





Abstract
TLRs are distributed in their characteristic cellular or subcellular compartments to efficiently recognize specific ligands and to initiate intracellular signaling. Whereas TLRs recognizing pathogen-associated lipids or proteins are localized to the cell surface, nucleic acid–sensing TLRs are expressed in endosomes and lysosomes. Several endoplasmic reticulum (ER)–resident proteins are known to regulate the trafficking of TLRs to the specific cellular compartments, thus playing important roles in the initiation of innate immune responses. In this study, we show that an ER-resident protein, Nogo-B (or RTN4-B), is necessary for immune responses triggered by nucleic acid–sensing TLRs, and that a newly identified Nogo-B–binding protein (glucosyltransferases, Rab-like GTPase activators and myotubularins [GRAM] domain containing 4 [GRAMD4]) negatively regulates the responses. Production of inflammatory cytokines in vitro by macrophages stimulated with CpG-B oligonucleotides or polyinosinic:polycytidylic acid was attenuated in the absence of Nogo-B, which was also confirmed in serum samples from Nogo-deficient mice injected with polyinosinic:polycytidylic acid. Although a deficiency of Nogo-B did not change the incorporation or delivery of CpG to endosomes, the localization of TLR9 to endolysosomes was found to be impaired. We identified GRAMD4 as a downmodulator for TLR9 response with a Nogo-B binding ability in ER, because our knockdown and overexpression experiments indicated that GRAMD4 suppresses the TLR9 response and knockdown of Gramd4 strongly enhanced the response in the absence of Nogo-B. Our findings indicate a critical role of Nogo-B and GRAMD4 in trafficking of TLR9.
Cells of eukaryotes include many organelles that partition the cytoplasm into compartments with specialized biological functions. The endoplasmic reticulum (ER) is one of the pivotal membrane-bounded organelles, where various biochemical processes take place, such as protein biosynthesis, folding and modifications, and calcium storage. Recent studies have indicated that ER-localized proteins regulate innate immunity and inflammatory responses by supporting receptor trafficking and transduction of intracellular signals (1, 2). In general, whereas pattern recognition receptors, including TLRs and cytosolic nucleic acid sensors such as retinoic acid–inducible protein I-like receptors, play critical roles in the innate immune system by detecting invading pathogens, each pattern recognition receptor molecule should be located in a specific subcellular compartment to adequately detect invading pathogens and to initiate its signaling to induce inflammatory responses (3, 4).
Currently, 10 and 12 TLRs have been identified in humans and mice, respectively. Each TLR recognizes unique microbial products such as lipids or proteins on the cell surface and nucleic acids within the cytosolic endosomal compartments (3–5). Recent studies have shown that some ER-localized proteins regulate TLR trafficking, albeit the mechanism by which different TLRs are transported to the plasma membrane or endosomal compartments following their biosynthesis in ER is not fully understood. For example, transportation of nucleic acid–sensing, intracellular TLRs is dependent on a transmembrane protein, UNC93B1 (6, 7). Nucleic acid–sensing TLRs, namely TLR3, 7, 8, and 9, bind to UNC93B1 after their synthesis in ER, whose binding is necessary for their posttranslational cleavage and delivery to endolysosomal compartments (8). A loss-of-function mutation of UNC93B1 resulted in decreased TLR3-, 7-, and 9-mediated signaling due to the lack of trafficking of these receptors (9). In addition to UNC93B1, ER-resident proteins gp96 and protein associated with TLR4 A (PRAT4A) also regulate the exit of all TLR members from ER to their respective subcellular compartments (10, 11). Another ER protein, stimulator of IFN genes, a translocon-associated transmembrane protein (12, 13), binds cytoplasmic nucleic acids, particularly dsDNA, and plays a critical role in innate immune responses in a somewhat different way from those described above. Stimulator of IFN genes was shown to bind to cyclic GMP-AMP, a second messenger, produced upon detection of cytosolic DNA by cyclic GMP-AMP synthase, and to activate downstream signaling molecules such as TBK1 for the induction of type I IFNs and other antiviral genes (14, 15). Thus, some ER proteins execute crucial functions to initiate innate immune responses.
Neurite outgrowth inhibitory protein (Nogo, also known as Reticulon 4 or RTN4) is a transmembrane protein localized preferentially in ER and was originally identified as an inhibitor of axonal regeneration in the CNS (16, 17). The Nogo isoforms A, B, and C are the major protein products translated from mRNA splice variants of the Rtn4 gene (16–18). These isoforms share a carboxyl terminal segment including a 66-aa sequence termed Nogo-66 flanked by two transmembrane regions (17). Although expression of Nogo-A and -C is found in the CNS, with Nogo-C being also detected in skeletal muscle, Nogo-B is expressed ubiquitously in most tissues (18). Nogo-A is expressed on the plasma membranes of oligodendrocytes and plays an inhibitory role in neurite outgrowth (17, 19). Studies of Nogo in the CNS suggested the presence of binding partners of the Nogo protein, one of which has been identified as Nogo receptor 1 (NgR1) (20). NgR1 forms a complex with other transmembrane proteins, LINGO1, p75, and TROY, for transduction of inhibitory signals in neurons. Through this protein complex, the Nogo–NgR1 interaction induces an increase in intracellular calcium and triggers activation of a small GTPase, RhoA, thereby leading to growth cone collapse (21, 22). Additionally, Paired Ig-like receptor (Pir)B identified in the immune system (23, 24) was recently found to bind to Nogo-66 in the CNS (25).
In addition to acting as an inhibitor of axonal sprouting, Nogo also contributes to maintain homeostasis in a nonneural system. For example, studies conducted using Nogo-A/B–deficient (nogo-A/B−⁄−) mice revealed that Nogo-B in lung epithelia regulates Th2-mediated inflammation, and endothelial Nogo-B regulates leukocyte transmigration to sites of inflammation (26–28). Although the links between Nogo and inflammatory responses are emerging, the role of Nogo in immune cells is virtually unknown.
In the present study, we show a regulatory role of Nogo-B in trafficking of the nucleic acid–sensing TLRs to endolysosomal compartments through interaction with a novel Nogo-B–binding, ER-resident protein (glucosyltransferases, Rab-like GTPase activators and myotubularins [GRAM] domain containing 4 [GRAMD4]). Nogo-A/B deficiency renders mice and cultured macrophages insensitive to stimulation with TLR9, TLR3, and TLR7 ligands. The attenuated innate responses were attributable to the loss of Nogo-B but not Nogo-A, because the A isoform was hardly detectable in immune cells in contrast to the abundant B isoform. Through mass spectrometry analysis, we identified GRAMD4 as a novel interacting partner of Nogo-B. GRAMD4 has been reported to be a proapoptotic protein (29, 30). Its function in immune responses, however, remained to be determined. Our knockdown and overexpression experiments on GRAMD4 in macrophages revealed inhibitory effects of GRAMD4 on TLR9- and TLR3-mediated responses. Our results suggest that Nogo-B and GRAMD4 are novel regulators of the nucleic acid–sensing TLR9 pathway and modulate innate immune responses potentially through the interaction between them.
Materials and Methods
Mice
C57BL/6 mice were purchased from CLEA Japan (Shizuoka, Japan). nogo-A/B−⁄− mice were developed by Stephen M. Strittmatter (Yale University). TLR9-deficient (Tlr9−⁄−) mice were purchased from Oriental BioService (Kyoto, Japan). Mice were maintained and bred in the animal facility of the Institute of Development, Aging and Cancer, Tohoku University, an environmentally controlled and specific pathogen-free facility, according to the guidelines for experimental animals defined by the university, and the animal protocols were reviewed and approved by the Animal Studies Committee of the university. All experiments were performed on 8- to 14-wk-old age-matched male and female mice.
