/dN analysis to assess the selection pressure, especially in ORF1 which is under a lower degree of selection pressure than the RT of ORF2. However, future comparative analysis of non-LTRs in closely related mosquitoes may provide answers to this question.
3′ Sequence Characteristics and Their Relation to Retrotransposition
One of the earliest characteristics defining non-LTRs was the presence of the canonical AATAAA polyadenylation signal and either poly(dA) or A-rich tandem repeats. Except for families in the CR1, Outcast, and RTE clades, tandem repeats are not A-rich. Most of the families found in A. gambiae have the polyadenylation signal, but only a small number of them have the poly (dA) tail associated with it in the genomic sequence. This is curious because it would be expected that RT would copy the poly (A) sequence from the RNA during retrotransposition resulting in poly (dA) tails in the genomic sequence. Such a disconnect between the polyadenylation signal and the presence of poly (dA) has been previously described in CM-gag, a non-LTR retrotransposon from the Culex pipiens mosquito (Bensaadi-Merchermek et al. 1997). Analysis of the transcript of CM-gag showed that it was polyadenylated immediately downstream of its TTGAA tandem repeat. It appears that in this case, reverse transcription is not initiated until the TTGAA repeat during retrotransposition. A recent study of the D. melanogaster I factor provided a similar conclusion (Chambeyron, Bucheton, and Busseau 2002). In contrast, some A. gambiae non-LTRs in the R1 clade do not have a canonical polyadenylation signal but have a poly (dA) tail in the genomic sequence. Several noncanonical poly (A) signal sequences have been documented (Graber et al. 1999; MacDonald and Redondo 2002), which could explain what is observed in these R1 families, although no particular candidate sequence could be found. In addition, the D. melanogaster I factor may have been using its TAA tandem repeat as a poly (A) signal (Chambeyron, Bucheton, and Busseau 2002). Finally, there are other non-LTR families in A. gambiae from the Jockey, L2, Outcast, and RTE clades that have neither poly(A) signals nor poly(dA) tails. If these families use cryptic polyadenylation signals, their poly(A) tails must have not been part of the reverse transcription reaction, or at least were not used as templates. Analysis of the 3′ regions of the transcripts from A. gambiae non-LTRs may help clarify RNA processing for these different groups, provided that transcripts of functional copies can be obtained. The human L1s are so far the only non-LTR families that are shown to be responsible for 3′ transduction and pseudogene formation because they recognize the poly(A) tail for reverse transcription (Esnault, Maestre, and Heidmann 2000; Wei et al. 2001). It will be interesting to determine whether A. gambiae L1s and other poly (dA) non-LTRs are capable of 3′ transduction or creating processed pseudogenes, which may determine whether poly (dA) non-LTRs had a broader genomic impact in species other than mammals.
The 3′ tandem repeat may originate from a telomerase-like activity of the retrotransposition machinery (Chaboissier, Finnegan, and Bucheton 2000). Such a mechanism could explain the conservation of the 3′ repeat sequence between some closely related non-LTR families (table 1 and Appendix A). However, there is also a high degree of variability of repeat sequences among some closely related families, as well as variability in the number of tandem repeats in different copies of a given family. The apparent exceptions to the conservation suggest that the specific tandem repeat sequences may not be required for retrotransposition, which is consistent with previous hypothesis by Chaboissier, Finnegan, and Bucheton (2000). However, the 3′ tandem repeats may influence retrotransposition in other ways. For example, Chambeyron, Bucheton, and Busseau (2002) showed that the TAA repeats of D. melanogaster I factor directed the precise initiation of reverse transcription. It was also suggested that the tandem repeats could play a role in target site specificity (Chaboissier, Finnegan, and Bucheton 2000).
Non-LTRs and Nonautonomous Retroelements
Many non-LTR families contain 5′ truncated copies, although these 5′ truncations most likely will not become a distinct unit of retrotransposition because of the lack of promoter. In this study we have discovered a non-LTR family Ag-Sponge that has a large internal deletion and that presumably is derived from Ag-CR1-3. We have shown that Ag-Sponge has been an intact unit of successful retrotransposition, probably by “borrowing” the protein machinery from Ag-CR1-3. This discovery underscores the potential for retrotransposition machinery to act in trans (Jensen et al. 1994; Wei et al. 2001). Two other nonautonomous non-LTRs, Het-A of D. melanogaster and CM-gag of Culex pipiens, have been reported which encode only the Gag-like protein (Biessmann et al. 1992; Bensaadi-Merchermek et al. 1997).
Short interspersed nuclear elements (SINEs) are another type of nonautonomous retrotransposons, which presumably also “borrow” the retrotransposition machinery from “partner” non-LTRs. However, SINEs are different from Ag-Sponge because they have a composite structure and use Pol III promoters. A highly repetitive SINE family named Ag-SINE200 that has been identified in A. gambiae has an apparent AAG tandem repeat at the 3′ end (Holt et al. 2002). Although several non-LTRs sharing the same 3′ tandem repeat have been identified (table 1 and appendix A), no significant similarity was found between Ag-SINE200 and the non-LTRs beyond the tandem repeats.
