




Abstract
Factor H controls proximal complement activation, and its dysfunction leads to diseases that often manifest in the kidney. Structural and functional analyses have identified 4 distinct functional segments: an N-terminal regulatory unit, a cell binding unit, a segment with combined low-affinity C3b and heparin sites, and a C-terminal recognition or sensor unit with overlapping C3b/C3d and heparin sites. Three segments are linked to diseases. The regulatory segment is affected in C3 glomerulopathy and antineutrophil cytoplasmic antibody–associated vasculitis. The second segment includes the Y402H polymorphism of age-related macular degeneration, is associated with different types of cancer, and is targeted by pathogens. The C-terminal sensor segment is involved in atypical hemolytic uremic syndrome, in FHR1:FHR3 deficient and autoantibody–positive hemolytic uremic syndrome form and is exploited by pathogens. Factor H function is modulated by Factor H like protein 1 and FHR1, 2 plasma proteins that share segments with Factor H. This interplay is critical for fine-tuning local complement. Understanding Factor H’s physiological role, as well as the impact of its absence, mutations, or autoantibody targeting, provides insights into disease mechanisms and provides opportunities for therapeutic intervention by using full-length Factor H, its fragments, or complement-modulatory compounds.
Introduction
Dysregulation of Factor H cause human diseases
Factor H is a key regulator of complement, an essential component of innate immunity responsible for homeostasis, and elimination of infectious microbes.1,2 Complement dysregulation can lead to diseases affecting almost every organ, including the kidneys, heart, blood, liver, brain, nervous system, and eyes, and increased susceptibility to infection.3
Deregulated complement is amenable to therapy.4 Several complement-targeting drugs are approved and currently used to treat diseases such as atypical hemolytic uremic syndrome (aHUS), paroxysmal nocturnal hemoglobinuria, generalized myasthenia gravis, geographic atrophy, age-related macular degeneration (AMD), neuromyelitis optica spectrum disorder, and Chapel's disease. Other inhibitors are under development and in advanced clinical trials. The challenge today is to identify more complement disorders that can be treated with existing or novel complement inhibitors. For each disorder, it is important to determine the affected component, pinpoint the specific defect in the complement cascade or effector system, understand the pathophysiology, identify the most suitable therapeutic target(s), and develop or select the most effective inhibitor.4
A closer look at complement
Appearing very early in evolution, complement proteins are integrated into the immune system, contributing to both innate and adaptive immune responses, while also playing important physiological and metabolic roles.5,6 Complement has 3 activation pathways and forms 2 major enzymatic platforms, namely the C3 and C5 convertases. Three major effector routes include opsonization and phagocytosis of altered self-particles and infectious agents, generation of inflammatory anaphylatoxins, and targeted cell lysis.7 The complement system consists of more than 100 proteins, including central components, regulators, activation fragments, and receptors interacting with specific components to induce specific effector function(s).8
Soluble components can assemble into active protein complexes and these can induce proteolysis of inert precursor proteins a process that is central to the function of this cascade system. Cleaved fragments can acquire biologic activity or bind to new ligands or receptors and induce cell activation and responses. New complement components are being continously integrated into this system, and also new functions for well-characterized complement proteins are frequently discovered. In addition, links to other cascade systems and metabolic functions are being identified.9
Complement acts in plasma and body fluids, which explains its ability to affect almost all tissues. Similarly, the transition from the fluid phase to surfaces is important for targeted complement action, with regulators and receptors displayed on cell surfaces. Factor H and many plasma-distributed proteins are primarily produced and secreted by the liver, forming the systemic complement. Extrahepatic sources of Factor H exist, including macrophages, leukocytes, or stimulated fibroblasts, which contribute to local effects and fine-tuning of complement action.10,11 Recent evidence suggests that Factor H plays intracellular roles, with novel, noncanonical functions, including metabolic and even nuclear effects.9
The alternative pathway: A self-propagating initiator and amplifier
The complement system, as a self-sustaining enzymatic cascade, is activated by 3 pathways: the alternative pathway (AP), the classical pathway (CP), and the lectin pathway (LP). The AP is active by default and always on. The other 2 pathways require triggers for activation.
The AP is initiated by tick-over activation of a C3(H2O) molecule in solution. Circulating C3 can spontaneously undergo a conformationally change to form C3(H2O), which adopts a C3b-like conformation. This tick-over activation is spontaneous, occurs at a low rate, and exposes a transient thioester bond. The initial C3(H2O) has a very short half-life and can bind nearby molecules, including surfaces. Surface-bound C3(H2O) can form an active convertase with Factor B in the presence of Factor D and initiate activation the amplification loop.12 This initial convertase, if not properly controlled (e.g. by Factor H), does trigger complement activation. Thus, at this initial phase control of convertase formation and amplification is essential, highlighting the relevance of regulators already at this step.7,12
The CP is activated by antibody-antigen complexes, and the LP is triggered when MBL (mannose binding lectin) binds to the terminal carbohydrate residues, such as mannose that are commonly found on microbial surfaces. In addition, ficolin-1, ficolin-2 and ficolin-3, as well as collectin-K1 and collectin-L1, can function as LP associated pattern recognition molecules by binding to acetylated surface entities including acetylated carbohydrate residues.13 Each activation pathway generates a C3 convertase that cleaves circulating C3 into C3a and C3b. C3a recruits immune cells, mediates inflammation, and has antimicrobial activity. C3b targets altered self or foreign particles for efficient elimination by C3b receptor–expressing immune cells.11,14
The AP convertase, C3bBb, Factor B binds to surface-deposited C3b and is then cleaved by Factor D to Bb, releasing Ba. CP and LP also form specific C3 convertases. In this case, C4b binds C2, consequently the serine protease C1s cleaves C2, releasing C2a, while C2b, a serine protease itself, remains bound to C4b to form the active C3 convertase C4bC2b. C3 convertases represent the initial enzymatic tier of complement and are primarily deposited on target surfaces.7 C3b generated by the both C3 convertases can induce a powerful AP-driven amplification loop that perpetuates activation.
When C3b binds to both AP or LP/CP C3 convertases, the cascade proceeds to the second major enzymatic step. The newly formed convertases change substrate specificity and cleave C5 producing C5a, the potent anaphylatoxin, and C5b.14 C5b assembles with C6, C7, C8, and C9 to form C5b-C9n (also named membrane attack complex or terminal complement complex), which generates a lytic pore.15
The initiated complement cascade generates potent effector components, many of which have chemotactic and cell-activating capacities, that orchestrate immune responses, while others are toxic and destructive. Regulation is central to complement action. A battery of regulators, found in the circulation or on cell surfaces, tightly control and modulate each single reaction.1,2 The self-propagating nature of the AP and the potent toxic effects underscore the need for regulation in time, space, and intensity. Destructive effects are directed to the appropriate sites, and the release and activity of inflammatory mediators are limited in time and space. At the same time, healthy cells and intact self-surfaces are protected from damage.
Regulatory proteins are distributed throughout the fluid phase, with some able to bind to surfaces and others integrated into cell membranes. Multiple regulators with overlapping functions work together to fine-tune complement activity, ensuring precise action at specific sites. As a result, regulators with overlapping profiles together adjust a precisely balanced activation and targeted effector functions.