Plasmid construction
pEF6/V5 (Invitrogen, San Diego, CA) was modified to an N-terminal FLAG or Myc-tagged vector (pEF6-FLAG, pEF6-Myc). Murine full-length Nogo-B2 cDNA was amplified by RT-PCR from total RNA prepared from splenocytes, and it was ligated into the cloning site of the pEF6-FLAG and pEGFP-N3 (C-terminal enhanced GFP tag, Clontech, Palo Alto, CA) vector. cDNA for murine GRAMD4 generated by PCR from cDNA derived from bone marrow–derived macrophages (BMMs) was ligated into the cloning site of the pEF6-Myc expression vector or pTagRFP-C (N-terminal red fluorescent protein [TagRFP] tag, Evrogen, Moscow, Russia). NF-κB reporter plasmid pELAM1-Luc (31) was constructed by inserting a fragment comprising +30 to −52 of the human E-selectin gene into pGL3-Basic (Promega, Madison, WI).
Cell culture
RAW264.7 (American Type Culture Collection, Manassas, VA) and 293T (RIKEN Cell Bank, Tsukuba, Japan) cells were maintained in DMEM (Sigma-Aldrich, St. Louis, MO) supplemented with penicillin/streptomycin and 10% FCS. For preparation of BMMs, bone marrow cells from femurs were cultured in DMEM containing penicillin/streptomycin, 10% FCS, and 20 ng/ml M-CSF (PeproTech, Rocky Hill, NJ). After 7 d, the adherent cells were used as BMMs. Mouse embryonic fibroblasts (MEFs) were isolated from 14.5-d-old embryos from wild-type mice. Briefly, the head and internal viscera were removed and thoroughly minced. The dissociated cells were cultured in complete medium (DMEM containing penicillin/streptomycin, 10% FCS, 2 mM GlutaMAX, 1 mM sodium pyruvate, and nonessential amino acids). After 2 d, adherent cells were harvested using a 0.25% trypsin-EDTA solution. To immortalize MEFs, cells were passaged continuously until the growth rate in a culture stabilized.
Serum cytokine measurements
Age- and sex-matched wild-type and nogo-A/B−⁄− mice were i.p. injected with polyinosinic:polycytidylic acid [poly(I:C)] (5 mg/kg body weight), LPS (15 mg/kg body weight), or saline. Serum samples were taken at 3 h after injection. IL-6 and IL-12p40 were determined with ELISA kits (BioLegend, San Diego, CA) according to the manufacturer’s specifications.
Identification of Nogo-B binding proteins
RAW264.7 cells stably expressing FLAG-Nogo-B2 or parental RAW264.7 cells were solubilized in lysis buffer (0.75% Brij 97, 50 mM HEPES [pH 7.4], 100 mM NaCl, and 10% glycerol) containing protease inhibitors (Sigma-Aldrich) and phosphatase inhibitors (Roche, Mannheim, Germany). Cell lysates were centrifuged at 12,000 × g for 10 min at 4°C. Each supernatant was incubated with anti-FLAG M2 agarose (Sigma-Aldrich). Beads were precipitated and washed with PBS, and the immunoprecipitate was eluted with 100 mM glycine-HCl (pH 2.7). To identify the proteins interacting with Nogo-B, a commercial isobaric tags for relative and absolute quantitation (iTRAQ) analysis service (Medical and Biological Laboratories, Nagoya, Japan) was used for mass spectrometric analysis. In brief, the eluents were concentrated and the buffer was exchanged for 50 mM triethylammonium acetate buffer. Protein mass was quantified with a BCA protein assay kit (Pierce, Rockford, IL). The reducing reagent and cysteine-blocking reagent were added, and then proteins (30 μg) present in the extract were digested with trypsin. The tryptic peptides were labeled with each isotope version in the iTRAQ reagents 4plex kit (AB Sciex, Framingham, MA). After iTRAQ labeling, the peptides were desalted and divided into eight fractions using AB Sciex CEX columns. Then iTRAQ-labeled peptides were analyzed by reverse-phase liquid chromatography (DiNa system, KYA Technologies, Tokyo, Japan) and mass spectrometry (TripleTOF 5600, AB Sciex).
Immunoblot analysis
BMMs were solubilized in lysis buffer (1% Nonidet P-40, 20 mM Tris-HCl [pH 7.3], 150 mM NaCl, 10 mM EDTA, and 10% glycerol) supplemented with protease inhibitors and phosphatase inhibitors. Cell lysates were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Each membrane was blotted with the specific Ab to the indicated protein and followed by visualization as chemiluminescence after incubation with ECL Plus Western blotting detection reagents (GE Healthcare, Piscataway, NJ). The bands were scanned and analyzed with an LAS-4000 mini image analyzer (Fujifilm, Tokyo, Japan).
Quantitative real-time PCR
Total RNA isolated with an RNeasy mini kit (Qiagen, Valencia, CA) was reverse transcribed with ReverTra Ace (Toyobo, Osaka, Japan). The cDNA was analyzed by quantitative PCR with SYBR GreenER qPCR SuperMix (Invitrogen) on a CFX Connect real-time PCR detection system (Bio-Rad, Hercules, CA). The primer sequences used in this study are presented in Table I.