Potential Applications of A. gambiae Non-LTRs
A. gambiae is the most important vector of malaria, a disease that is responsible for more than a million deaths every year. Vector control, a major component of malaria control strategies, is hampered by increasing insecticide resistance and the genetic heterogeneity of the vector complex. One novel approach is actively being pursued which aims to replace vector mosquitoes in wild populations with genetically modified mosquitoes that are incompetent disease vectors. Our analysis of A. gambiae non-LTRs may contribute to the genetic strategy to control mosquito-borne diseases by providing transformation tools, gene-driving mechanisms, and genetic markers for population studies. One major challenge to establishing sophisticated vector control programs and meaningful epidemiological studies has been the genetic complexities in A. gambiae populations (Powell et al. 1999). Several recent studies using a number of genetic markers have made significant progress toward illustrating this complexity while pointing to the need for more extensive research (e.g., Black and Lanzaro 2001; della Torre et al. 2001). The development of new population genomic approaches is needed because conflict exists between results obtained using different types of genetic markers and markers at different genomic locations (Besansky, 1999). Because many non-LTR families are interspersed throughout the genome, and because relatively high levels of insertion polymorphisms are expected on the basis of evidence for recent retrotransposition, non-LTRs are great sources of polymorphic markers across different regions of the A. gambiae genome. Like the human Alu elements, these polymorphic insertion sites provide co-dominant markers when sequences flanking a TE at the specific locus are used as primers for PCR amplification of genomic DNA isolated from an individual sample (e.g., Batzer et al. 1994; Stoneking et al. 1997; Roy-Engel et al. 2001). Like Alu, inserted non-LTR copies are not subject to excision by transposition, which makes them ideal for use as markers in population genetics because the ancestral state is known and is non-insertion. Furthermore, non-LTRs can be exploited as gene vectors. For example, the human L1 has been used to make an retrotransposon-adenovirus hybrid vector capable of efficient delivery of stably intergrated transgenes (Soifer et al. 2001). Because non-LTRs transpose through a mechanism completely different from DNA-mediated TEs that are currently pursued as major vectors for the genetic manipulation of mosquitoes, non-LTRs can possibly be developed as alternative vectors with different features. For example, retrotransposed copies are not subject to further excision, which could make them more stable than DNA-mediated TE vectors. As mosquito biology is entering the post-genomic era, these potential new tools of population analysis and genetic manipulation will undoubtedly contribute to a better understanding of these important disease vectors and help toward an informed and sustainable control strategy.
Thomas Eickbush, Associate Editor
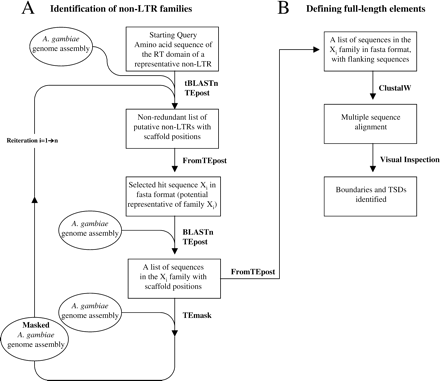
Strategy for the identification and characterization of all non-LTR retrotransposons in the A. gambiae genome. A. Identification of non-LTR families. B. Defining full-length elements. Circles indicate databases used for searches. Rectangles indicate input/output files. Programs used are written in bold beside arrows. See Materials and Methods for details such as E-value and other parameters. TSDs, target site duplications. Note that some non-LTRs are not associated with TSDs
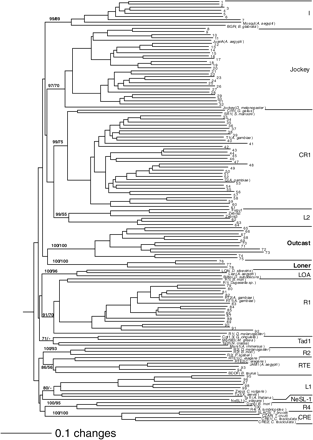
Phylogenetic analysis classifies A. gambiae non-LTRs into two new clades and eight previously defined clades. The two new clades, Loner and Outcast, are in bold. Shown here is the Neighbor-Joining tree constructed using alignment of approximately 260 amino acids of the RT domain from non-LTR retrotransposons of A. gambiae, A. aegypti, and divergent non-mosquito and non-insect species. The tree was rooted using RTs of three prokaryotic group II introns (not shown, Malik, Burke, and Eickbush 1999). Maximum parsimony was also used, and it produced a similar phylogenetic tree (not shown). Confidence of the groupings was estimated using 500 bootstrap replications for both methods. Each Arabic numeral at the base of a node is the bootstrap value that represents percent of