Factor H: The major regulator of the alternative pathway
Factor H is the major regulator of the alternative pathway, controlling proximal complement at the C3 level (Fig. 1A).1,10,11
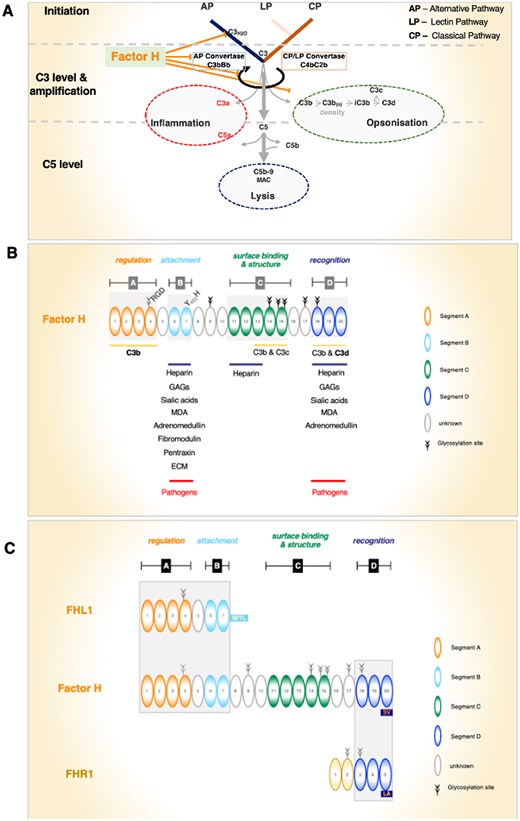
(A) Factor H controls multiple steps of proximal complement. The central steps of complement activation: the AP, LP, and CP induce C3 convertase and C5 convertases. Activation results in 3 major effector functions: inflammation by the activation products C3a and C5a, opsonization by surface-deposited C3b and by C3b split products, and cell lysis is induced by C5b-9n, also termed membrane attack complex (MAC) or terminal complement complex. Factor H (i) controls C3(H2O) AP tick-over activation of the AP; (ii) targets C3 activation; (iii) controls the activity of the AP C3 convertase, C3bBb (decay accelerating activity); (iv) adjusts the AP amplification loop; and (v) assists Factor I in the progression and inactivation of C3b (cofactor activity). (B) Elongated Factor H domain structure and binding segments. Factor H domain structure with the 4 binding and functional units (i.e. segments). Factor H has 3 C3-interacting segments, each of which has a preference for different C3 variants and binds with different affinities to the C3 fragments. Factor H has 3 heparin binding regions. The binding sites and identified ligands are shown. Segment A has regulatory functions, Segment B has a role in attachment, the function of segment C is not defined in detail so far, and segment D is involved in surface recognition. Please note that segments A and B bind single ligands, either C3 or surface constituents, and segments C and D bind both C3 and surface ligands. (C) FHL1 and FHR1 share identical and related domains with Factor H. FHL1 is derived from the Factor H gene and FHR1 is encoded by a separate unique gene. FHL1 shares the 7 N-terminal SCR domains with Factor H including the N-terminal regulatory and the attachment recognition region. FHR1, which forms dimers in solution, shares the 3 C-terminal SCRs (i.e. the attachment and sensor region D). Shared functional segments and SCR domains are highlighted by darker background boxes.
The Factor H gene and the Factor H-FHR gene cluster
The human Factor H gene encodes Factor H and Factor H-like protein 1 (FHL1), the alternatively spliced FHL1.16 The Factor H gene has 23 exons, and the first encodes the signal peptide. The remaining 22 exons, for the most part encode single short consensus repeat (SCR) domains, the exceptions being SCR2, which is encoded by split exons 3 and 4, and exon 10, which defines the alternatively spliced exon encoding the unique C-terminal part of FHL1.17 The Factor H gene is located on human chromosome 1q32, in the Factor H-FHR gene cluster. The genes of this cluster are arranged in the order Factor H, FHR3, FHR1, FHR4, FHR2, and FHR5. This cluster contains several highly homologous long interspersed duplicated segments that are prone to chromosomal instability. Recombination events can generate hybrid genes, chromosomal deletions, and duplications.18 Factor H is the best characterized member of this protein family, which also includes FHL1, and the 5 Factor H–related proteins.
Structure of Factor H–ligand interaction and functional regions
Factor H, with the signal peptide removed, is a secreted 150 kDa single-chain 1,213-amino-acid plasma glycoprotein assembled into 20 SCR domains. Each SCR represents a unit with 4 cysteine residues. Disulfide bonds give a globular structure. The concentration of Factor H in human plasma is approximately 500 µg/mL. In the literature, its concentration in plasma ranges from approximately 150 µg to 700 µg/mL. Such variations may depend on the experimental assay used, particularly in enzyme-linked immunosorbent assays by the antibodies used for capture and detection. Historically, FHR crossreacting antibodies have been used. In addition, common genetic variants of Factor H and FHRs also influence Factor H plasma levels.
The protein sequence of Factor H includes 8 consensus sites for N-linked glycosylation. FHL1 is not glycosylated. Glycosylation does not seem important for complement control but likely affects protein stability. The half-life of Factor H in plasma is approximately 6 d.19
Factor H controls proximal complement at the C3 convertase level and influences the fate of C3. (Fig. 1A), and it (i) regulates AP tick-over activation in the fluid phase; (ii) controls the transition from the fluid phase to surfaces; (iii) regulates the formation, stability, and activity of C3bBb convertases, through decay-accelerating activity, and modulates C3b density on surfaces; (iv) also influences the fate and processing of surface-deposited C3b by acting as a cofactor for the further degradation and processing of C3b by the protease Factor I; (v) modulates the strength of the AP amplification loop; and (vi) can discriminate between intact and damaged self-surfaces and non-self foreign surfaces,
Factor H acts in the fluid phase and on surfaces and membranes under immune attack and also interacts with foreign particles including pathogenic microbes. In addition, intracellular and nuclear roles of Factor H are emerging.20
Structure-function analyses, along with disease-associated genetic alterations and autoantibodies, highlight 4 functional segments in the Factor H protein (Fig. 1B):
Segment A: the N-terminal regulatory segment, encompassing SCR1-4, contains a high-affinity C3b binding region, and includes in SCR4 and RGD integrin binding motif.
Segment B: the N-terminal attachment segment, located by SCR6-7, binds to heparin, to glycosaminoglycans (GAGs), sialic acids, and pattern recognition molecules.
Segment C: the middle element, which is functionally less well characterized, contains overlapping, binding sites for C3b, C3c, and heparin, and likely contains a structural element (SCR10-15).
Segment D: SCR18-20, the C-terminal surface recognition region, uses overlapping binding sites for C3b/C3d heparin, GAGs, sialic acids, and pattern recognition molecules. This recognition region directs Factor H to C3b/C3d-decorated self-surfaces and thereby acts as a central sensor element.
For segment A, the N-terminus of Factor H, with SCR1–4, represents the central regulatory segment and includes a high-affinity binding element for C3b/C3d.1,10 This region binds to native C3 with a Kd of 0.4 µM and to C3b with a Kd of 0.08 µM.21 Complement regulatory functions include decay accelerating activity for AP C3-convertase (C3bBb) and cofactor activity for the C3b cleaving protease Factor I. Decay accelerating activity controls C3 convertase assembly, activity, stability, and dissociation of this convertase. Cofactor-mediated activity includes inactivation of surface-deposited C3b and processing to iC3b and C3d. Structural analyses show that SCR1–4 binds almost diagonally and covers the entire length of C3b.22 In addition, the SCR4 domain contains RGD residues, which represent a consensus motif for integrin receptors such as CR3 (CD11b/CD18) and CR4 (CD11c/CD18) (Fig. 1B).23
Segment B, formed by SCR6–7, represents a surface attachment element and includes high-affinity binding sites for heparin, GAGs, and sialic acids. This segment binds to heparin with high-affinity, Kd 1.2 µM21 and binds to other surface moieties including polyanionic GAGs, sialic acids, and surface components, such as monomeric C-reactive protein (mCRP), malondialdehyde (MDA), fibromodulin, adrenomedullin, pentraxin III, and extracellular matrix (ECM) components.24,25 Importantly, this recognition segment does neither bind C3b nor C3d. This interaction region modulates Factor H and FHL1 binding to modified self-cells including necrotic and apoptotic cells. In addition, many pathogenic microbes bind host Factor H and FHL1 through this segment.