Primer sequences for quantitative real-time PCR analysis
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| nogo-A | GCAGAGCTGAATAAAACTTCAG | GTTCACATGACCAAGAGCAG |
| nogo-B | GCAGGGGCTCGGGCTCAGTG | GTTCACATGACCAAGAGCAG |
| nogo-C | ATCAGAAGAAACGTTGGAAGG | GTTCACATGACCAAGAGCAG |
| Il6 | CCACGGCCTTCCCTAC | AGTGCATCATCGTTGTTC |
| Tnf | AAAATTCGAGTGACAAGCCTGTAG | CCCTTGAAGAGAACCTGGGAGTAG |
| Il12b | TGGTTTGCCATCGTTTTGCTG | ACAGGTGAGGTTCACTGTTTCT |
| Il1b | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| Il10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| Cxcl1 | ACTGCACCCAAACCGAAGTC | TGGGGACACCTTTTAGCATCTT |
| Ifnb | CAGCTCCAAGAAAGGACGAAC | GGCAGTGTAACTCTTCTGCAT |
| Gramd4 | CTCCCCGTCATGTCCAGGTG | CTGTTGAAGTCCTGCACGC |
| Unc93b1 | GGCTCCCTTGGACGAACTC | CACGTTCTTGACCACTCCGAG |
| Hprt1 | TCAGTCAACGGGGGACATAAA | GGGGCTGTACTGCTTAACCAG |
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| nogo-A | GCAGAGCTGAATAAAACTTCAG | GTTCACATGACCAAGAGCAG |
| nogo-B | GCAGGGGCTCGGGCTCAGTG | GTTCACATGACCAAGAGCAG |
| nogo-C | ATCAGAAGAAACGTTGGAAGG | GTTCACATGACCAAGAGCAG |
| Il6 | CCACGGCCTTCCCTAC | AGTGCATCATCGTTGTTC |
| Tnf | AAAATTCGAGTGACAAGCCTGTAG | CCCTTGAAGAGAACCTGGGAGTAG |
| Il12b | TGGTTTGCCATCGTTTTGCTG | ACAGGTGAGGTTCACTGTTTCT |
| Il1b | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| Il10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| Cxcl1 | ACTGCACCCAAACCGAAGTC | TGGGGACACCTTTTAGCATCTT |
| Ifnb | CAGCTCCAAGAAAGGACGAAC | GGCAGTGTAACTCTTCTGCAT |
| Gramd4 | CTCCCCGTCATGTCCAGGTG | CTGTTGAAGTCCTGCACGC |
| Unc93b1 | GGCTCCCTTGGACGAACTC | CACGTTCTTGACCACTCCGAG |
| Hprt1 | TCAGTCAACGGGGGACATAAA | GGGGCTGTACTGCTTAACCAG |
Primer sequences for quantitative real-time PCR analysis
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| nogo-A | GCAGAGCTGAATAAAACTTCAG | GTTCACATGACCAAGAGCAG |
| nogo-B | GCAGGGGCTCGGGCTCAGTG | GTTCACATGACCAAGAGCAG |
| nogo-C | ATCAGAAGAAACGTTGGAAGG | GTTCACATGACCAAGAGCAG |
| Il6 | CCACGGCCTTCCCTAC | AGTGCATCATCGTTGTTC |
| Tnf | AAAATTCGAGTGACAAGCCTGTAG | CCCTTGAAGAGAACCTGGGAGTAG |
| Il12b | TGGTTTGCCATCGTTTTGCTG | ACAGGTGAGGTTCACTGTTTCT |
| Il1b | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| Il10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| Cxcl1 | ACTGCACCCAAACCGAAGTC | TGGGGACACCTTTTAGCATCTT |
| Ifnb | CAGCTCCAAGAAAGGACGAAC | GGCAGTGTAACTCTTCTGCAT |
| Gramd4 | CTCCCCGTCATGTCCAGGTG | CTGTTGAAGTCCTGCACGC |
| Unc93b1 | GGCTCCCTTGGACGAACTC | CACGTTCTTGACCACTCCGAG |
| Hprt1 | TCAGTCAACGGGGGACATAAA | GGGGCTGTACTGCTTAACCAG |
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| nogo-A | GCAGAGCTGAATAAAACTTCAG | GTTCACATGACCAAGAGCAG |
| nogo-B | GCAGGGGCTCGGGCTCAGTG | GTTCACATGACCAAGAGCAG |
| nogo-C | ATCAGAAGAAACGTTGGAAGG | GTTCACATGACCAAGAGCAG |
| Il6 | CCACGGCCTTCCCTAC | AGTGCATCATCGTTGTTC |
| Tnf | AAAATTCGAGTGACAAGCCTGTAG | CCCTTGAAGAGAACCTGGGAGTAG |
| Il12b | TGGTTTGCCATCGTTTTGCTG | ACAGGTGAGGTTCACTGTTTCT |
| Il1b | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| Il10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| Cxcl1 | ACTGCACCCAAACCGAAGTC | TGGGGACACCTTTTAGCATCTT |
| Ifnb | CAGCTCCAAGAAAGGACGAAC | GGCAGTGTAACTCTTCTGCAT |
| Gramd4 | CTCCCCGTCATGTCCAGGTG | CTGTTGAAGTCCTGCACGC |
| Unc93b1 | GGCTCCCTTGGACGAACTC | CACGTTCTTGACCACTCCGAG |
| Hprt1 | TCAGTCAACGGGGGACATAAA | GGGGCTGTACTGCTTAACCAG |
Cell staining and flow cytometry assays
For the staining of BMMs, cells were preincubated with anti-CD16/32 (clone 2.4G2) and stained with the indicated Abs in ice-cold PBS containing 2% FCS. Intracellular staining of TLR9 was performed as described previously (32). The fluorescence signal was detected by flow cytometry (FACSCalibur, BD Biosciences, San Jose, CA) and analyzed with FlowJo software (Tree Star, Ashland, OR).
Uptake and translocation of CpG-B
BMMs were cultured on a 24-well plate at 1.25 × 105 cells/well and stimulated with CpG-B–Cy5 at 37°C for 10 min. After washing and trypsin treatment, detached BMMs were subjected to flow cytometry for measurement of Cy5 fluorescence. For detection of colocalization of internalized CpG-B–Cy5 and an early endosome marker (Rab5), BMMs were stimulated with 0.5 μM CpG-B–Cy5 at 37°C for 10 min. After washing, the cells were fixed with 4% paraformaldehyde for 15 min at room temperature (RT), permeabilized with PBS containing 0.2% Triton X-100 for 5 min at RT, treated with anti-Rab5 Abs (no. 2413, Cell Signaling Technology, Beverly, MA) in PBS containing 1% BSA for 60 min at RT, and then stained with the Alexa Fluor 488–conjugated anti-rabbit IgG (Invitrogen) in PBS containing 1% BSA for 60 min at RT. For detection of colocalization of CpG-B and lysosomes, BMMs were incubated with 0.2 μM Alexa Fluor 647–labeled CpG-B and LysoTracker Red DND-99 (Invitrogen) for 1 h. Fluorescence signals were observed under a confocal laser-scanning microscope (FluoView FV1000, Olympus, Tokyo, Japan).
RNA interference
Silencing of Gramd4 and leucine-rich repeats and calponin homology domain containing 4 (Lrch4) expression was performed by application of small interfering RNA (siRNA) oligonucleotides (Dharmacon, Lafayette, CO) targeted to the cDNA sequence of the mouse Gramd4 or Lrch4 gene. RAW264.7 cells were cultured on a 6-well plate at 5.0 × 105 cells/well and BMMs were cultured on a 48-well plate at 7.5 × 104 cells/well. Cells were transfected with 3 nM (RAW264.7) or 20 nM (BMMs) negative control siRNA (Dharmacon) or the siRNA targeting mouse Gramd4 or Lrch4 gene using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturers’ instructions. The effect of RNA interference was examined by real-time PCR at 48 or 60 h following transfection.
Luciferase reporter assay
RAW264.7 cells were cultured on a six-well plate at 1.0 × 106 cells/well and transfected with an expression vector and reporter plasmid using Lipofectamine LTX according to the manufacturer’s instructions. Two days after transfection, the cells were stimulated for 6 h as indicated and then lysed for the luciferase assay. Luciferase activity was measured with a Dual-Luciferase reporter assay system (Promega) according to the manufacturer’s instructions using LMAX II384 (Molecular Devices, Sunnyvale, CA) and SoftMax Pro v5 (Molecular Devices). Firefly luciferase activity was divided by the Renilla luciferase activity of the pGL4.74 vector plasmid (Promega).