times out of 500 bootstrap re-samplings that branches were grouped together at a particular node. The first and second numbers at a particular node represent the bootstrap values derived from Neighbor-Joining and maximum parsimony analysis, respectively. Only the values for the major groupings (clades) that are above 50% are shown. The scale at bottom left indicates amino acid divergence. The names of elements from previously established clades are given, but names of new A. gambiae non-LTR families are omitted to save space (see table 1 and Appendix A for family name). Previously reported A. gambiae non-LTRs in the tree are Q, T1, RT1, and RT2 (Besansky 1990; Besansky et al. 1992; Besansky, Bedell, and Mukabayire 1994). Non-LTRs from other mosquito species are MosquI, Lian, JAM1, and JuanA from Aedes aegypti (Mouches, Bensaadi, and Salvado 1992; Warren, Hughes, and Crampton 1997; Tu, Isoe, and Guzova 1998; Tu and Hill 1999). Amino acid sequences from previously reported non-LTRs used in this phylogenetic analysis were obtained from an alignment produced by Malik, Burke, and Eickbush (1999). Accession numbers with corresponding positions for other sequences are as follows: Fugu1 (AJ459419, 1929–1123); Zebra2 (AL591210, 72154–72957); Zebra3 (AL831768, 51653–52456). Representatives of the recently described Ingi and Rex1 clades are not included in the tree. We performed phylogenetic analysis including representatives of the above two clades, and the resulting groupings were unchanged (not shown)
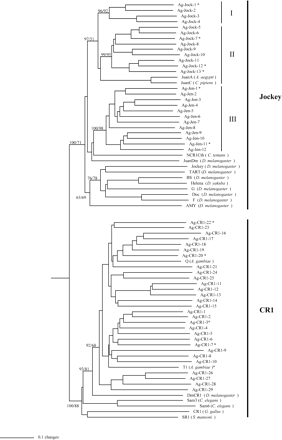
The Jockey and CR1 clades contain a high level of intracladal diversity. Shown here is the phylogeny derived from the Neighbor-Joining analysis of non-LTRs in the Jockey and CR1 clades in A. gambiae and other species. The tree was rooted using elements of the CRE clade (see fig. 1). All other phylogenetic analysis parameters are as in figure 1. Asterisks indicate A. gambiae families that have evidence of recent activity based on sequence analysis and hits to A. gambiae ESTs (see table 1). Bootstrap values for 500 replicates are displayed for Neighbor-Joining and maximum parisomony methods, respectively. Values are shown only for major branches. Amino acid sequences from previously reported non-LTRs used in this phylogenetic analysis were obtained from an alignment produced by Malik, Burke, and Eickbush (1999). Accession numbers for the sequences of JuanDm and DmCR1 were obtained from Berezikov, Bucheton, and Busseau (2000)
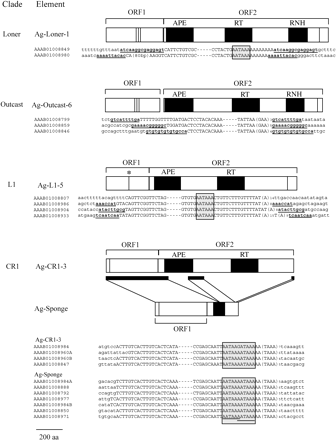
Schematic showing structural features of selected non-LTR elements of interest. See Malik, Burke, and Eickbush (1999) for comparison to elements from other clades. Open boxes represent open reading frames (ORFs) and filled boxes represent domains (APE, RT, and RNH, indicate AP endonuclease, reverse transcriptase, and RNase H, respectively). Vertical bars indicate the location of cysteine-hystidine motifs typical of nucleic acid–binding domains. An asterisk is located above the cysteine-histidine motif in ORF1 of Ag-L1-5 to highlight this unique feature for this family. TSDs are in bold and underlined. The canonical polyadenylation signal is shaded in gray, and two potential overlapping signals in the CR1/Sponge elements are underlined. TSDs were not identified for Ag-CR1-3 or Ag-Sponge. The inability to find TSDs in mosquito elements of the CR1 clade has been previously noted (Besansky 1990; Besansky, Bedell, and Mukabayire 1994). Flanking sequence letters are in lowercase. Filled bars under Ag-CR1-3 indicate regions of this element that are retained in Sponge. In the alignments, dashes indicate omitted internal sequence. Accession numbers with coordinates corresponding to the full-length elements in the figure are as follows: AAAB01008849, 2633065–2639407; AAAB01008980, 2507563–2513883; AAAB01008799, 2183752–2190172; AAAB01008859, 5480435–5486855; AAAB01008846, 1481833–1488240; AAAB01008807, 10233605–10238070; AAAB01008986, 2250740–2255203; AAAB01008904, 1312097- 1316560; AAAB01008933, 2055049–2059525; AAAB01008984, 7831634–7836275; AAAB01008960A, 3142131–3146786; AAAB01008960B, 6376559–6381153; AAAB01008847, 2204110–2208769; AAAB01008984A, 9556504–9558865; AAAB01008888, 1828526–1830888; AAAB01008792, 7850–10207; AAAB01008977, 60152–62512; AAAB01008984B, 2896406–2898768; AAAB01008850, 376190–378552; AAAB01008971, 196324–198686
Classification and Characteristics of Recently Active Non-LTRs in A. gambiae.
