





Abstract
Silver-Russell syndrome (SRS) is a clinical and molecular heterogeneous disorder associated with short stature, typical facial gestalt, and body asymmetry. Though molecular causes of SRS can be identified in a significant number of patients, about one-half of patients currently remain without a molecular diagnosis. However, determination of the molecular cause is required for a targeted treatment and genetic counselling.
The aim of this study was to corroborate the role of HMGA2 as an SRS-causing gene and reevaluate its mode of inheritance.
Patients were part of an ongoing study aiming on SRS-causing genes. They were classified according to the Netchine-Harbison clinical scoring system, and DNA samples were investigated by whole exome sequencing. Common molecular causes of SRS were excluded before.
Three novel pathogenic HMGA2 variants were identified in 5 patients from 3 SRS families, and fulfilling diagnostic criteria of SRS. For the first time, homozygosity for a variant in HMGA2 could be identified in a severely affected sibpair, whereas parents carrying heterozygous variants had a mild phenotype. Treatment with recombinant growth hormone led to a catch-up growth in 1 patient, whereas all others did not receive growth hormone and stayed small. One patient developed type 2 diabetes at age 30 years.
Identification of novel pathogenic variants confirms HMGA2 as an SRS-causing gene; thus, HMGA2 testing should be implemented in molecular SRS diagnostic workup. Furthermore, inheritance of HMGA2 is variable depending on the severity of the variant and its consequence for protein function.
Silver-Russell syndrome (SRS, OMIM 180860) is a congenital growth retardation disorder that is clinically and genetically heterogeneous (1). In addition to severe intrauterine and postnatal growth retardation, SRS is characterized by relative macrocephaly at birth, feeding difficulties, body asymmetry, and a typical facial gestalt with protruding forehead and triangular face during early childhood (2). Molecular alterations are detectable in about 60% of cases, with loss of methylation (LOM) of the imprinting center 1 (IC1, H19/IGF2:IG-DMR) in 11p15.5 as the most common alteration, accounting for 40% of patients. It is followed by maternal uniparental disomy for chromosome 7 (upd(7)mat) and chromosome 14q32 disturbances with a frequency of 7% to 10% each (1). In single cases, pathogenic variants in different genes involved in human growth and development have been reported (IGF2, CDKN1C, PLAG1, HMGA2) (3-5).
Recently, pathogenic variants within the HMGA2 gene (OMIM 600698) have been identified in growth retarded patients with the clinical diagnosis of SRS but lacking the typical molecular genetic defects of SRS (5-8) (Table 1). The patients exhibited heterozygosity of HMGA2 variants, and the familial segregation in these pedigrees suggested an autosomal dominant inheritance. HMGA2 is a transcription-regulating protein contributing to embryonal and neoplastic processes. It has been postulated to play a role in growth and development as a common HMGA2 single nucleotide variant (rs1042725) has been associated with height in the general population (9).
Comparison of the NH-CSS Results of the Reported Patients Carrying HMGA2 Variants
| P1 From This Study | Father of P1 From This Study | P3 From This Study | P3 From This Study | Brother of P3 From This Study | Plachy et al., (8) | Leszinski et al., (6) | Abi Habib et al., (5) | Abi Habib et al., (5) | De Crescenzo et al., (7) | De Crescenzo et al., (7) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HMGA2 variant | Deletion of exon 1 to 3 | Deletion of exon 1 to 3 | NM_003483. 4:c.111 + 1G>T, p.? | NM_003483. 4:c.239C>T, p.Pro80Leu | NM_003483. 4:c.239C>T, p.Pro80Leu | c.223C>T, p.Arg75Trp | 7,3kb deletion in 12q14.3 including exons 2 and 3 | c.193C>T, p.Gln65* | c.189del, p.Ala64 Leufs*102 | 7 bp deletion in exon 5 | 7 bp deletion in exon 5 |
| 1. Small for gestational age | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | NA |
| 2. Postnatal growth retardation | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| 3. Relative macrocephaly at birth | No | Yes | No | NA | NA | NA | No | Yes | Yes | Yes | NA |