Materials
Poly(I:C) (low m.w.), R837, Pam3CSK4, and FSL-1 were obtained from InvivoGen (San Diego, CA). Nuclease-stable phosphorothioate-modified CpG-B 1826 (5′-TCCATGACGTTCCTGACGTT-3′) was purchased from Hokkaido System Science (Sapporo, Japan). Cy5-labeled CpG-B 1826 and Alexa Fluor 647–labeled CpG-B 1826 were purchased from Nihon Gene Research Laboratory (Sendai, Japan) and Japan Bio Services (Saitama, Japan), respectively. LPS, Polybrene, biotin-tagged anti–FLAG-M2, agarose-conjugated anti–FLAG-M2 Abs, and anti–conjugated Abs were purchased from Sigma-Aldrich. Anti–Nogo-A/B Abs were purchased from Imgenex (San Diego, CA). Anti-Myc Abs were purchased from Wako (Osaka, Japan). Anti–IκB-α (no. 9242), anti–phospho-p38 (no. 9211), anti-p38 (no. 9212), anti–phospho-ERK1/2 (no. 4370), anti–phospho-STAT1 (no. 9171), and anti-STAT1 (no. 9172) were purchased from Cell Signaling Technology. Anti-IRAK1 (H-273), anti-TRAF6 (H-274), and anti-ERK1/2 (K-23) Abs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-MyD88 was purchased from QED Bioscience (San Diego, CA). Polyclonal anti-PirB and anti-PRAT4A Abs were purchased from R&D Systems (Minneapolis, MN). HRP-linked anti-rabbit IgG and anti-mouse IgG were purchased from Amersham Pharmacia Biotech (Buckinghamshire, U.K.). Monoclonal anti-PirA/B (6C1) and PE-conjugated anti-mouse CD11b (M1/70) were obtained from BD Biosciences. Alexa Fluor 647–conjugated anti-mouse F4/80 (A3-1), PE-conjugated anti-mouse TLR4 (MTS510), and PE-conjugated anti-mouse IgG (Poly4053) were obtained from BioLegend. Monoclonal anti-TLR9 (B33A4 and J15A7) was developed by K. Miyake (University of Tokyo). Anti-LAMP1 Ab (1D4B) was obtained from Millipore (Billerica, MA).
Confocal imaging
To assesse the localization of Nogo-B and GRAMD4, MEFs were cultured on a six-well plate at 8.0 × 104 cells/well and transfected with Nogo-B2–GFP or GRAMD4-TagRFP expression vector using Xfect transfection reagent (Takara Bio, Otsu, Japan) according to the manufacturer’s instructions. One day after transfection, the cells were reseeded to μ-Dishes (Ibidi, Munich, Germany). The next day, the cells were washed with HBSS and then incubated with BODIPY FL– or BODYPY TR–conjugated ER-Tracker (Invitrogen) to stain ERs. After staining, MEFs were fixed in 4% paraformaldehyde for 2 min. Immunofluorescence staining of endogenous TLR9 was performed as previously described (32). Confocal microscopy was performed under a confocal laser-scanning microscope (FluoView FV100). Pearson correlation coefficients were used for the statistical analysis of colocalization.
Statistical analysis
The statistical significance of differences in mean values was analyzed with an unpaired, two-tailed Student t test. The p values < 0.05 were considered statistically significant.
Results
Nogo-B is indispensable for full activation of nucleic acid–sensing TLR responses in macrophages
To assess the expression pattern of Nogo isoforms in immune cells, nogo-A/B−⁄− mice and a specific Ab that recognizes Nogo-A and -B were used. Western blotting analysis using BMM lysates demonstrated the expression of Nogo-B (Fig. 1A, 1C), corresponding to the Nogo-B1 (∼42 kDa) and -B2 (∼46 kDa) isoforms as reported (33, 34). In brain tissues, expression of Nogo-A isoform was detectable in both mRNA and protein levels. However, only a low level of Nogo-A mRNA expression was observed in BMMs (Fig. 1B, 1C, Table I). Immunoblot analysis of whole-cell lysates of splenocytes, T cells, B cells, bone marrow derived–dendritic cells, and BMMs from wild-type mice revealed the preferential expression of the Nogo-B2 isoform in BMMs, as well as its absence, together with the B1 isoform, in BMMs from nogo-A/B−⁄− mice (Fig. 1A, 1C). Cells isolated from bone marrow of nogo-A/B−⁄− mice differentiated normally into macrophages in the presence of M-CSF in vitro, as judged by the cell growth (not shown) and the cell surface expression of CD11b and F4/80 (Supplemental Fig. 1A). Furthermore, the deficiency of Nogo-B did not affect the phagocytic capacity of Polystyrene beads in BMMs (Supplemental Fig. 1B).

Nogo-B promotes production of inflammatory cytokines triggered by nucleic acid–sensing TLRs in macrophages. (A) CD3+ T cells and B220+ B cells from the spleens from wild-type mice were purified with a magnetic-activated cell sorter. Bone marrow–derived dendritic cells (BMDCs) and BMMs were differentiated from bone marrow cells isolated from wild-type mice. Whole-cell lysates were subjected to Western blot analysis using specific Abs for Nogo-A/B and loading control β-actin. (B) Brain tissues and BMMs from wild-type mice were subjected to quantitative real-time PCR using a forward primer specific for each of the Nogo isoforms and a reverse primer located in a common region. Relative mRNA expression levels were normalized to Hprt1. (C) Whole-cell lysates of brain tissues and BMMs from wild-type and nogo-A/B−⁄− mice were subjected to Western blot analysis using specific Abs for Nogo-A/B and loading control Erk1/2. (D and E) BMMs from wild-type and nogo-A/B−⁄− mice were stimulated with 10 ng/ml LPS, 100 ng/ml FSL-1, 0.1 μM CpG-B, 10 μg/ml poly(I:C), 100 ng/ml Pam3CSK4, or 200 ng/ml R837 for indicated times. The culture supernatants were collected and subjected to IL-6 measurement by ELISA (D). The mRNA level of IL-6 was determined by real-time RT-PCR and relative mRNA expression levels were normalized to Hprt1 (E). (F–H) BMMs from wild-type and nogo-A/B−⁄− mice were treated with 0.3 μM CpG-B (F), 3 μg/ml poly(I:C) (G), or 1 ng/ml LPS (H) for the indicated times and then analyzed for the activation of NF-κB, p38 MAPK, Erk, and STAT1 by Western blotting. The data shown are representative results from three independent experiments (A, C, and F–H), the mean ± SEM of three independent experiments (B), and mean ± SEM of triplicate samples of one representative of at least three independent experiments (D and E).
We then examined the role of Nogo in the cytokine responses of macrophages upon stimulation with various TLR ligands. Although BMMs from both wild-type and nogo-A/B−⁄− mice responded similarly to LPS, Pam3CSK4, and FSL-1 (ligands for TLR4, TLR2/1, and TLR2/6, respectively), we found a marked reduction of IL-6 production upon stimulation with CpG-B oligonucleotide (TLR9 ligand), poly(I:C) (TLR3 ligand), and R837 (TLR7 ligand) (Fig. 1D, 1E). Similarly, nogo-A/B−⁄− BMMs exhibited significantly lower induction of proinflammatory cytokine and chemokine mRNAs in response to CpG-B than did cells from wild-type mice (Supplemental Fig. 2). We further investigated the effects of a deficiency of Nogo-B on the TLR signal pathway in macrophages. To assess the functional integrity of the intracellular signal pathway in nogo-A/B−⁄− macrophages, we monitored the activation of NF-κB and MAPKs induced by TLR ligands. We observed impaired degradation of IκBα and phosphorylation of p38 MAPK in BMMs stimulated with CpG-B in nogo-A/B−⁄− BMMs (Fig. 1F). Similar results were obtained with poly(I:C)-stimulated nogo-A/B−⁄− BMMs (Fig. 1G). Additionally, consistent with the normal response shown in Fig. 1D and 1E, the activation of signal molecules in nogo-A/B−⁄− cells stimulated with LPS was comparable to that in control cells (Fig. 1H). Considering that the Nogo-A protein was undetectable in macrophages, the attenuated TLR3, TLR9, and TLR7 responses observed in nogo-A/B−⁄− BMMs were suggested to be caused primarily by the lack of Nogo-B protein.