| Clade | Family | # in fig. 2 | 3′ Repeat | Poly A Signalb | Poly A Tail | Length (kb) | TSDs–bpc (# copies) | # Copies with Intact ORFs/FLe Copies | % Identity of FLe Copies Compared)f | Copy numberg |
|---|---|---|---|---|---|---|---|---|---|---|
| I | Ag-I-2 | 2 | CAA | Yes | No | 5.6 | 10(1), 12(1), 13(2) | 4/4 | 99.9(4) | 48 |
| Jockey | Ag-Jock-1 | 8 | Not present | Yes | Yes | 4.7 | 13(2), 15(1) | 3/3 | 99.9(3) | 19 |
| Ag-Jock-7 | 14 | CAAT | Yes | No | 4.3 | 12(2) | 2/2 | 99.9(2)h | 16 | |
| Ag-Jock-12 | 19 | AACT | No | No | 4.3 | 13(1), 14(1), 15(1) | 3/3 | 99.8–99.9(3) | 27 | |
| Ag-Jock-13a | 20 | AT | Yes | No | 4.6 | 12(1), 14(1) | 1/1 | 99.9(4)i | 29 | |
| Ag-Jen-1 | 21 | CTA | Yes | No | 4.3 | 12(1), 13(1), 15(2), 16(1) | 5/5 | 99.7–99.9(5) | 25 | |
| Ag-Jen-11 | 31 | TTAC | Yes | No | 4.2 | 12(1), 14(1), 15(1) | 0/3 | 97.2–99.8(3) | 25 | |
| CR1 | Ag-CR1-3a | 35 | TAAA | Yes | No | 4.7 | NDd | 2/4 | 99.3–99.6(4) | 228 |
| Ag-CR1-7 | 39 | TATGAA | Yes | No | 4.3 | NDd | 2/3 | 98.9–99.8(3) | 209 | |
| T1a | TGAAA | Yes | No | 4.6 | NDd | 7/9 | 99.4–99.9(9) | 301 | ||
| Ag-CR1-20a | 52 | CAAATAA | Yes | No | 4.5 | NDd | 4/4 | 99.5–99.9(4) | 184 | |
| Ag-CR1-22 | 54 | CAAATTAAA | Yes | No | 4.5 | NDd | 1/4 | 99.3–99.8(4)j | 92 | |
| L2 | Ag-L2-1 | 62 | TCA | No | No | 5.2 | NDd | 0/2 | 99.9(2) | 86 |
| Outcast | Ag-Outcast-2 | 66 | GAA | No | No | 5.4 | 12(1), 15(1) | 0/2 | 99.4(2) | 14 |
| Ag-Outcast-5 | 69 | AAG | No | No | 5.0 | 12(3) | 2/3 | 99.8–99.9(3) | 27 | |
| Ag-Outcast-6 | 70 | GAA | No | No | 6.4 | 12(2), 14(2) | 3/3 | 99.9–100(4)k | 24 | |
| Loner | Ag-Loner-1a | 76 | Not present | Yes | Yes | 6.3 | 10(1), 15(1) | 0/2 | 99.3(2) | 86 |
| RTE | Ag-JAMMIN-1 | 93 | AAG | No | No | 3.4 | 10(1), 11(1), 12(2) | 3/4 | 96.4–99.9(4)l | 519 |
| Ag-JAMMIN-2a | 94 | TAAG | No | No | 3.3 | 9(2), 10(2), 13(1), 14(2) | 5/7 | 97.8–99.6(7)m | 1950 | |
| L1 | Ag-L1-2 | 96 | Not present | Yes | Yes | 4.9 | 6(2) | 1/2 | 99.7(2) | 59 |
| Ag-L1-5a | 99 | Not present | Yes | Yes | 4.5 | 7(1), 8(1), 9(1) | 3/4 | 99.7–99.9(4)n | 27 |
| Clade | Family | # in fig. 2 | 3′ Repeat | Poly A Signalb | Poly A Tail | Length (kb) | TSDs–bpc (# copies) | # Copies with Intact ORFs/FLe Copies | % Identity of FLe Copies Compared)f | Copy numberg |
|---|---|---|---|---|---|---|---|---|---|---|
| I | Ag-I-2 | 2 | CAA | Yes | No | 5.6 | 10(1), 12(1), 13(2) | 4/4 | 99.9(4) | 48 |
| Jockey | Ag-Jock-1 | 8 | Not present | Yes | Yes | 4.7 | 13(2), 15(1) | 3/3 | 99.9(3) | 19 |
| Ag-Jock-7 | 14 | CAAT | Yes | No | 4.3 | 12(2) | 2/2 | 99.9(2)h | 16 | |
| Ag-Jock-12 | 19 | AACT | No | No | 4.3 | 13(1), 14(1), 15(1) | 3/3 | 99.8–99.9(3) | 27 | |
| Ag-Jock-13a | 20 | AT | Yes | No | 4.6 | 12(1), 14(1) | 1/1 | 99.9(4)i | 29 | |
| Ag-Jen-1 | 21 | CTA | Yes | No | 4.3 | 12(1), 13(1), 15(2), 16(1) | 5/5 | 99.7–99.9(5) | 25 | |
| Ag-Jen-11 | 31 | TTAC | Yes | No | 4.2 | 12(1), 14(1), 15(1) | 0/3 | 97.2–99.8(3) | 25 | |
| CR1 | Ag-CR1-3a | 35 | TAAA | Yes | No | 4.7 | NDd | 2/4 | 99.3–99.6(4) | 228 |
| Ag-CR1-7 | 39 | TATGAA | Yes | No | 4.3 | NDd | 2/3 | 98.9–99.8(3) | 209 | |
| T1a | TGAAA | Yes | No | 4.6 | NDd | 7/9 | 99.4–99.9(9) | 301 | ||
| Ag-CR1-20a | 52 | CAAATAA | Yes | No | 4.5 | NDd | 4/4 | 99.5–99.9(4) | 184 | |
| Ag-CR1-22 | 54 | CAAATTAAA | Yes | No | 4.5 | NDd | 1/4 | 99.3–99.8(4)j | 92 | |
| L2 | Ag-L2-1 | 62 | TCA | No | No | 5.2 | NDd | 0/2 | 99.9(2) | 86 |