| 4. Protruding forehead | Yes | Yes | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | Yes |
| 5. Body asymmetry | No | No | No | Uncertain | Uncertain | NA | No | No | No | No | NA |
| 6. Feeding difficulties | Yes | NR | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | Yes |
| NH-CSS | 5/6 | 4/5 | 4/6 | 4/4 | 4/4 | 1/2 | 4/6 | 5/6 | 5/6 | 5/6 | 3/3 |
| Clinical diagnosis of SRS | Confirmed | Confirmed | Not confirmed | Not confirmed | Not confirmed | Not confirmed | Not confirmed | Confirmed | Confirmed | Confirmed | Not confirmed |
| P1 From This Study | Father of P1 From This Study | P3 From This Study | P3 From This Study | Brother of P3 From This Study | Plachy et al., (8) | Leszinski et al., (6) | Abi Habib et al., (5) | Abi Habib et al., (5) | De Crescenzo et al., (7) | De Crescenzo et al., (7) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HMGA2 variant | Deletion of exon 1 to 3 | Deletion of exon 1 to 3 | NM_003483. 4:c.111 + 1G>T, p.? | NM_003483. 4:c.239C>T, p.Pro80Leu | NM_003483. 4:c.239C>T, p.Pro80Leu | c.223C>T, p.Arg75Trp | 7,3kb deletion in 12q14.3 including exons 2 and 3 | c.193C>T, p.Gln65* | c.189del, p.Ala64 Leufs*102 | 7 bp deletion in exon 5 | 7 bp deletion in exon 5 |
| 1. Small for gestational age | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | NA |
| 2. Postnatal growth retardation | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| 3. Relative macrocephaly at birth | No | Yes | No | NA | NA | NA | No | Yes | Yes | Yes | NA |
| 4. Protruding forehead | Yes | Yes | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | Yes |
| 5. Body asymmetry | No | No | No | Uncertain | Uncertain | NA | No | No | No | No | NA |
| 6. Feeding difficulties | Yes | NR | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | Yes |
| NH-CSS | 5/6 | 4/5 | 4/6 | 4/4 | 4/4 | 1/2 | 4/6 | 5/6 | 5/6 | 5/6 | 3/3 |
| Clinical diagnosis of SRS | Confirmed | Confirmed | Not confirmed | Not confirmed | Not confirmed | Not confirmed | Not confirmed | Confirmed | Confirmed | Confirmed | Not confirmed |
Abbreviations: NH-CSS, Netchine-Harbison clinical scoring system; NA, not assessed; NR, not reported; SRS, Silver-Russell syndrome.
Comparison of the NH-CSS Results of the Reported Patients Carrying HMGA2 Variants
| P1 From This Study | Father of P1 From This Study | P3 From This Study | P3 From This Study | Brother of P3 From This Study | Plachy et al., (8) | Leszinski et al., (6) | Abi Habib et al., (5) | Abi Habib et al., (5) | De Crescenzo et al., (7) | De Crescenzo et al., (7) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HMGA2 variant | Deletion of exon 1 to 3 | Deletion of exon 1 to 3 | NM_003483. 4:c.111 + 1G>T, p.? | NM_003483. 4:c.239C>T, p.Pro80Leu | NM_003483. 4:c.239C>T, p.Pro80Leu | c.223C>T, p.Arg75Trp | 7,3kb deletion in 12q14.3 including exons 2 and 3 | c.193C>T, p.Gln65* | c.189del, p.Ala64 Leufs*102 | 7 bp deletion in exon 5 | 7 bp deletion in exon 5 |
| 1. Small for gestational age | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | NA |
| 2. Postnatal growth retardation | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| 3. Relative macrocephaly at birth | No | Yes | No | NA | NA | NA | No | Yes | Yes | Yes | NA |
| 4. Protruding forehead | Yes | Yes | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | Yes |
| 5. Body asymmetry | No | No | No | Uncertain | Uncertain | NA | No | No | No | No | NA |
| 6. Feeding difficulties | Yes | NR | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | Yes |
| NH-CSS | 5/6 | 4/5 | 4/6 | 4/4 | 4/4 | 1/2 | 4/6 | 5/6 | 5/6 | 5/6 | 3/3 |
| Clinical diagnosis of SRS | Confirmed | Confirmed | Not confirmed | Not confirmed | Not confirmed | Not confirmed | Not confirmed | Confirmed | Confirmed | Confirmed | Not confirmed |