Nogo-B promotes cytokine responses induced by poly(I:C) in mice
Next we examined whether a deficiency of Nogo decreases the innate immune responses triggered by TLR ligands in vivo. Flow cytometry revealed that the number of F4/80+ macrophages among resident peritoneal cells did not differ between wild-type and nogo-A/B−⁄− mice (Fig. 2A). nogo-A/B−⁄− mice challenged with poly(I:C) showed lower levels of IL-6 and IL-12p40 in their sera (Fig. 2B). This finding was confirmed by the reduced expression of IL-6 and type 1 IFN mRNAs in splenocytes from the same animals (Fig. 2C). In contrast, there was no substantial difference between wild-type and nogo-A/B−⁄− mice in the cytokine responses triggered by LPS challenge (Fig. 2D). These results indicated that Nogo is required for full activation of innate immune responses mediated by nucleic acid–sensing TLRs in vivo.
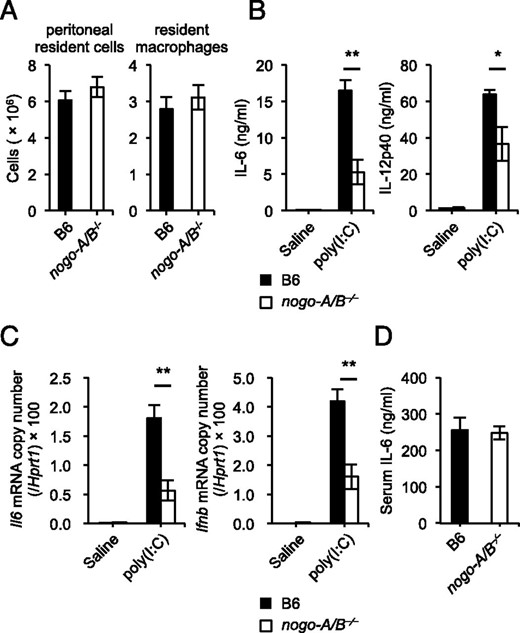
Deficiency of Nogo attenuates cytokine responses triggered by TLR3 stimulation. (A) Numbers of resident cells and F4/80+ macrophages in peritoneal cavities harvested from wild-type and nogo-A/B−⁄− mice (n = 4 mice/group). (B–D) Age- and sex-matched wild-type and nogo-A/B−⁄− mice (n = 5–7 mice/group) were i.p. injected with poly(I:C) (5 mg/kg body weight), LPS (15 mg/kg body weight), or saline, and then sera were taken at 3 h after injection. The serum concentrations of IL-6 (B and D) and IL-12p40 (B) were measured by ELISA. Whole splenocytes were harvested at the same time, and then total RNA was isolated for quantitative real-time PCR to determine the mRNA levels of IL-6 and IFN-β (C). *p < 0.05, **p < 0.01.
Deficiency of Nogo-B does not influence incorporation and intracellular transport of nucleic acid ligands
We then attempted to determine whether Nogo-B regulates the delivery of nucleic acid ligands to endolysosomal compartments, where they activate intracellular TLRs. Nucleic acid–sensing TLRs, which are synthesized in ER, are transported to endolysosomes to recognize their ligands via the binding to UNC93B1 on the ER membrane (6, 7). CpG-B is rapidly internalized via a clathrin-dependent endocytotic pathway (35, 36). To determine possible effects of Nogo on the uptake and delivery of nucleic acid ligands, we examined the fluorescence intensity and subcellular localization of fluorescently labeled CpG-B internalized in macrophages using flow cytometry and fluorescence microscopy. BMMs were incubated for 10 or 60 min at 37°C in the presence of fluorescently labeled CpG-B, and then the cells were washed and analyzed. In BMMs from nogo-A/B−⁄− mice, there was no substantial defect in the incorporation of the CpG-B–Cy5, and the localization of the internalized CpG-B in Rab5-marked early endosomes and LysoTracker-positive lysosomes was similar to that in the case of wild-type cells (Fig. 3A–E). Nogo bound to its receptor complex on neuronal cell surface is reported to be internalized through Pincher- and Rac1-dependent endocytosis, and this complex is then transported to the cell bodies to trigger activation of signal molecules for the inhibition of axonal outgrowth (37). Our results, however, indicate that Nogo-B is dispensable for the uptake and traffic of a nucleic acid ligand for TLR9. We also noted that neither intracellular expression level of TLR9 nor cell surface trafficking of TLR4 was altered in nogo-A/B−⁄− BMMs compared with in wild-type cells (Fig. 3F). These data suggest that the attenuated innate immune responses observed in nogo-A/B−⁄− mice in vivo and macrophages in vitro following stimulation of the intracellular TLRs are not due to defects in the transport of TLR ligands.
![Nogo is dispensable for uptake and delivery of nucleic acid ligands. (A) BMMs from wild-type and nogo-A/B−⁄− mice were incubated with or without 1 μM Cy5-conjugated CpG-B. Ten minutes after incubation, BMMs were detached from culture dishes and Cy5 fluorescence was measured by flow cytometry. The column graph indicates induction of mean fluorescence intensity (MFI). (B and C) BMMs were stimulated with 0.5 μM CpG-B–Alexa Fluor 647. After 10 min stimulation, cells were fixed, and then stained with rabbit anti-Rab5 Abs and Alexa Fluor 488–conjucated anti-rabbit-IgG to visualize early endosomes. (D and E) BMMs were cultured with 0.2 μM CpG-B–Alexa Flour 647 and 60 nM LysoTracker Red for 1 h. (F) BMMs from wild-type, nogo-A/B−⁄− and Tlr9−⁄− mice were stained with anti-TLR9 (clone B33A4), anti-TLR4 Abs, or isotype control Abs and subjected to flow cytometric analysis. For staining TLR9, cells were fixed and permeabilized before staining. Data shown are from three independent experiments [(A), mean ± SEM] or are representative of three independent experiments with similar results (B–F). Acquisition of images was performed under confocal laser scanning microscopy. The histograms show cross-line scans of the fluorescence intensities of the merged panels. To quantify the extent of endolysosomal localization of CpG-B, fluorescence intensities on the lines drawn on the major axes of cells were measured. Then Pearson correlation coefficients were calculated for 60 cells per sample (B–E). Original magnification ×300. Scale bars, 10 μm. Statistical analyses were performed using a Student t test.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jimmunol/194/11/10.4049_jimmunol.1402006/2/m_ji_1402006_f3.jpeg?Expires=1750157979&Signature=qXclmCzQEUoquLA4RAu5rsdfAul2n9wrlotWKrSmuxXCEBe07FmFMTiM3OE6B2CTY4QpeC2KF1SJwz1qNLu3NLyCkPLJEO-PvFaIhV32Rtqo6lQE8E8Nxrrt91GWa-cM3O19gOAArbPpH-sG3n11wxDfpeSUdZ7aRyheda0XdojUxFIs9wCAIa56pE3oT20N-c0YZ~SIykvn71Yd2Oawbes8dp3mtcv0tNM3sbeM9~x0Sk5O-QNHGL22cJ36AegUzn0BDLHo0E80jqog70RrpIUsxj0YlRNbIDk61tFkgw2dFcYR8jNsZMcbg6rigYx1xAyn2a2IuXt03K1hyJ4zPQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Nogo is dispensable for uptake and delivery of nucleic acid ligands. (A) BMMs from wild-type and nogo-A/B−⁄− mice were incubated with or without 1 μM Cy5-conjugated CpG-B. Ten minutes after incubation, BMMs were detached from culture dishes and Cy5 fluorescence was measured by flow cytometry. The column graph indicates induction of mean fluorescence intensity (MFI). (B and C) BMMs were stimulated with 0.5 μM CpG-B–Alexa Fluor 647. After 10 min stimulation, cells were fixed, and then stained with rabbit anti-Rab5 Abs and Alexa Fluor 488–conjucated anti-rabbit-IgG to visualize early endosomes. (D and E) BMMs were cultured with 0.2 μM CpG-B–Alexa Flour 647 and 60 nM LysoTracker Red for 1 h. (F) BMMs from wild-type, nogo-A/B−⁄− and Tlr9−⁄− mice were stained with anti-TLR9 (clone B33A4), anti-TLR4 Abs, or isotype control Abs and subjected to flow cytometric analysis. For staining TLR9, cells were fixed and permeabilized before staining. Data shown are from three independent experiments [(A), mean ± SEM] or are representative of three independent experiments with similar results (B–F). Acquisition of images was performed under confocal laser scanning microscopy. The histograms show cross-line scans of the fluorescence intensities of the merged panels. To quantify the extent of endolysosomal localization of CpG-B, fluorescence intensities on the lines drawn on the major axes of cells were measured. Then Pearson correlation coefficients were calculated for 60 cells per sample (B–E). Original magnification ×300. Scale bars, 10 μm. Statistical analyses were performed using a Student t test.