| Outcast | Ag-Outcast-2 | 66 | GAA | No | No | 5.4 | 12(1), 15(1) | 0/2 | 99.4(2) | 14 |
| Ag-Outcast-5 | 69 | AAG | No | No | 5.0 | 12(3) | 2/3 | 99.8–99.9(3) | 27 | |
| Ag-Outcast-6 | 70 | GAA | No | No | 6.4 | 12(2), 14(2) | 3/3 | 99.9–100(4)k | 24 | |
| Loner | Ag-Loner-1a | 76 | Not present | Yes | Yes | 6.3 | 10(1), 15(1) | 0/2 | 99.3(2) | 86 |
| RTE | Ag-JAMMIN-1 | 93 | AAG | No | No | 3.4 | 10(1), 11(1), 12(2) | 3/4 | 96.4–99.9(4)l | 519 |
| Ag-JAMMIN-2a | 94 | TAAG | No | No | 3.3 | 9(2), 10(2), 13(1), 14(2) | 5/7 | 97.8–99.6(7)m | 1950 | |
| L1 | Ag-L1-2 | 96 | Not present | Yes | Yes | 4.9 | 6(2) | 1/2 | 99.7(2) | 59 |
| Ag-L1-5a | 99 | Not present | Yes | Yes | 4.5 | 7(1), 8(1), 9(1) | 3/4 | 99.7–99.9(4)n | 27 |
Note.—Numbers are excluded for the previously described non-LTR T1 (Besansky 1990). Only families having full-length copies with at least 99% identity were included.
aFamily that showed a significant hit to an EST.
bA poly (A) signal was considered present when the canonical sequence AATAAA was found conserved in the 3′ end of a family.
cColumn describing target site duplications (TSDs) lists length of TSD and number of copies in parentheses.
dND, not determined. It has been noted that TSDs could not be found in A. gambiae non-LTRs T1 and Q of the CR1 clade (Besansky 1990; Besansky, Bedell, and Mukabayire 1994). This likely explains the inability to find TSDs in other families of the CR1 clade and the closely related L2 clade.
eFL, full-length.
fPercent identities are listed with number of copies in parenthesis and identity values represent percent identity to the consensus when more than two sequences were used.
gCopy numbers were determined using nucleotide sequences of full-length elements or the longest obtainable sequence for a family (including the 3′ terminus when possible).
hA stretch of 20Ns are in the 3′ UTR of one sequence used for comparison.
iOnly one putative full-length sequence was found having two intact ORFs. The other three sequences were 5′ truncated due to presumably incomplete reverse transcription or the presence of ambiguous sequence denoted by Ns.
jOne sequence has a stretch of 61 Ns.
kOne sequence used for comparison was not used to construct the consensus because it is 5′ truncated and has a stretch of 496 Ns.
lOne sequence has a stretch of 59 Ns.
mTwo sequences have Ns (20 and 33).
nTSDs could not be found for one full-length copy.
Classification and Characteristics of Recently Active Non-LTRs in A. gambiae.
| Clade | Family | # in fig. 2 | 3′ Repeat | Poly A Signalb | Poly A Tail | Length (kb) | TSDs–bpc (# copies) | # Copies with Intact ORFs/FLe Copies | % Identity of FLe Copies Compared)f | Copy numberg |
|---|---|---|---|---|---|---|---|---|---|---|
| I | Ag-I-2 | 2 | CAA | Yes | No | 5.6 | 10(1), 12(1), 13(2) | 4/4 | 99.9(4) | 48 |
| Jockey | Ag-Jock-1 | 8 | Not present | Yes | Yes | 4.7 | 13(2), 15(1) | 3/3 | 99.9(3) | 19 |
| Ag-Jock-7 | 14 | CAAT | Yes | No | 4.3 | 12(2) | 2/2 | 99.9(2)h | 16 | |
| Ag-Jock-12 | 19 | AACT | No | No | 4.3 | 13(1), 14(1), 15(1) | 3/3 | 99.8–99.9(3) | 27 | |
| Ag-Jock-13a | 20 | AT | Yes | No | 4.6 | 12(1), 14(1) | 1/1 | 99.9(4)i | 29 | |
| Ag-Jen-1 | 21 | CTA | Yes | No | 4.3 | 12(1), 13(1), 15(2), 16(1) | 5/5 | 99.7–99.9(5) | 25 | |
| Ag-Jen-11 | 31 | TTAC | Yes | No | 4.2 | 12(1), 14(1), 15(1) | 0/3 | 97.2–99.8(3) | 25 | |