For segment C, the middle segment of Factor H contains low-affinity interaction sites for C3b, C3c, and heparin and appears important for protein conformation. An initial study identified an interaction site for C3b and C3c within SCR12–14.26 Subsequent analyses determined for SCR13–15 a Kd of 15 μM for C3b.21 A heparin binding region was also identified within SCR13 and for SCR11–13, the affinity for heparin was determined to Kd 17 μM. This middle segment has overlapping low-affinity binding sites for both C3b and heparin. In addition, SCR10-14, represents a kink element that probably induces a loop structure, with the regulatory segment A positioned next to or close to the C-terminal recognition segment D.27 The exact role of this region for Factor H conformation and surface action remains to be defined.
For segment D, the C-terminal recognition and attachment element of Factor H is represented by SCR18–20. This primary surface sensor combines C3b/C3d binding sites with heparin/GAG binding elements. The segment binds to C3b and C3d but not to native C3. In addition, segment D interacts with heparin as well as surface-exposed GAGs, sialic acids, and pattern ligands such as mCRP, MDA, ECM, pentraxin, and adrenomedullin.25,26 This C-terminal segment contacts surface-deposited C3b and C3d, with affinities of 1.7 μM and 1.9 μM, respectively.21 Structural studies show that SCR19–20 contacts a short region in C3d, that is also accessible in C3b.28,29 This C-terminal segment is shared with FHR1 (Fig. 1C).30 The same segment binds heparin with a Kd of 4.9 μM. In addition, there is evidence that this C-terminal region contains discontinuous heparin/GAG binding sites.
All 3 SCRs contribute to the interaction, but their roles differ. SCR18 supports the interaction and can likely influence protein conformation. SCR19 contains the major binding sites for C3b/C3d and heparin. Variations in this domain influence selectivity and alter the cooperation of Factor H with FHR1, driving selective recognition and the discriminatory action of Factor H. SCR20 also supports this interaction (see Factor H never acts alone: FHL1 and FHR1 share domains with Factor H).
Segment D is unique in that it contains overlapping high-affinity binding sites for C3b, iC3b, C3d, heparin, GAGs, sialic acids, and pattern recognition proteins. The central role of this element in surface discrimination is highlighted by aHUS-associated gene mutations that cluster in segment D of Factor H. In the autoimmune form, homozygous FHR1-FHR3 deficiency and autoantibody positive form of HUS (DEAP-HUS), autoantibodies target this C-terminal region and block Factor H surface binding. This highlights its role as a sensor and discriminator.
Full-length Factor H also binds additional proteins, including annexin, DNA, lipids, ECM components, and neutrophil extracellular traps (NETs). However, the binding sites have not yet been mapped.
Noncanonical functions of Factor H
The Factor H-FHR gene cluster evolved early in evolution, long before the emergence of the adaptive immune system, which is dominated by T and B lymphocytes.30 As a result, Factor H, FHL1, and the FHRs are integrated into key biological steps. Today, 65 yr after the isolation of Factor H, new functions and new action sites are discovered.31
Factor H controls proximal complement at multiple sites in the fluid phase, directs the transition from circulation to surfaces, and acts on surfaces. In addition to these established canonical roles, noncanonical roles have been identified. Factor H induces monocyte chemotaxis, thromboxane and prostaglandin E release from macrophages, and phagocytosis, and also copurifies with thrombospondin and binds to human leukocytes. Factor H promotes the accumulation of mononuclear phagocytes at sites of inflammation thereby inhibiting resolution of inflammation.32
Factor H also induces monocyte differentiation, into immunosuppressive CD14-positive macrophages.32 Interestingly, the C-terminal segment D is responsible for these morphological changes. In addition, in monocytes extracellular Factor H drives intracellular C3 expression. Macrophage-derived Factor H reduces the resolution of inflammation affecting efferocytosis and plaque size in a model of atherosclerosis.
Recently, Factor H was identified as a ligand for inducible T cell costimulator, which is expressed on regulatory T cells.33 Factor H promoted the survival of regulatory T cells in a glioma microenvironment. In addition, intracellular Factor H has been described in tumor cells and platelets34,35 and nuclear Factor H interacts with the cell cycle transcription factor E2F3.36 Also a signaling role of Factor H during apoptosis is reported.37
Factor H has also been described to act as the regulator of the CP.38 This role could be significant, though its direct role in AP convertase regulation is pending. An alternative explanation can be that Factor H and the classical pathway initiator C1q bind to related surface constituents including anionic phospholipids, lipid A, and Escherichia coli and thereby compete for ligand binding and interaction. Showing that the pattern recognition roles of Factor H and of the other complement proteins and pattern recognition molecules is an important field to follow.
Factor H never acts alone: FHL1 and FHR1 share domains with Factor H
Factor H’s action is modulated by FHL1 and FHR1, 2 human plasma proteins that share SCR domains with Factor H. Both proteins can cooperate with Factor H, but also oppose its binding thereby influencing and modulating local complement. Tracking the interplay between Factor H, its related regulator FHL1, and the modulator FHR1 is essential for understanding the physiology of Factor H and the dynamics of the local action profile.
FHL1 shares the N-terminal SCR1-7 with Factor H, including segment A and segment B. Thus, FHL1 combines the regulatory C3b interaction region with attachment element B (Fig. 1C). While FHL1 and Factor H cooperate in fluid phase control, they differ in length and conformation, which explains differences in surface binding and in surface control. FHL1 most likely adopts a linear extended conformation. This allows FHL1 to discriminate for C3b decorated surfaces in a manner distinct from Factor H.39 In contrast, Factor H in its condensed conformation, exposes the C3b/C3d and GAG interacting segment D, while segments A, B and C are likely masked (Fig. 2A for Factor H in solution).40

The conformation of Factor H influences the action profile. (A) Factor H conformation in solution. The protein most likely has a compact, condensed, and folded structure. (B–E) Factor H has different possibilities to bind to deposited C3b (as well as to iC3b, and C3d), surface-exposed heparin, GAGs, sialic acids, and pattern recognition molecules. The flexibility of Factor H allows for different interactions on surfaces. For an elongated linear Factor H protein, single-point contacts orient the other protein segments outward, toward the environment. Surface attachment of full-length Factor H via single segments orients and exposes different parts of the protein and makes them available for ligand binding and function. Factor H can adopt multiple conformations. (F) Factor H attached via C-terminal segment D to surface-deposited C3b or C3d allows control of a larger surface area and large hemispheres. (G) Factor H folded back can interact with the two segments, the N-terminal segment A and also via the C-terminal D to the same C3b molecule. (H) Elongated Factor H can also interact via segment A and segment D with 2 separate surface-bound C3b proteins.