Trafficking of TLR9 to endolysosomes is attenuated in Nogo-deficient macrophages
Proper localization of sensors to endolysosomes is crucial to ensure the encounter of nucleic acid–sensing TLRs with their respective ligands. To analyze the behavior of intracellular TLR, we tracked subcellular localization of endogenous TLR9 using an mAb against TLR9 and Tlr9−⁄− BMMs as a negative control. Recent studies demonstrated that a significant portion of TLR9 was already processed and localized to endolysosomal compartments in resting primary B cells, dendritic cells, and macrophages (32, 38). Confocal images with the statistical analysis showed that, in unstimulated nogo-A/B−⁄− BMMs, colocalization of TLR9 with LAMP1+ lysosomes was decreased in comparison with that of wild-type cells (Fig. 4A, 4B). One hour of CpG-B stimulation upregulated TLR9 colocalization with LAMP1 in wild-type BMMs, although only slightly, whereas the elevation was not detectable in nogo-A/B−⁄− cells (Fig. 4B). We next compared the distribution of TLR9 in CpG-B+ compartments in wild-type and nogo-A/B−⁄− BMMs. After 20 min stimulation with CpG-B–Alexa Fluor 647, TLR9 showed lower colocalization with CpG-B in nogo-A/B−⁄− BMMs than that of wild-type cells (Fig. 4C, 4D). We further confirmed that there was no substantial difference between nogo-A/B−⁄− and wild-type cells in the expression levels of TLR-specific chaperon Unc93b1 and PRAT4A, and intracellular signal molecules including MyD88, IRAK1, or TRAF6 (Fig. 4D). Collectively, these data suggested that insufficient translocation of TLR9 to endolysosomes resulted in the impaired responses to nucleic acid ligands in nogo-A/B−⁄− cells.
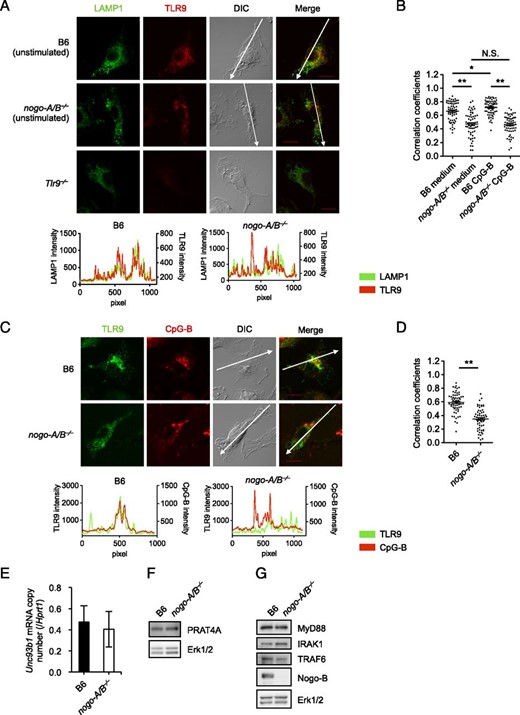
Nogo is required for the localization of TLR9 to endolysosomes. (A and B) BMMs from wild-type, nogo-A/B−⁄−, and Tlr9−⁄− mice were stimulated with 0.3 μM CpG-B. After 1 h stimulation, cells were fixed and then stained with anti-LAMP1 and anti-TLR9 Abs. (C and D) BMMs were stimulated with 0.2 μM CpG-B–Alexa Fluor 647 for 20 min followed by staining for TLR9. We chose 20 min of stimulation because a longer reaction period such as 1 h yielded a less sharp CpG signal. (E) BMMs from wild-type and nogo-A/B−⁄− mice were subjected to quantitative real-time PCR. Relative mRNA expression level of Unc93B1 was normalized to Hprt1. (E–G) BMMs from wild-type and nogo-A/B−⁄− mice were lysed and analyzed by Western blot analysis using specific Abs for PRAT4A, MyD88, IRAK1, TRAF6, Nogo-A/B, and loading control Erk1/2. The data shown are representative results from two (C and D), three (A, B, F, and G), or four (E) independent experiments and represent the mean ± SEM. The histograms show cross-line scans of the fluorescence intensities of the merged panels. To quantify the extent of colocalization, fluorescence intensities on the lines drawn on the major axes of cells were measured. Then Pearson correlation coefficients were calculated for 60 cells per sample (B and D). Original magnification ×300. Scale bars, 10 μm. *p < 0.05, **p < 0.01.
GRAMD4 binds to Nogo-B and inhibits nucleic acid–induced TLR responses
Next we aimed at clarifying the mechanisms underlying the involvement of Nogo in the enhancement of the TLR trafficking. Nogo-A regulates redistribution of the ER-resident chaperone protein disulfide isomerase and contributes the proper function of protein disulfide isomerase (39). Studies have shown that some RTN family molecules could also function in protein transport from ER to other membrane compartments, including the plasma membrane, Golgi, and endosomes (40–42). For example, RTN2B enhances trafficking of glutamate transporter EAAC1 from the ER to the cell surface. Although they are likely to promote exit and appropriate trafficking of target proteins from the ER by the interaction with their ER binding partners, the precise mechanisms are so far unclear.
Preceding studies by others indicated that, in the CNS, NgR1 and PirB interact with Nogo-66. NgR1 forms a complex with proteins, including presumed signal transducers p75 and TROY on neural cells (17, 19). However, we could not detect coimmunoprecipitation of endogenous NgR1 and Nogo in macrophages (data not shown). Moreover, macrophages from NgR1-deficient mice responded normally to TLR ligands and produced IL-6 at a comparable level to that by cells from wild-type mice (data not shown). It is well known that PirB is highly expressed in myeloid-lineage immune cells and plays a crucial role in the inhibition of immune responses via recruitment of phosphatase SHP-1 and transducing a negative signal, and we previously demonstrated that PirB inhibits the TLR9 pathway in vivo and in vitro (43–45). Although abundant expression of PirB protein was observed in BMMs, Nogo-B was not coprecipitated with PirB (Supplemental Fig. 3A). Similarly, we could not detect the binding of Nogo-B to PirB in human embryonic kidney 293T cells, in which murine Nogo-B and PirB were ectopically expressed (Supplemental Fig. 3B, 3C). Thus, at least in macrophages, we considered that Nogo binds to neither NgR1 nor PirB and uses other effector molecules to regulate the TLR pathways.