| CR1 | Ag-CR1-3a | 35 | TAAA | Yes | No | 4.7 | NDd | 2/4 | 99.3–99.6(4) | 228 |
| Ag-CR1-7 | 39 | TATGAA | Yes | No | 4.3 | NDd | 2/3 | 98.9–99.8(3) | 209 | |
| T1a | TGAAA | Yes | No | 4.6 | NDd | 7/9 | 99.4–99.9(9) | 301 | ||
| Ag-CR1-20a | 52 | CAAATAA | Yes | No | 4.5 | NDd | 4/4 | 99.5–99.9(4) | 184 | |
| Ag-CR1-22 | 54 | CAAATTAAA | Yes | No | 4.5 | NDd | 1/4 | 99.3–99.8(4)j | 92 | |
| L2 | Ag-L2-1 | 62 | TCA | No | No | 5.2 | NDd | 0/2 | 99.9(2) | 86 |
| Outcast | Ag-Outcast-2 | 66 | GAA | No | No | 5.4 | 12(1), 15(1) | 0/2 | 99.4(2) | 14 |
| Ag-Outcast-5 | 69 | AAG | No | No | 5.0 | 12(3) | 2/3 | 99.8–99.9(3) | 27 | |
| Ag-Outcast-6 | 70 | GAA | No | No | 6.4 | 12(2), 14(2) | 3/3 | 99.9–100(4)k | 24 | |
| Loner | Ag-Loner-1a | 76 | Not present | Yes | Yes | 6.3 | 10(1), 15(1) | 0/2 | 99.3(2) | 86 |
| RTE | Ag-JAMMIN-1 | 93 | AAG | No | No | 3.4 | 10(1), 11(1), 12(2) | 3/4 | 96.4–99.9(4)l | 519 |
| Ag-JAMMIN-2a | 94 | TAAG | No | No | 3.3 | 9(2), 10(2), 13(1), 14(2) | 5/7 | 97.8–99.6(7)m | 1950 | |
| L1 | Ag-L1-2 | 96 | Not present | Yes | Yes | 4.9 | 6(2) | 1/2 | 99.7(2) | 59 |
| Ag-L1-5a | 99 | Not present | Yes | Yes | 4.5 | 7(1), 8(1), 9(1) | 3/4 | 99.7–99.9(4)n | 27 |
| Clade | Family | # in fig. 2 | 3′ Repeat | Poly A Signalb | Poly A Tail | Length (kb) | TSDs–bpc (# copies) | # Copies with Intact ORFs/FLe Copies | % Identity of FLe Copies Compared)f | Copy numberg |
|---|---|---|---|---|---|---|---|---|---|---|
| I | Ag-I-2 | 2 | CAA | Yes | No | 5.6 | 10(1), 12(1), 13(2) | 4/4 | 99.9(4) | 48 |
| Jockey | Ag-Jock-1 | 8 | Not present | Yes | Yes | 4.7 | 13(2), 15(1) | 3/3 | 99.9(3) | 19 |
| Ag-Jock-7 | 14 | CAAT | Yes | No | 4.3 | 12(2) | 2/2 | 99.9(2)h | 16 | |
| Ag-Jock-12 | 19 | AACT | No | No | 4.3 | 13(1), 14(1), 15(1) | 3/3 | 99.8–99.9(3) | 27 | |
| Ag-Jock-13a | 20 | AT | Yes | No | 4.6 | 12(1), 14(1) | 1/1 | 99.9(4)i | 29 | |
| Ag-Jen-1 | 21 | CTA | Yes | No | 4.3 | 12(1), 13(1), 15(2), 16(1) | 5/5 | 99.7–99.9(5) | 25 | |
| Ag-Jen-11 | 31 | TTAC | Yes | No | 4.2 | 12(1), 14(1), 15(1) | 0/3 | 97.2–99.8(3) | 25 | |
| CR1 | Ag-CR1-3a | 35 | TAAA | Yes | No | 4.7 | NDd | 2/4 | 99.3–99.6(4) | 228 |
| Ag-CR1-7 | 39 | TATGAA | Yes | No | 4.3 | NDd | 2/3 | 98.9–99.8(3) | 209 | |
| T1a | TGAAA | Yes | No | 4.6 | NDd | 7/9 | 99.4–99.9(9) | 301 | ||
| Ag-CR1-20a | 52 | CAAATAA | Yes | No | 4.5 | NDd | 4/4 | 99.5–99.9(4) | 184 | |
| Ag-CR1-22 | 54 | CAAATTAAA | Yes | No | 4.5 | NDd | 1/4 | 99.3–99.8(4)j | 92 | |
| L2 | Ag-L2-1 | 62 | TCA | No | No | 5.2 | NDd | 0/2 | 99.9(2) | 86 |
| Outcast | Ag-Outcast-2 | 66 | GAA | No | No | 5.4 | 12(1), 15(1) | 0/2 | 99.4(2) | 14 |
| Ag-Outcast-5 | 69 | AAG | No | No | 5.0 | 12(3) | 2/3 | 99.8–99.9(3) | 27 | |
| Ag-Outcast-6 | 70 | GAA | No | No | 6.4 | 12(2), 14(2) | 3/3 | 99.9–100(4)k | 24 | |
| Loner | Ag-Loner-1a | 76 | Not present | Yes | Yes | 6.3 | 10(1), 15(1) | 0/2 | 99.3(2) | 86 |
| RTE | Ag-JAMMIN-1 | 93 | AAG | No | No | 3.4 | 10(1), 11(1), 12(2) | 3/4 | 96.4–99.9(4)l | 519 |
| Ag-JAMMIN-2a | 94 | TAAG | No | No | 3.3 | 9(2), 10(2), 13(1), 14(2) | 5/7 | 97.8–99.6(7)m | 1950 | |
| L1 | Ag-L1-2 | 96 | Not present | Yes | Yes | 4.9 | 6(2) | 1/2 | 99.7(2) | 59 |
| Ag-L1-5a | 99 | Not present | Yes | Yes | 4.5 | 7(1), 8(1), 9(1) | 3/4 | 99.7–99.9(4)n | 27 |
Note.—Numbers are excluded for the previously described non-LTR T1 (Besansky 1990). Only families having full-length copies with at least 99% identity were included.