| P1 From This Study | Father of P1 From This Study | P3 From This Study | P3 From This Study | Brother of P3 From This Study | Plachy et al., (8) | Leszinski et al., (6) | Abi Habib et al., (5) | Abi Habib et al., (5) | De Crescenzo et al., (7) | De Crescenzo et al., (7) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HMGA2 variant | Deletion of exon 1 to 3 | Deletion of exon 1 to 3 | NM_003483. 4:c.111 + 1G>T, p.? | NM_003483. 4:c.239C>T, p.Pro80Leu | NM_003483. 4:c.239C>T, p.Pro80Leu | c.223C>T, p.Arg75Trp | 7,3kb deletion in 12q14.3 including exons 2 and 3 | c.193C>T, p.Gln65* | c.189del, p.Ala64 Leufs*102 | 7 bp deletion in exon 5 | 7 bp deletion in exon 5 |
| 1. Small for gestational age | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | NA |
| 2. Postnatal growth retardation | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| 3. Relative macrocephaly at birth | No | Yes | No | NA | NA | NA | No | Yes | Yes | Yes | NA |
| 4. Protruding forehead | Yes | Yes | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | Yes |
| 5. Body asymmetry | No | No | No | Uncertain | Uncertain | NA | No | No | No | No | NA |
| 6. Feeding difficulties | Yes | NR | Yes | Yes | Yes | NA | Yes | Yes | Yes | Yes | Yes |
| NH-CSS | 5/6 | 4/5 | 4/6 | 4/4 | 4/4 | 1/2 | 4/6 | 5/6 | 5/6 | 5/6 | 3/3 |
| Clinical diagnosis of SRS | Confirmed | Confirmed | Not confirmed | Not confirmed | Not confirmed | Not confirmed | Not confirmed | Confirmed | Confirmed | Confirmed | Not confirmed |
Abbreviations: NH-CSS, Netchine-Harbison clinical scoring system; NA, not assessed; NR, not reported; SRS, Silver-Russell syndrome.
In this paper, we report on the identification of 3 novel likely pathogenic variants in the HMGA2 gene associated with growth retardation and SRS. To the best of our knowledge, for the first time we describe a sibpair homozygous for a HMGA2 variant who exhibit a more severe phenotype than heterozygous HMGA2 variant carriers.
Study cohort
The patients and their families were ascertained as part of an ongoing study of ~60 patients aiming on the identification of genetic factors associated with growth retardation and SRS features. All patients received routine molecular testing according to the first international consensus statement for diagnosis and management of SRS (1). This screening comprised testing for LOM of IC1, upd(7)mat, 14q32 disturbances. Additionally, maternal uniparental disomy of chromosomes 16 and 20 as well as point mutations in IGF2 and CDKN1C were analyzed. All tests gave negative results. By single nucleotide polymorphism microarray analysis (CytoScan HD, Affymetrix, Santa Clara, CA), rare copy number variations > 50 kb were excluded as well. Variants in other differential diagnostic genes (e.g., BLM, TRIM28, PIK3R1) were not identified by whole exome next-generation sequencing analysis. Clinical characterization of the patients was performed using the Netchine-Harbison clinical scoring system (NH-CSS) (2).
The study was approved by the ethical committee of the Medical Faculty, RWTH Aachen University (EK303-18).
Material and Methods
DNA was extracted from peripheral blood lymphocytes using a simple salting-out procedure or a commercially available DNA extraction kit (QIAamp DNA Mini Kit, Qiagen, Hilden, Germany).
Whole exome enrichment was conducted by using the Illumina Rapid Capture method (Nextera Rapid Capture Exome v1.2, 45.33 Mb target size, Illumina Inc. San Diego, CA) and sequenced on a NextSeq500/550 High Output Kit with 2 × 75 cycles (Illumina Inc.) with an average yield of ~10 Gb per sample. Data analysis was performed with an in-house pipeline based on Seqmule (10) with standard parameters. Three different variant callers were applied that integrated data to the final variant file, if at least 2 of them called the variant. Variant filtering annotation was performed using KGGSeq (v1.0, 20 June 2018) discarding variants with a minor allele frequency higher than 0.75% in public databases (i.e., gnomAD, 1000 Genome Project, Exome Variant Server). Variant prioritization and evaluation of pathogenicity was based on different prediction tools (CADD, PolyPhen, SIFT, Mutation Taste) and variant frequency in public databases. Variant confirmation and segregational analysis in the families were done by Sanger sequencing. Multiplex ligation-dependent probe amplification in family 1 was carried out according to the manufacturer’s protocols (kit MLPA P323-B1, MRC Holland, Amsterdam, Netherlands).