To find any molecules associated with Nogo-B in immune cells, we generated RAW264.7 murine macrophage-like cells stably expressing an exogenous Nogo-B2–FLAG fusion protein, hereafter referred to as RAW–Nogo-B cells. Recent technological advances in mass spectrometry (MS)–based proteomics and peptide-labeling strategies using iTRAQ have enabled the large-scale identification of protein–protein interactions (46). In this study, we applied iTRAQ coupled with MS to identify the interacting partners of Nogo-B that have a regulatory function in the intracellular TLR pathways. Whole-cell lysates of RAW–Nogo-B cells in the steady-state and parental RAW264.7 cells as a control were subjected to immunoprecipitation with anti-FLAG mAb and subsequent iTRAQ-coupled liquid chromatography (LC) tandem MS (LC-MS/MS) analysis. Quantitative information on identified proteins was generated by calculating the relative ratios of peak areas for the iTRAQ marker ions in the MS/MS spectra. We then obtained some (∼40) potentially interacting proteins exhibiting high ratios and confidence of protein identification (see Materials and Methods), but we did not detect coimmunoprecipitation of any TLRs, their signaling molecules, UNC93B1, PRAT4A, and gp96 with Nogo-B. After applying these criteria, we addressed whether the LC-MS/MS–detected binding could be confirmed by coimmunoprecipitation of FLAG-tagged Nogo-B with Myc-tagged, potentially interacting proteins from dually transfected 293T cells. Coimmunoprecipitation experiments showed that a few proteins, including GRAMD4 and LRCH4, which was reported previously as a Nogo-binding protein (47), could bind to Nogo-B in 293T cells (Fig. 5A, Supplemental Fig. 4A).
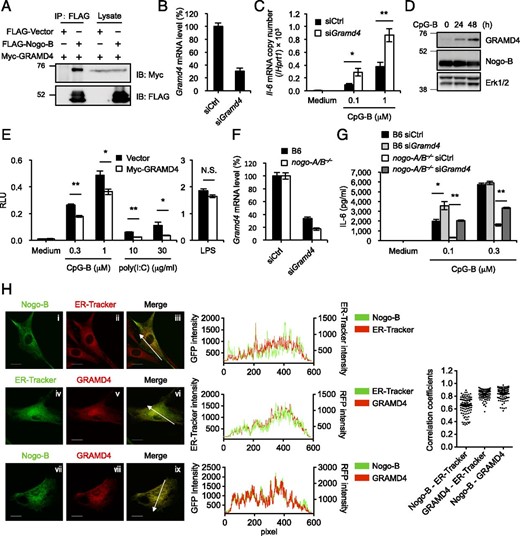
GRAMD4 binds to Nogo-B and negatively regulates immune responses initiated by nucleic acid–sensing TLRs. (A) Western blot analysis of 293T cells 48 h after cotransfection of FLAG-tagged Nogo-B or the FLAG empty vector plus Myc-tagged GRAMD4, followed by immunoprecipitation (IP) with anti-FLAG. (B) RAW264.7 cells were treated with siRNA specifically targeting the Gramd4 gene or nontargeting control siRNA for 60 h, and then evaluated for the mRNA level of Gramd4 by real-time RT-PCR. Data are expressed relative to negative control. (C) IL-6 mRNA level in siRNA-transfected RAW264.7 cells was measured following 4 h stimulation with 0.1 or 1 μM CpG-B by real-time RT-PCR. Relative mRNA expression levels were normalized to Hprt1. (D) BMMs were stimulated with 0.2 μM CpG-B for the indicated times and then analyzed for the expression of Gramd4 and Nogo-B by Western blotting. (E) Luciferase reporter plasmids (pELAM1-Luc and pGL4.74-TK-Luc) were cotransfected into RAW264.7 cells with Myc-tagged GRAMD4. Cells were stimulated with CpG-B, poly(I:C), or LPS for 6 h after 60 h transfection. Transfected ELAM1 promoter-driven luciferase activity was normalized to Renilla luciferase expression. (F) BMMs from wild-type and nogo-A/B−⁄− mice were treated with siRNA specific for Gramd4 or control siRNA for 48 h and then evaluated for the mRNA level of Gramd4 by real-time RT-PCR. Data are expressed relative to negative control. (G) IL-6 in supernatants of siRNA-transfected BMMs was measured following 6 h stimulation with 0.1 or 0.3 μM CpG-B by ELISA. (H) MEFs from wild-type mice were transfected with GFP-tagged Nogo-B (i–iii, vii–ix) or TagRFP-tagged GRAMD4 (iv–ix). Forty-eight hours after transfection, MEFs were stained with ER-Tracker Red (i–iii) or ER-Tracker Green (iv–vi). Samples were examined under a FluoView FV1000 confocal laser-scanning microscope. The histograms show cross-line scans of the fluorescence intensities of the merged panels. Pearson correlation coefficients were calculated for at least 100 cells per sample. Original magnification ×120. Scale bars, 20 μm. Data shown are representative of three (A and D) or two (H) independent experiments, mean ± SEM of three independent experiments (B and C), or mean ± SEM for triplicate samples of one representative of three independent experiments with similar results (E–G). *p < 0.05, **p < 0.01.
We then investigated the functional properties of these proteins in the TLR pathways. Because RAW264.7 cells express Nogo-B endogenously, we used these cells to assess the knockdown effects of these two genes on TLR responses. Treatment with Gramd4 siRNA consistently reduced the Gramd4 level by 70% (Fig. 5B), as determined by real-time PCR analysis. When the Gramd4 knockdown RAW264.7 cells were stimulated with CpG-B, IL-6 mRNA was expressed on an ∼2-fold higher level than that in control cells (Fig. 5C). In contrast, treatment with siRNA against Lrch4 caused no change in IL-6 mRNA induction triggered by CpG-B (Supplemental Fig. 4B, 4C). To examine the function of GRAMD4 in NF-κB activation induced by TLR stimulation, we transiently transfected RAW264.7 cells with an expression plasmid encoding Gramd4 and studied the effect by luciferase reporter assaying. The activation of NF-κB reporter pELAM1-Luc (see Materials and Methods) induced by CpG-B and poly(I:C) was inhibited by GRAMD4 overexpression without affecting TLR4 response (Fig. 5E). Immunoblot analysis of lysates from wild-type BMMs showed GRAMD4 expression in unstimulated cells, and treatment with CpG-B led to the induction of GRAMD4 protein. Of note, the expression levels of Nogo-B proteins were unaltered (Fig. 5D). Although we found that Nogo-B and GRAMD4 affect TLR responses and GRAMD4 interacts with Nogo-B, each of them might function separately in TLR pathway. To test this possibility, we examined whether the negative function of GRAMD4 is canceled in Nogo-deficient conditions. We observed the effect of knockdown of GRAMD4 on IL-6 production induced by CpG-B in BMMs from wild-type and nogo-A/B−⁄− mice (Fig. 5F, 5G). When GRAMD4 expression was silenced in nogo-A/B−⁄− cells, the enhancing effect on IL-6 production was much greater than that in wild-type cells (Fig. 5G). These results suggested that GRAMD4 exerts its negative function on the nucleic acid–sensing TLR pathway without interference by Nogo-B in nogo-A/B−⁄− cells, whereas GRAMD4-mediated negative function is suppressed by Nogo-B in wild-type cells. We also examined the subcellular distribution of Nogo-B and GRAMD4 expressed as the transgenes by confocal microscopy of MEFs transiently expressing Nogo-B2–enhanced GFP and TagRFP-GRAMD4. Nogo-B moderately colocalized with the ER marker ER-Tracker (r = 0.647), but Pearson correlation coefficients between GRAMD4 versus ER-Tracker and Nogo-B versus GRAMD4 showed strong correlations (r = 0.829 and 0.841, respectively) (Fig. 5H), indicating that the interaction between Nogo-B and GRAMD4 mostly occurred in the ER. Taken together, these results indicate that Nogo-B and GRAMD4 have positive and negative function in the response to nucleic acid stimulation, respectively, and they interact with each other in the ER.