FHR1, which is encoded by a separate gene, shares recognition segment D, the 3 C-terminal SCRs, with Factor H.41 Composed of 5 SCRs, FHR1 has a plasma concentration of approximately 70 to 100 µg/mL and exists in 2 glycosylated isoforms of 37 and 42 kDa. FHR1 can form homodimers, and heterodimers with FHR2 and FHR5, and interacts with Factor H on apoptotic surfaces.42,43
FHR1 and Factor H share segment D, ie the 3 C-terminal SCRs, with the C3b/C3d and heparin/GAG binding sites. However, in the most C terminal domain, FHR1 and Factor H differ by 2 amino acids. FHR1 uses LA (FHR1L290A296) in SCR5, whereas Factor H uses SV (Factor HS1191V1197) in SCR20. These changes have functional consequences. An amino acid swap resulting in either FHR1S290V296 with the 2 residues of Factor H or Factor HL1191A1197 with the residues of FHR1 causes aHUS pathology. This highlights that LA versus SV residues in the most C-terminal SCRs affect surface attachment and likely influence the regulatory action and fate of C3b and C3d decorated surfaces.44,45
FHR1 influences Factor H binding to C3b decorated surfaces and affects local complement control. Because FHR1 blocks access of Factor H to these surfaces, it has been argued that FHR1 acts as a competitor for Factor H, reducing complement control at the endothelial surface. However, because FHR1 has a modulatory function of its own the situation appears more complex.46 Although both Factor H and FHR1 bind to closely related surfaces under complement attack, each protein prefers different C3 decorated surfaces and has distinct functions. FHR1 prefers C3d decorated surfaces and Factor H has a preference for C3b decorated surfaces.45 In addition, surface-bound FHR1 mediates the silent clearance of particles promoting inflammation resolution. Thus, FHR1 can compete with surface-bound Factor H and support inflammatory effects. Despite these differences, the 2 proteins cooperate in surface control. Factor H assists in C3 convertase regulation, C3b inactivation and processing, while FHR1 bound to C3d decorated surfaces can induce inflammatory clearance.46
The Factor H:FHR1 activity balance
Factor H, the regulator, and FHR1, the modulator, tune and balance the progression and effector roles of complement. Homozygous combined FHR1-FHR3 deficiency confirms the relevance of the tight interplay in for surface complement control. This combined deficiency is protective and thus beneficial in AMD, IgA nephropathy (IgAN) and antineutrophil cytoplasmic antibody–associated vasculitis (AAV).47,48 But the same deficiency increases the risk for DEAP-HUS and systemic lupus erythematosus (SLE).49–51 In the protective setting, efficient local Factor H mediated proximal complement control appears beneficial, with FHR1 action being less relevant. In the risk setting, FHR1 modulated inflammatory control is necessary, while Factor H dominated complement regulation appears less relevant or detrimental.
C3b and surface binding segments have unique interaction profiles
Full-length Factor H can bind to C3 or processed C3b products via 2 high-affinity and 1 low-affinity binding segments. Similarly, Factor H contacts heparin or sialic acids via 3 distinct sites.
Importantly, both N-terminal segments bind to individual ligands with high affinities. Segment A binds to C3b, but not to heparin, while segment B binds to heparin but not to C3. In contrast, segments C and D, combine lower affinity sites with overlapping sites for C3b/C3d and heparin/GAG interaction.
Each C3-interacting segment is unique in contacting intact C3 or C3 isoforms. Segment A contacts native C3, C3b, and iC3b but binds to neither C3c nor C3d. Segment C binds to C3b with 2,000-fold lower affinity than segment A and also binds to C3c (Table 1).26 Segment D contacts C3b with approximately 20-fold lower intensity and binds C3d and iC3b but not native C3. Thus, each segment contacts different regions of C3 and different isoforms.
Binding affinities of Factor H segments and full-length Factor H to C3 and to heparin.
| Segment A: SCR1–4 (regulation) | Segment B: SCR6–7 (attachment) | Segment C: SCR11–15 (C3b-surface binding and structure) | Segment D: SCR18–20 (recognition) | Factor H | ||
|---|---|---|---|---|---|---|
| C3 | C3 | 0.4 µM | — | — | NBD | |
| C3b | 0.08 µM | — | 15.0 µM | 1.7 µM | 0.1 µM | |
| iC3b | 5.0 µM | — | — | 2.8 µM | ||
| C3c | NBD | — | — | NBD | ||
| C3d | NBD | — | — | 1.9 µM | ||
| Heparin | — | — | 1.2 µM | 17.0 µM | 4.9 µM | 0.4 µM |
| Segment A: SCR1–4 (regulation) | Segment B: SCR6–7 (attachment) | Segment C: SCR11–15 (C3b-surface binding and structure) | Segment D: SCR18–20 (recognition) | Factor H | ||
|---|---|---|---|---|---|---|
| C3 | C3 | 0.4 µM | — | — | NBD | |
| C3b | 0.08 µM | — | 15.0 µM | 1.7 µM | 0.1 µM | |
| iC3b | 5.0 µM | — | — | 2.8 µM | ||
| C3c | NBD | — | — | NBD | ||
| C3d | NBD | — | — | 1.9 µM | ||
| Heparin | — | — | 1.2 µM | 17.0 µM | 4.9 µM | 0.4 µM |
Factor H uses 3 segments to bind to C3 or to C3 fragments and also 3 segments to bind to heparin. The two N-terminal segments bind either to C3 (segment A) or to heparin (segment B), while the two other units, segment C and segment D, include overlapping binding sites that bind to both C3 and heparin. These fragments and their binding affinities have been identified by several expert groups. To minimize the experimental variations and effects, we here present the data from one group.25 However, variations in the experimental setup, use of different factor fragments, and combinations of SCR domains add larger variations, making a direct comparison more difficult. In addition, the expression and purification of the fragments and the type of immobilization did vary. Overall, all these data show related results and interaction profiles. An em dash indicates that the relationship was not evaluated.
NBD, no binding detected.
Binding affinities of Factor H segments and full-length Factor H to C3 and to heparin.
| Segment A: SCR1–4 (regulation) | Segment B: SCR6–7 (attachment) | Segment C: SCR11–15 (C3b-surface binding and structure) | Segment D: SCR18–20 (recognition) | Factor H | ||
|---|---|---|---|---|---|---|
| C3 | C3 | 0.4 µM | — | — | NBD | |
| C3b | 0.08 µM | — | 15.0 µM | 1.7 µM | 0.1 µM | |
| iC3b | 5.0 µM | — | — | 2.8 µM | ||
| C3c | NBD | — | — | NBD | ||
| C3d | NBD | — | — | 1.9 µM | ||
| Heparin | — | — | 1.2 µM | 17.0 µM | 4.9 µM | 0.4 µM |
| Segment A: SCR1–4 (regulation) | Segment B: SCR6–7 (attachment) | Segment C: SCR11–15 (C3b-surface binding and structure) | Segment D: SCR18–20 (recognition) | Factor H | ||
|---|---|---|---|---|---|---|
| C3 | C3 | 0.4 µM | — | — | NBD | |
| C3b | 0.08 µM | — | 15.0 µM | 1.7 µM | 0.1 µM | |
| iC3b | 5.0 µM | — | — | 2.8 µM | ||
| C3c | NBD | — | — | NBD | ||
| C3d | NBD | — | — | 1.9 µM | ||
| Heparin | — | — | 1.2 µM | 17.0 µM | 4.9 µM | 0.4 µM |
Factor H uses 3 segments to bind to C3 or to C3 fragments and also 3 segments to bind to heparin. The two N-terminal segments bind either to C3 (segment A) or to heparin (segment B), while the two other units, segment C and segment D, include overlapping binding sites that bind to both C3 and heparin. These fragments and their binding affinities have been identified by several expert groups. To minimize the experimental variations and effects, we here present the data from one group.25 However, variations in the experimental setup, use of different factor fragments, and combinations of SCR domains add larger variations, making a direct comparison more difficult. In addition, the expression and purification of the fragments and the type of immobilization did vary. Overall, all these data show related results and interaction profiles. An em dash indicates that the relationship was not evaluated.