aFamily that showed a significant hit to an EST.
bA poly (A) signal was considered present when the canonical sequence AATAAA was found conserved in the 3′ end of a family.
cColumn describing target site duplications (TSDs) lists length of TSD and number of copies in parentheses.
dND, not determined. It has been noted that TSDs could not be found in A. gambiae non-LTRs T1 and Q of the CR1 clade (Besansky 1990; Besansky, Bedell, and Mukabayire 1994). This likely explains the inability to find TSDs in other families of the CR1 clade and the closely related L2 clade.
eFL, full-length.
fPercent identities are listed with number of copies in parenthesis and identity values represent percent identity to the consensus when more than two sequences were used.
gCopy numbers were determined using nucleotide sequences of full-length elements or the longest obtainable sequence for a family (including the 3′ terminus when possible).
hA stretch of 20Ns are in the 3′ UTR of one sequence used for comparison.
iOnly one putative full-length sequence was found having two intact ORFs. The other three sequences were 5′ truncated due to presumably incomplete reverse transcription or the presence of ambiguous sequence denoted by Ns.
jOne sequence has a stretch of 61 Ns.
kOne sequence used for comparison was not used to construct the consensus because it is 5′ truncated and has a stretch of 496 Ns.
lOne sequence has a stretch of 59 Ns.
mTwo sequences have Ns (20 and 33).
nTSDs could not be found for one full-length copy.
The A. gambiae Genome Contains a Greater Diversity of Non-LTRs Than Reported for Any Other Genome.
| Organism | Haploid Genome Size (MB) | Number of Clades Found | Clades |
|---|---|---|---|
| Giardia lambliaa | 12 | 1 | Close to NeSL-1/R2 |
| Candidas albicansb | 12 | 1 | L1 |
| Saccharomyces cerevisiaec | 12 | 0 | |
| Caenorhabditis elegansd | 100 | 3 | CR1, NeSL, RTE |
| Arabidopsis thalianae | 100 | 1 | L1 |
| Drosophila melanogasterf | 165 | 6 | R2, CR1, LOA, R1, Jockey, I |
| Anopheles gambiae | 278 | 10 | CR1, L1, R4, RTE, R1, Jockey, I, L2, Outcast, Loner |
| Fugu rubripesg | 365 | 4 | L1, L2, R4, RTE, Rex1 |
| Homo sapiensh | 3,400 | 2 | L1, L2 |
| Organism | Haploid Genome Size (MB) | Number of Clades Found | Clades |
|---|---|---|---|
| Giardia lambliaa | 12 | 1 | Close to NeSL-1/R2 |
| Candidas albicansb | 12 | 1 | L1 |
| Saccharomyces cerevisiaec | 12 | 0 | |
| Caenorhabditis elegansd | 100 | 3 | CR1, NeSL, RTE |
| Arabidopsis thalianae | 100 | 1 | L1 |
| Drosophila melanogasterf | 165 | 6 | R2, CR1, LOA, R1, Jockey, I |
| Anopheles gambiae | 278 | 10 | CR1, L1, R4, RTE, R1, Jockey, I, L2, Outcast, Loner |
| Fugu rubripesg | 365 | 4 | L1, L2, R4, RTE, Rex1 |
| Homo sapiensh | 3,400 | 2 | L1, L2 |
aArkhipova and Morrison 2001;
bGoodwin et al. 2001;
cKim et al. 1998;
dMalik and Eickbush 2000, Marin et al. 1998, and Youngman 1996;
eWright et al. 1996;
The A. gambiae Genome Contains a Greater Diversity of Non-LTRs Than Reported for Any Other Genome.
| Organism | Haploid Genome Size (MB) | Number of Clades Found | Clades |
|---|---|---|---|
| Giardia lambliaa | 12 | 1 | Close to NeSL-1/R2 |
| Candidas albicansb | 12 | 1 | L1 |
| Saccharomyces cerevisiaec | 12 | 0 | |
| Caenorhabditis elegansd | 100 | 3 | CR1, NeSL, RTE |
| Arabidopsis thalianae | 100 | 1 | L1 |
| Drosophila melanogasterf | 165 | 6 | R2, CR1, LOA, R1, Jockey, I |
| Anopheles gambiae | 278 | 10 | CR1, L1, R4, RTE, R1, Jockey, I, L2, Outcast, Loner |
| Fugu rubripesg | 365 | 4 | L1, L2, R4, RTE, Rex1 |
| Homo sapiensh | 3,400 | 2 | L1, L2 |
| Organism | Haploid Genome Size (MB) | Number of Clades Found | Clades |
|---|---|---|---|
| Giardia lambliaa | 12 | 1 | Close to NeSL-1/R2 |
| Candidas albicansb | 12 | 1 | L1 |
| Saccharomyces cerevisiaec | 12 | 0 | |
| Caenorhabditis elegansd | 100 | 3 | CR1, NeSL, RTE |
| Arabidopsis thalianae | 100 | 1 | L1 |
| Drosophila melanogasterf | 165 | 6 | R2, CR1, LOA, R1, Jockey, I |
| Anopheles gambiae | 278 | 10 | CR1, L1, R4, RTE, R1, Jockey, I, L2, Outcast, Loner |
| Fugu rubripesg | 365 | 4 | L1, L2, R4, RTE, Rex1 |
| Homo sapiensh | 3,400 | 2 | L1, L2 |
aArkhipova and Morrison 2001;
bGoodwin et al. 2001;
cKim et al. 1998;
dMalik and Eickbush 2000, Marin et al. 1998, and Youngman 1996;
eWright et al. 1996;
TEpost, TEcombine, TEmask, and FromTEpost were implemented by Feng Zhang of the Department of Computer Science at Virginia Tech. We thank Chunhong Mao at the Virginia Bioinformatics Institute for help with the programming and design. This work was supported by National Institutes of Health grant AI42121, by a grant from the Jeffres Foundation to Z. T., and by the Agricultural Experimental Station at Virginia Tech.