Discussion
Nogo protein is expressed in various tissues and cells, but the function in the immune system remains largely unknown. In the present study, we identified Nogo-B as a positive regulator of immune responses triggered by nucleic acid–sensing TLRs. This was confirmed in vivo, in that we observed attenuated cytokine responses in nogo-A/B−⁄− mice stimulated with poly(I:C). In nogo-A/B−⁄− macrophages, there was no defect in the uptake or transport of nucleic acid ligands to endosomes and lysosomes, which are essential steps in intracellular TLR activation (4). In contrast, a deficiency of Nogo-B prevented the localization of TLR9 to endolysosomal compartments, and thereby activation of NF-κB and MAPKs following stimulation with nucleic acids was attenuated. Based on previous studies showing that Nogo plays an inhibitory role in axonal regeneration through coupling to its binding partners (20, 25), we hypothesized that Nogo-B regulates TLR trafficking via interactions with other effector molecules. To find the interacting partners of Nogo-B, we conducted LC-MS/MS analysis and identified GRAMD4 as a novel Nogo-B–binding protein (Fig. 5). Interestingly, GRAMD4 negatively regulated the TLR9 and TLR3 pathways, but it failed to affect the TLR4 pathway. Our observations led us to speculate that Nogo-B could bind to GRAMD4 to attenuate its inhibitory function, leading to full activation of nucleic acid–initiated innate immune responses. Of note, GRAMD4 induced by TLR stimulation might terminate inflammatory signaling. Taken together, our results suggested a fine-tuning mechanism of intracellular TLR pathways regulated by Nogo-B and GRAMD4.
The expression pattern of Nogo isoforms differs among tissues and cell types. Using Nogo-A– and Nogo-B–specific Abs, we found that Nogo-B was highly expressed in immune cells, especially in macrophages. In contrast, as previously reported, we could not detect the Nogo-A protein in macrophages (Fig. 1A, 1C). Therefore, the imperfect immune responses in nogo-A/B−⁄− mice and cells are attributed to the loss of Nogo-B protein in the immune system. In the CNS, previous studies showed that NgR1 and PirB could bind not only to all three Nogo isoforms through the Nogo-66 loop region, but also to other axonal outgrowth inhibitory proteins, namely MAG and OMgp (18, 48–50). NgR1 forms a protein complex including LINGO1, p75, or TROY, leading to growth cone collapse in neural cells (51). Because the search for binding partners of Nogo and the actions of the receptor components have been technically difficult, the molecular mechanisms in the CNS as well as in the immune system are still poorly understood.
We previously reported an inhibitory role of PirB in regulation of the TLR9 signaling pathway in murine B-1 cells (45). However, we could not detect the interaction of Nogo with PirB in immunoprecipitation experiments using lysates from BMMs or 293T cells overexpressing both proteins (Supplemental Fig. 3). In the CNS, Nogo-A is also expressed on the cell surface and binds to some cell surface receptors such as PirB. Although we have no Ab to stain Nogo for flow cytometry and immunofluorescence analysis, Nogo-B may not be expressed on the plasma membrane of immune cells and may not be able to interact with PirB. NgR1 is a known receptor for Nogo, but the tissue distribution of mRNA expression for NgR1 involved the brain, heart, and kidney, but not other peripheral tissues (20). Therefore, the function of Nogo as a regulator of the TLR pathways is likely to be independent of PirB and NgR1. In the present study, GRAMD4 was found to interact with Nogo-B and to play an inhibitory role in the nucleic acid–sensing TLR pathways. GRAMD4 was reported to interact with Bcl-2 and to promote Bax oligomerization on mitochondria (30). Other than its apoptogenic role, there was no known physiological role of GRAMD4. Our current finding provides an insight into the immunoregulatory function of GRAMD4, albeit that the molecular mechanism by which GRAMD4 suppresses the TLR pathways is unknown.
Studies have shown that RTN family molecules could function in protein transport from the ER to other membrane compartments, including the plasma membrane, Golgi, and endosomes (40–42). For example, RTN2B enhances trafficking of glutamate transporter EAAC1 from the ER to the cell surface. Alternatively, the delivery from the ER to endolysosomes or cell surface of TLRs is essential for their proper ligand recognition and activation. ER-resident proteins such as UNC93B1, gp96, and PRAT4A regulate this translocation (6, 7, 10, 11). The molecular mechanisms that control each step of TLR trafficking from cargo sorting, vehicle formation, to docking are obscure. However, studies showed the role of small GTPase Rab family proteins on TLR4 trafficking to the plasma membrane and lysosomes (52, 53). Recently, Lee et al. (54) reported that UNC93B1 directly controls the packaging of intracellular TLRs into COPII vesicles, which mediates the transport of cargos between the ER and Golgi. Rab1 is reported to recruit vesicle tethering factor p115, which is required for the docking of ER-derived COPII-coated vesicles with Golgi membranes (55, 56). Alternatively, the GRAM domain is present in several Rab-like GTPase activators, although this domain is not likely to be essential for GTPase activation (57). Given these reports, one may speculate that the binding of Nogo-B and GRAMD4 mediates activation of Rab1. Further studies are needed to clarify a potential involvement of Rab1 in membrane trafficking of TLRs and the molecular mechanisms of nucleic acid–sensing TLR-specific regulation by Nogo-B and GRAMD4.
Acknowledgements
We thank Stephen M. Strittmatter (Yale University School of Medicine) for providing mutant mice and Nicholas Halewood for editorial assistance.
Disclosures
The authors have no financial conflicts of interest.
References
Footnotes
This work was supported by the Core Research for Evolutional Science and Technology Program of the Japan Science and Technology Agency (to T.T.), a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to T.K. and T.T.), and by a grant from the Global Center of Excellence Program for Innovative Therapeutic Development Towards the Conquest of Signal Transduction Diseases with Network Medicine (to T.T.).
The online version of this article contains supplemental material.
Abbreviations used in this article:
- BMM
bone marrow–derived macrophage
- ER
endoplasmic reticulum
- GRAM
glucosyltransferases, Rab-like GTPase activators and myotubularins
- GRAMD4
GRAM domain containing 4
- iTRAQ
isobaric tags for relative and absolute quantitation
- LC
liquid chromatography
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- LRCH4
leucine-rich repeats and calponin homology domain containing 4
- MEF
mouse embryonic fibroblast
- MS
mass spectrometry
- NgR1
Nogo receptor 1
- Pir
paired Ig-like receptor
- poly(I:C)
polyinosinic:polycytidylic acid
- PRAT4A
protein associated with TLR4 A
- RFP
red fluorescent protein
- RT
room temperature
- siRNA
small interfering RNA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}