NBD, no binding detected.
Segment B binds to heparin, GAGs, and sialic acids. Segments B and D have similar binding profiles for heparin, GAGs, sialic acids and surface ligands and pattern recognition molecules, such as mCRP, MDA, pentraxin, and ECM components. Both segments also bind to necrotic surfaces and to pathogenic microbes.
Factor H; the other family members (FHL1, FHR1, FHR2, FHR3, FHR4, FHR5); multiple fluid phase complement proteins, including C4bp, C1q, MBL, ficolin-1, ficolin-2, ficolin-3, collectins (CL-K1, CL-L1), and pentraxin III; and the many other pattern recognition proteins bind to related ligands. Thus a large variety of plasma pattern recognition molecules bind the same or highly related surface targets. This shows a tightly regulated, fine-tuned homeostatic role of complement in pattern and surface recognition, which is essential for self- vs non–self-discrimination and the innate immune response.52,53
Factor H conformation influences target interaction
Factor H, with its “bead-like structure,” shows extreme flexibility, allowing multiple conformations and dynamic interactions with C3, C3b, iC3b, and C3d, as well as with heparin, GAGs, and sialic acids. In solution, Factor H adopts a folded, and compact, likely zinc-depended conformation and has the C-terminus exposed.54,55 The other interaction sites are either masked or hidden (Fig. 2A). Factor H can also form homodimers and complexes with FHR1.42,43,45
Full-length Factor H and segment A exhibit nearly identical affinity for C3b, indicating no apparent cooperation among the sites. With respect to heparin binding, intact Factor H shows approximately 3-fold higher affinity compared with segment B alone, 42-fold higher binding compared with segment C, and 12-fold higher binding than segment D (Table 1). This indicates a cooperation among the 3 heparin/GAG sites.
Factor H conformation affects the accessibility of the binding sites
In solution, condensed Factor H primarily targets a surface via segment D (Fig. 2A). Upon binding, Factor H undergoes a conformational change, making the 4 segments with all 6 interaction sites accessible. Thus unfolded Factor H can interact with deposited C3 via 3 sites and surfaces via 3 separate segments.55,56
Single versus multipoint interaction
Factor H, when bound via the C-terminal segment D to a single C3b molecule, remains flexible and controls a relatively large surface area, or 3-dimensional hemisphere (Fig. 2B–E). However, when bound more tightly to C3b and heparin, Factor H has less flexibility, resulting in a smaller regulated area or volume. With all 6 binding sites engaged, Factor H makes stable contacts and adopts a rigid conformation (Fig. 2F–H). In addition, the bound segment defines the orientation and accessibility of the other sites, and the controlled the surface area or the 3-dimensional hemisphere. When Factor H is bound to C3b via segment D, segment A remains accessible to bind either the same C3b molecule (Fig. 2G), or alternatively, segment A can bind a second neighboring C3b assisting in its inactivation (Fig. 2H).
FHL1 influences Factor H contact via segment B, while FHR1 influences contact via segment D. Thus, FHL1 or FHR1 can fine-tune local Factor H complement control.
The surface influences Factor H binding
The surface itself exposes Factor H ligands, such as C3b, C3b degradation products, GAGs, sialic acids, and pattern recognition molecules. The combination and density of these ligands affect Factor H binding strength, determine which domains are bound and thereby affect protein conformation. This, in turn, influences which interaction segments become exposed and accessible. Thus, multiple ligands precisely influence Factor H positioning and ultimately determine timing and duration of local control.54,57,58
The surface plays a key role in Factor H binding and thereby can adjust for the regulator density and activity, and also the controlled area or volume.45 C3b deposition is indiscriminate in its initial steps, and subsequently regulators, such as Factor H, adjust the self- and non–self-distinction. On intact self-surfaces, complement activation and toxic effector functions are undesirable and prohibited. On modified self-surfaces (i.e. apoptotic or necrotic particles), moderate regulated activation allows controlled inflammation and phagocytic clearance. This opsonphagocytosis by recruited immune cells is highly advantageous for the removal of self-debris and the elimination of infectious microbes.57,58 Thus, the density and type surface ligands modulate Factor H binding, regulate complement action, and ultimately discriminate between intact self, modified self, and non-self (foreign) surfaces.
Factor H–mediated diseases: Role in pathophysiology
Factor H alterations are associated with human diseases, highlighting the global effects of this multifunctional inhibitor in tissues.59–61 Pathogenic scenarios include gene mutations, chromosomal deletions, and copy number variations in the Factor H-FHR gene cluster and even common gene polymorphisms. At the protein level, these alterations result in mutant Factor H, absent protein(s), Factor H::FHR hybrid proteins, and altered Factor H:FHR1 plasma levels. Factor H binding autoantibodies also cause diseases.62–64
Factor H–associated diseases include (i) C3G, (ii) aHUS/DEAP-HUS, (iii) AMD, (iv) IgAN, (v) AAV, (vi) cancer, and (vii) infections by pathogens (Table 2).61,65–68 These diseases manifest in different organs and affect different tissues and cell types. Segment A is affected in C3G and AAV,69,70 segment B in AMD and cancer, and the C-terminal segment D in aHUS/DEAP-HUS and cancer as well (Fig. 3). This highlights the importance and specific roles of each segment. In addition, during evolution, pathogenic microbes have adopted strategies to use Factor H, FHL1, and FHR1 for complement evasion. Many pathogenic microbes exploit Factor H and FHL1 via segment B, and they also bind Factor H and FHR1 via segment D.61,65
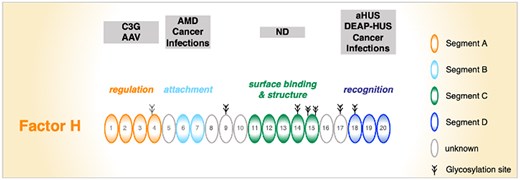
Specific segments or functional units of Factor H are targeted in human diseases. Segment A is mainly affected in C3G and AAV. Segment B contains the AMD-relevant Y402H polymorphism. Both variations affect Factor H and FHL1. Segment C has not been associated with disease so far. Segment D is often altered in aHUS and is the major target for autoantibodies in DEAP-HUS. In addition, pathogenic microbes attach Factor H (and also FHL1) to their surface via segment B, and they also acquire Factor H and FHR1 via segment D. ND, not determined.