Literature Cited
Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman.
Aparicio, S., J. Chapman, E. Stupka, N. Putnam, J. M. Chia, P. Dehal, A. Christoffels, S. Rash, S. Hoon, and A. Smit, et al.
Bao, Z., and S. R. Eddy.
Batzer, M. A., M. Stoneking, M. Alegria-Hartman, H. Bazan, D. H. Kass, T. H. Shaikh, G. E. Novick, P. A. Ioannou, W. D. Scheer, and R. J. Herrera, et al.
Bensaadi-Merchermek, N., C. Cagnon, I. Desmons, J. C. Salvado, S. Karama, F. D'Amico, and C. Mouches.
Berezikov, E., A. Bucheton, and I. Busseau.
Besansky, N. J.
Besansky, N. J.
Besansky, N. J., J. A. Bedell, and O. Mukabayire.
Besansky, N. J., S. M. Paskewitz, D. M. Hamm, and F. H. Collins.
Biessmann, H., K. Valgeirsdottir, A. Lofsky, C. Chin, B. Ginther, R. W. Levis, and M. L. Pardue.
Black, W. C. T., and G. C. Lanzaro.
Brosius, J.
Chaboissier, M. C., D. Finnegan, and A. Bucheton.
Chambeyron, S., A. Bucheton, and I. Busseau.
Della Torre, A., C. Fanello, M. Akogbeto, J. Dossou-yovo, G. Favia, V. Petrarca, and M. Coluzzi.
Doolittle, W. F., and C. Sapienza.
Eickbush, T. H., and H. S. Malik.
Esnault, C., J. Maestre, and T. Heidmann.
Finnegan, D. J.
Goodier, J. L., E. M. Ostertag, and H. H. Kazazian, Jr.
Graber, J. H., C. R. Cantor, S. C. Mohr, and T. F. Smith.
Hill, S. R., S. S. Leung, N. L. Quercia, D. Vasiliauskas, J. Yu, I. Pasic, D. Leung, A. Tran, and P. Romans.
Holmes, S. E., M. F. Singer, and G. D. Swergold.
Holt, R. A., G. M. Subramanian, and A. Halpern, et al. (123 co-authors).
Jensen, S., L. Cavarec, O. Dhellin, and T. Heidmann.
Kidwell, M. G., and D. R. Lisch.
Lander, E. S., L. M. Linton, and B. Birren, et al. (252 co-authors).
Lovsin, N., F. Gubensek, and D. Kordi.
Luan, D. D., M. H. Korman, J. L. Jakubczak, and T. H. Eickbush.
MacDonald, C. C., and J. L. Redondo.
Malik, H. S., W. D. Burke, and T. H. Eickbush.
Malik, H. S., and T. H. Eickbush.
Martin, S. L., and F. D. Bushman.
Mathews, D. H., A. R. Banerjee, D. D. Luan, T. H. Eickbush, and D. H. Turner.
Moran, J. V., R. J. DeBerardinis, and H. H. Kazazian, Jr.
Mouches, C., N. Bensaadi, and J. C. Salvado.
Pickeral, O. K., W. Makalowski, M. S. Boguski, and J. D. Boeke.
Powell, J. R., V. Petrarca, A. della Torre, A. Caccone, and M. Coluzzi.
Roy-Engel, A. M., M. L. Carroll, E. Vogel, R. K. Garber, S. V. Nguyen, A. H. Salem, M. A. Batzer, and P. L. Deininger.
Soifer, H., C. Higo, H. H. Kazazian, Jr., J. V. Moran, K. Mitani, and N. Kasahara.
Stoneking, M., J. J. Fontius, S. L. Clifford, H. Soodyall, S. S. Arcot, N. Saha, T. Jenkins, M. A. Tahir, P. L. Deininger, and M. A. Batzer.
Swofford, D. L.
Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins.
Thompson, J. D., D. G. Higgins, and T. J. Gibson.
Tu, Z., and J. J. Hill.
Tu, Z., J. Isoe, and J. A. Guzova.
Warren, A. M., M. A. Hughes, and J. M. Crampton.

{kind=link}
{kind=link}
{kind=link}
{kind=link}