Factor H, FHL1, and FHR1 in human diseases
| Disease | Role of complement | Details | Affected organ and tissue | Localization Factor H mutations | Autoantibody binding domain | FHR1-FHR3 | Comment | |
|---|---|---|---|---|---|---|---|---|
| 1 | C3G | Proximal complement; AP | Chronic | Kidney glomerular environment; GBM | Region A; Factor H and FHL1 | Region A | ND | — |
| 2 | aHUS, DEAP-HUS | Distal complement; terminal pathway | Acute | Kidney endothelial cells of blood vessels | Region D; Factor H and FHR1 | Region D | Risk | — |
| 3 | AMD | Likely proximal complement; AP | Chronic, and age | Retinal, epithelial cells, Bruch’s membrane | Region B; Factor H and FHL1 | ND | Protective | — |
| 4 | IgAN | Proximal complement; AP | — | Glomerulus | ND | ND | Protective | — |
| 5 | AAV | Inflammatory complement | — | Neutrophils | ND | Not to Factor H | Protective | — |
| 6 | Cancer | Extracellular and intracellular complement | Hepato-cellular carcinoma in aged complement Factor H knockout mice | Multiple |
| Not to Factor H | ND |
|
| 7 | Pathogenic microbes | AP, LP, and CP; all levels of complement | Acute infections | Immune evasion—infection | Regions B and D; Factor H, FHL1, and FHR1 | No antibodies | RISK Neisseria meningitidis |
| Disease | Role of complement | Details | Affected organ and tissue | Localization Factor H mutations | Autoantibody binding domain | FHR1-FHR3 | Comment | |
|---|---|---|---|---|---|---|---|---|
| 1 | C3G | Proximal complement; AP | Chronic | Kidney glomerular environment; GBM | Region A; Factor H and FHL1 | Region A | ND | — |
| 2 | aHUS, DEAP-HUS | Distal complement; terminal pathway | Acute | Kidney endothelial cells of blood vessels | Region D; Factor H and FHR1 | Region D | Risk | — |
| 3 | AMD | Likely proximal complement; AP | Chronic, and age | Retinal, epithelial cells, Bruch’s membrane | Region B; Factor H and FHL1 | ND | Protective | — |
| 4 | IgAN | Proximal complement; AP | — | Glomerulus | ND | ND | Protective | — |
| 5 | AAV | Inflammatory complement | — | Neutrophils | ND | Not to Factor H | Protective | — |
| 6 | Cancer | Extracellular and intracellular complement | Hepato-cellular carcinoma in aged complement Factor H knockout mice | Multiple |
| Not to Factor H | ND |
|
| 7 | Pathogenic microbes | AP, LP, and CP; all levels of complement | Acute infections | Immune evasion—infection | Regions B and D; Factor H, FHL1, and FHR1 | No antibodies | RISK Neisseria meningitidis |
The table depicts Factor H gene mutations, copy number variations in the Factor H-FHR gene cluster, and autoantibodies in human complement mediated diseases.
ND, not described.
Factor H, FHL1, and FHR1 in human diseases
| Disease | Role of complement | Details | Affected organ and tissue | Localization Factor H mutations | Autoantibody binding domain | FHR1-FHR3 | Comment | |
|---|---|---|---|---|---|---|---|---|
| 1 | C3G | Proximal complement; AP | Chronic | Kidney glomerular environment; GBM | Region A; Factor H and FHL1 | Region A | ND | — |
| 2 | aHUS, DEAP-HUS | Distal complement; terminal pathway | Acute | Kidney endothelial cells of blood vessels | Region D; Factor H and FHR1 | Region D | Risk | — |
| 3 | AMD | Likely proximal complement; AP | Chronic, and age | Retinal, epithelial cells, Bruch’s membrane | Region B; Factor H and FHL1 | ND | Protective | — |
| 4 | IgAN | Proximal complement; AP | — | Glomerulus | ND | ND | Protective | — |
| 5 | AAV | Inflammatory complement | — | Neutrophils | ND | Not to Factor H | Protective | — |
| 6 | Cancer | Extracellular and intracellular complement | Hepato-cellular carcinoma in aged complement Factor H knockout mice | Multiple |
| Not to Factor H | ND |
|
| 7 | Pathogenic microbes | AP, LP, and CP; all levels of complement | Acute infections | Immune evasion—infection | Regions B and D; Factor H, FHL1, and FHR1 | No antibodies | RISK Neisseria meningitidis |
| Disease | Role of complement | Details | Affected organ and tissue | Localization Factor H mutations | Autoantibody binding domain | FHR1-FHR3 | Comment | |
|---|---|---|---|---|---|---|---|---|
| 1 | C3G | Proximal complement; AP | Chronic | Kidney glomerular environment; GBM | Region A; Factor H and FHL1 | Region A | ND | — |
| 2 | aHUS, DEAP-HUS | Distal complement; terminal pathway | Acute | Kidney endothelial cells of blood vessels | Region D; Factor H and FHR1 | Region D | Risk | — |
| 3 | AMD | Likely proximal complement; AP | Chronic, and age | Retinal, epithelial cells, Bruch’s membrane | Region B; Factor H and FHL1 | ND | Protective | — |
| 4 | IgAN | Proximal complement; AP | — | Glomerulus | ND | ND | Protective | — |
| 5 | AAV | Inflammatory complement | — | Neutrophils | ND | Not to Factor H | Protective | — |
| 6 | Cancer | Extracellular and intracellular complement | Hepato-cellular carcinoma in aged complement Factor H knockout mice | Multiple |
| Not to Factor H | ND |
|
| 7 | Pathogenic microbes | AP, LP, and CP; all levels of complement | Acute infections | Immune evasion—infection | Regions B and D; Factor H, FHL1, and FHR1 | No antibodies | RISK Neisseria meningitidis |
The table depicts Factor H gene mutations, copy number variations in the Factor H-FHR gene cluster, and autoantibodies in human complement mediated diseases.
ND, not described.
The role of Factor H and complement in disease pathology is well understood for several disorders. In C3G and aHUS, gene mutations and Factor H binding autoantibodies target the same segments (i.e. segment A and D, respectively). In addition, altered plasma Factor H:FHR1 ratios contribute to disease pathology. However homozygous FHR1-FHR3 deficiency, which alters the Factor H:FHR1 ratio, defines both risk and protective scenarios (Table 2).47–51
C3 glomerulopathy
C3G is an ultra-rare chronic kidney disease due to persistent AP activation, with pathology developing over years.60,66 C3G is a multifactorial disease characterized by C3b deposition along the glomerular basement membrane, hypercellularity, cell damage, and inflammation.59,60,66
Mutations often involve segment A of Factor H and FHL1 and affect regulatory functions.66 In other cases, mutations disrupt framework Cys residues, blocking protein secretion and resulting in the absence of both Factor H and FHL1 in plasma. Most mutations are homozygous, although compound heterozygous mutations exist. Additionally, FHR1::FHR1 and FHR31–2::FHR1 hybrid proteins, in context of an intact Factor H gene and normal-acting Factor H protein cause C3G.63 Highlighting a relevant function of FHR1, which is independent of complement control (e.g. by mediating inflammatory clearance). Most of autoantibodies target the N-terminal regulatory regions of Factor H and FHL1.67,68 Yorkshire pigs deficient for Factor H represented the first animal model for C3G with L493V in SCR9 and I1166R SCR20.71 In addition, Factor H knockout mice provide a valuable model for studying C3G.72
aHUS/DEAP-HUS
aHUS, which is characterized by microangiopathic hemolytic anemia, thrombocytopenia, and acute renal failure, has genetic and autoimmune causes.73 Most Factor H mutations cluster in the C-terminal segment D, the recognition region, resulting in defective C3b/C3d and surface binding. Other genetic scenarios include Factor H mutants that incorporate the C-termini of FHR1-FHR3, as seen in Factor H::FHR1, Factor H::FHR3 hybrid proteins, and also FHR1::Factor H hybrids, the latter have the specific residues of Factor H. In the autoimmune form, DEAP-HUS, most autoantibodies also target segment D of Factor H, similarly impairing C3b/C3d and surface binding. Homozygous FHR1: FHR3 deficiency is a risk factor for the development of DEAP-HUS antibodies.74,75
Age-related macular degeneration
AMD is a chronic inflammatory disease in developed countries and the leading cause of vision loss in the elderly (>50 yr). AMD, which affects approximately 20.0 million people in the United States, manifests as a neovascular (wet) and a dry, atrophic form.76 In both cases, changes occur in the outer retina, where damaged retinal pigment epithelial (RPE) cells and a compromised Bruch's membrane contribute to retinal deterioration. In the neovascular form, these changes also stimulate the growth of abnormal blood vessels, contributing to damage and to vision loss.
AMD has several genetic causes.77 Likely the most significant is a Factor H T-to-C polymorphism (rs1061170), which results in a Y402H exchange in segment B in SCR7.78 This polymorphism significantly influences AMD risk: individuals homozygous for the HH402 variant have a 12-fold increased risk, while those heterozygous for HY402 have a 2.5-fold increased risk compared with the protective YY402 variants.79 The Y402H polymorphism affects Factor H and FHL1-mediated complement homeostasis by affecting binding to heparin, mCRP, and apoptotic and necrotic surfaces.80 These changes highlight the role of segment B in surface control.
In addition, homozygous deletion of the FHR1-FHR3 genes is protective in AMD, pointing to the relevance of the Factor H:FHR1 balance for complement action in the retina.47,81 Emerging evidence show noncanonical functions of Factor H in AMD. Intracellular Factor H has been implicated in maintaining metabolic homeostasis and protecting RPE cells from oxidative stress.82 In microglial cells, Factor H inhibits CD47-mediated resolution of local inflammation, with the AMD risk HH402 variant acting as a more potent inhibitor.32 In addition, vascular epithelial growth factor induced Factor H expression in the retina.83
IgA nephropathy
IgAN is a leading cause of chronic kidney disease and occurs in several subgroups.84 IgAN has a multihit pathogenesis, wherein mesangial deposition of galactose-deficient IgA1 immune complexes seem to trigger complement activation via the AP. This leads to complement glomerular deposition, inflammation, and cell damage.85
High Factor H plasma levels, a mean 428.2 µg/mL, were reported in patients with microangiopathy compared with 364.6 µg/mL in the nonmicroangiopathy group.85 In addition, genetic Factor H variants have been reported in IgAN.86
Genome-wide association studies showed that homozygous FHR1-FHR3 deficiency is protective in IgAN.87 This protective effect underscores the role of altered Factor H:FHR1 ratios for disease progression, as they correlate with declining eGFR (estimated glomerular filtration rate). Circulating FHR1 (and FHR5) plasma levels also correlate with disease activity.88,89 Elevated FHR1 levels at sites of tissue damage are thought to reduce Factor H binding, thereby affecting proximal complement regulation, increasing C3 fragment deposition, and exacerbating renal injury.90
ANCA-associated vasculitis
AAV is a systemic autoimmune disease characterized by necrotizing inflammation in small blood vessels, by the presence of circulating myeloperoxidase (MPO) and PK3 antibodies and the formation of NETs.91 AAV is an AP-mediated disease, but shows little complement deposition in the kidneys (Pauci immune). Most tissue damage in AAV is caused by the inflammatory effects mediated by the anaphylatoxin C5a.92
The role for Factor H in AAV is based on low plasma factor H levels in patient serum and impaired plasma complement function.93 A recent AAV cohort study showed that plasma Factor H levels correlate with disease severity.94 Low Factor H levels also correlated with the FHR1-FHR3 genotype.95 FHR1-FHR3 homozygous patients had higher plasma Factor H levels (261 µg/mL) compared with heterozygous patients (216 µg/mL) and those with 2 copies of FHR1-FHR3 (199 µg/mL). While AP was activated, no effects of CP or LP were observed. In the active phase sC5b-9 as a marker for complement activation was elevated.
During inflammation, NETs are formed and mCRP is deposited on the surface of altered self-cells. Both are AP-activating components that bind Factor H.58 An in vitro study showed that Factor H binds to MPO via SCR1–4 with a Kd of 5.5 nM. MPO inhibited Factor H binding to C3b and blocked decay accelerating activity.96
Cancer
Factor H and complement influence the fate of tumors and cancer cells. Factor H has a dual role: the protein can limit tumor growth or can induce tumor proliferation and cancer progression. The tumor microenvironment modulates these opposing effects, it influences how Factor H interacts with tumor cells and thereby affects cancer outcome.
The role of complement in cancer is summarized in excellent reviews.97,98 Here, we focus on Factor H expression and on noncanonical roles of Factor H. In cancer cells, Factor H can induce the differentiation of regulatory T cells and determine the fate of macrophages. Intracellular and nuclear Factor H localizations have been also reported for cancer cells. These locations highlight the complex role of Factor H in cancer and its potential impact on cancer immunology and therapy.
In general, the complement system exerts antitumor effects through opsonophagocytosis and complement-mediated cytotoxicity. However elevated factor H levels may counteract the beneficial effects of complement in eliminating tumor cells. High levels of Factor H (and of FHL1) have been reported in renal cell carcinoma, lung cancer cells, glioma cells, cutaneous squamous cell carcinoma, breast cancer cells, lung adenocarcinoma, and ovarian tumor cells,99 suggesting a critical role for Factor H (and FHL1) in tumor progression.
Intracellular Factor H has recently been identified in clear cell renal cell carcinoma and lung cancer cells, where it drives tumor progression. It is also associated with poor prognosis.100 Factor H has been localized to the cytoplasm and even to the nucleus. Cytoplasmic Factor H interacts with the mitochondrial protein NARS2 and can enhance oxidative phosphorylation in the Krebs cycle, a process that supports metabolism.101 Segment B appears to be involved in this intracellular tumor activity. Nuclear Factor H binds to the transcription factor E2F3. In addition, Factor H has an immunosuppressive role in breast cancer cells. The regulator also promotes the formation of immunosuppressive macrophages, and this effect is mediated by segment D, in particular by SCR19–20.32
Aged Factor H–deficient mice develop spontaneous tumors in the liver. These findings have been correlated with human hepatocellular carcinoma. Individuals with elevated Factor H messenger RNA levels showed improved survival.102 Autoantibodies to Factor H have been described in patients with non-small cell lung cancer and identified at an early stage of the disease (see also Table 3).103
Factor H autoantibodies in human diseases.
| Disease | Reference(s) | |
|---|---|---|
| 1 | C3G | Current paper (The Factor H gene and the Factor H-FHR gene cluster) |
| 2 | DEAP-HUS | Current paper (Structure of Factor H–ligand interaction and functional regions) |
| 3 | AMD | 103,104 |
| 4 | Non-small cell lung cancer | 106 |
| 5 | SLE and lupus nephritis | 107–109 |
| 6 | Antiphospholipid syndrome and thrombosis | 110–112 |
| 7 | Rheumatoid arthritis | 112,113 |
| 8 | Atopic gastritis and rheumatoid arthritis | 114 |
| 9 | Hematopoietic stem cell transplant–associated TMA | 115 |
| 10 | Membranous nephropathy | 116–118 |
| 11 | Multisystemic inflammatory syndrome | 119 |
| 12 | Neuromyelitis optica spectrum disorders | 120 |
| 13 | Pregnancy and preeclampsia | 121 |
| Disease | Reference(s) | |
|---|---|---|
| 1 | C3G | Current paper (The Factor H gene and the Factor H-FHR gene cluster) |
| 2 | DEAP-HUS | Current paper (Structure of Factor H–ligand interaction and functional regions) |
| 3 | AMD | 103,104 |
| 4 | Non-small cell lung cancer | 106 |
| 5 | SLE and lupus nephritis | 107–109 |
| 6 | Antiphospholipid syndrome and thrombosis | 110–112 |
| 7 | Rheumatoid arthritis | 112,113 |
| 8 | Atopic gastritis and rheumatoid arthritis | 114 |
| 9 | Hematopoietic stem cell transplant–associated TMA | 115 |
| 10 | Membranous nephropathy | 116–118 |
| 11 | Multisystemic inflammatory syndrome | 119 |
| 12 | Neuromyelitis optica spectrum disorders | 120 |
| 13 | Pregnancy and preeclampsia | 121 |
TMA, thrombotic angiopathy.
Factor H autoantibodies in human diseases.
| Disease | Reference(s) | |
|---|---|---|
| 1 | C3G | Current paper (The Factor H gene and the Factor H-FHR gene cluster) |
| 2 | DEAP-HUS | Current paper (Structure of Factor H–ligand interaction and functional regions) |
| 3 | AMD | 103,104 |
| 4 | Non-small cell lung cancer | 106 |
| 5 | SLE and lupus nephritis | 107–109 |
| 6 | Antiphospholipid syndrome and thrombosis | 110–112 |
| 7 | Rheumatoid arthritis | 112,113 |
| 8 | Atopic gastritis and rheumatoid arthritis | 114 |
| 9 | Hematopoietic stem cell transplant–associated TMA | 115 |
| 10 | Membranous nephropathy | 116–118 |
| 11 | Multisystemic inflammatory syndrome | 119 |
| 12 | Neuromyelitis optica spectrum disorders | 120 |
| 13 | Pregnancy and preeclampsia | 121 |
| Disease | Reference(s) | |
|---|---|---|
| 1 | C3G | Current paper (The Factor H gene and the Factor H-FHR gene cluster) |
| 2 | DEAP-HUS | Current paper (Structure of Factor H–ligand interaction and functional regions) |
| 3 | AMD | 103,104 |
| 4 | Non-small cell lung cancer | 106 |
| 5 | SLE and lupus nephritis | 107–109 |
| 6 | Antiphospholipid syndrome and thrombosis | 110–112 |
| 7 | Rheumatoid arthritis | 112,113 |
| 8 | Atopic gastritis and rheumatoid arthritis | 114 |
| 9 | Hematopoietic stem cell transplant–associated TMA | 115 |
| 10 | Membranous nephropathy | 116–118 |
| 11 | Multisystemic inflammatory syndrome | 119 |
| 12 | Neuromyelitis optica spectrum disorders | 120 |
| 13 | Pregnancy and preeclampsia | 121 |
TMA, thrombotic angiopathy.
Neurodegenerative diseases
In neurological diseases, deregulated complement causes pathologic inflammation in neurons and the brain. These include neurological and neurodegenerative diseases such as Alzheimer's disease, myasthenia gravis, Parkinson's disease, and schizophrenia.122 The regulatory and anti-inflammatory role of Factor H is consistent with an inhibitory role in neuroinflammation. Factor H expression has been demonstrated on astrocytes, microglial cells, neurons, and oligodendrocytes.123 The exact mechanisms by which Factor H mediates complement regulation and noncanonical cellular effects in the central nervous system and neuroinflammation are subject of ongoing research.
In multiple sclerosis (MS), higher levels of Factor H have been found in the serum and cerebrospinal fluid of MS patients, especially those with progressive disease or during active relapses, compared with patients with stable relapsing MS.
Infections with pathogenic microbes
Many pathogenic microbes use Factor H, FHL1, and FHR1 for immune evasion.124 The acquired host regulators block complement attack prevent the formation of toxic effector fragments on the pathogen surface. Numerous binding proteins for Factor H, FHL1, and FHR1 have been identified in Gram-positive and Gram-negative bacteria (including Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis) and fungi (e.g. Candida albicans, Aspergillus fumigatus) and from multicellular parasites (e.g. Plasmodium falciparum).125–127 The vast majority of these pathogen-encoded Factor H binding proteins bind Factor H primarily via segments B and D. In addition, after immunization with Factor H binding peptide from N. meningitidis serotype B, some subjects developed a mild and transient increase in serum anti-Factor H antibody reactivity.128
Autoantibodies to Factor H
The role of autoantibodies to Factor H in C3G and DEAP-HUS pathology is well established and described in the sections “The Factor H gene and the Factor H-FHR gene cluster” and “aHUS/DEAP-HUS.” However, autoantibodies to Factor H have also been identified in other diseases (Table 3). It will be of interest to define their binding profiles and to determine autoantibody titers in larger, well-defined patient cohorts. This will help to explain how exactly these antibodies contribute to disease pathology and also provide a rationale for diagnosis and therapy.
Factor H binding antibodies have been detected in approximately 10% of patients with lupus nephritis characterized by polyepitopes and a predominance of IgG2 subclass. Patients with Factor H antibodies had milder kidney damage, and purified autoantibodies enhanced C3b binding and factor I–mediated cofactor activity of Factor H in vitro.106,107 A genetic study linked FHR1-FHR3 deficiency to this complement disorder demonstrating that homozygous deletion of FHR1 and FHR3 has a protective role in SLE.
Outlook
Factor H is the primary regulator of AP-mediated proximal complement. Disease-associated mutations in the Factor H gene, mapped binding sites of pathologic autoantibodies, and structure-function analyses define 4 functional segments in the Factor H protein, each composed of multiple SCR domains. These functional units include 2 N-terminal segments that bind single ligands with high affinity (i.e. the regulatory C3b binding segment A and segment B, the attachment segment, which binds to heparin GAGs and sialic acids). Both segment C and segment D bind 2 overlapping ligands, C3b and C3 isoforms, and also heparin, GAGs, and sialic acids, and they bind with lower affinity. Segment C has overlapping binding sites for C3b and heparin/GAGs and includes a structural kink element. Such multicontact binding allows a dynamic action profile of the regulator, which is consistent with the multiple roles of Factor H in complement control.
Factor H regulates proximal complement in the fluid phase, controls the transition from the fluid phase to the surface, and mediates complement surface control. It also cooperates with FHL1 and FHR1, 2 human plasma proteins that share functional segments with Factor H. FHL1 cooperates with Factor H, and FHR1 and Factor H bind to the same or closely related modified self-surfaces under complement attack. Apparently, Factor H and the inflammatory modulator FHR1 induce distinct effector pathways to determine the fate of modified self particles. Together, Factor H and FHL1 or FHR1 fine-tune local complement action, cascade progression, and effector activity.
Altered Factor H plasma levels and disturbed Factor H:FHR1 plasma ratios contribute to pathology in several diseases, demonstrating the importance of this interplay in local and spatiotemporal complement action. Altered plasma Factor H and FHR1 levels may correlate with disease severity and may have prognostic value for disease progression, as reported in AAV.
In addition to the well-established role in complement regulation, new roles and new locations of Factor H are emerging, including intracellular and nuclear presence. New functions include the induction of regulatory macrophages, driving intracellular C3 expression, exhibiting tumorigenic effects, and influencing the development of regulatory T cells.129 The integration of intracellular Factor H into cytoplasmic and nuclear pathways suggests additional functional facets for this evolutionarily conserved complement protein. These direct regulatory roles of Factor H in balancing complement action makes it an attractive target for therapeutic intervention.
Author contributions
P.F.Z.: Conceptualization, generation of figures and tables original draft resources, writing, review & editing of text, project administration. K.H.: Conceptualization, generation of figures and tables—original draft, resources, writing—review & editing, project administration.
Funding
P.F.Z. has received funding from the DFG priority program SFB 1192, Immune-mediated glomerular diseases project B6, and Kidneeds.
Conflicts of interest
K.H. is employed by Eleva GmbH. P.F.Z. has received consulting fees from Alnylam, Bayer, Novartis, Samsung Bioepis, Alexion/AstraZeneca Rare Disease, CSL Vifor, and Eleva GmbH.
Data availability
This is a review and no original data are used. All information is presented in the references cited. The figures are new and prepared specifically for this review article.

{kind=link}
{kind=link}
{kind=link}