Results
In 5 patients referred for genetic SRS testing from 3 families, 3 new HMGA2 germline variants could be identified (Fig. 1). They all showed characteristic SRS features, with NH-CSS of at least 4 out of 6 parameters (Tables 1 and 2).
Overview on the Main Clinical Features in the 5 Patients
| P1 | Father of P1 | P2 | P3 | Brother of P3 | ||
|---|---|---|---|---|---|---|
| Small for gestational age | Gestational weeks | 39 + 6 | 42 | 39 | 39 | 38 |
| DOB length in cm (SDS) | 46 (-2.82) | 45 (-3.5) | 47 (-2.13) | 36 (-6.86) | NA | |
| DOB weight in g (SDS) | 2365 (-2.79) | 2280 (-3.27) | 2360 (-2.6) | 1400 (-4.65) | 1050 (-5.18) | |
| Postnatal growth retardation | Age at investigation (year) | 2 | 32 | 3 8/12 | 3 | 1 1/12 |
| Length at investigation in cm (SDS) | 67 (-6.21) | 160 (-2.77) | 88 (-3.24) | 70 (-6.73) | 57 (-6.19) | |
| Weight at investigation in g (SDS) | 6990 (-4.57) | NA | 10000 (-3.93) | 6200 (-7.09) | 4100 (-6.76) | |
| Relative macrocephaly at birth | Head circumference at birth (SDS) | 33 (-1.97) | 34.5 (-1.15) | 31 (-3.31) | NA | NA |
| Protruding forehead | Yes | Yes | Yes | Yes | Yes | |
| Body asymmetry | No | No | No | Uncertain | Uncertain | |
| Feeding difficulties | BMI at investigation (SDS) | 15.6 (-0.33) | NR | 12.9 (-2.47) | 12.7 (-2.9) | 12.6 (-4.14) |
| Triangular face | Yes | Yes | Yes | Yes | Yes | |
| Further symptoms | Type 2 diabetes mellitus since age of 30 y | Café au lait spots, genu valgum, teeth abnormalities | Delayed teething, deep set eyes, clinodactyly V, small nose and ears, craniofacial disproportion, narrow palate | Clinodactyly V, midface hypoplasia, small nose and ears, craniofacial disproportion, narrow palate |
| P1 | Father of P1 | P2 | P3 | Brother of P3 | ||
|---|---|---|---|---|---|---|
| Small for gestational age | Gestational weeks | 39 + 6 | 42 | 39 | 39 | 38 |
| DOB length in cm (SDS) | 46 (-2.82) | 45 (-3.5) | 47 (-2.13) | 36 (-6.86) | NA | |
| DOB weight in g (SDS) | 2365 (-2.79) | 2280 (-3.27) | 2360 (-2.6) | 1400 (-4.65) | 1050 (-5.18) | |
| Postnatal growth retardation | Age at investigation (year) | 2 | 32 | 3 8/12 | 3 | 1 1/12 |
| Length at investigation in cm (SDS) | 67 (-6.21) | 160 (-2.77) | 88 (-3.24) | 70 (-6.73) | 57 (-6.19) | |
| Weight at investigation in g (SDS) | 6990 (-4.57) | NA | 10000 (-3.93) | 6200 (-7.09) | 4100 (-6.76) | |
| Relative macrocephaly at birth | Head circumference at birth (SDS) | 33 (-1.97) | 34.5 (-1.15) | 31 (-3.31) | NA | NA |
| Protruding forehead | Yes | Yes | Yes | Yes | Yes | |
| Body asymmetry | No | No | No | Uncertain | Uncertain | |
| Feeding difficulties | BMI at investigation (SDS) | 15.6 (-0.33) | NR | 12.9 (-2.47) | 12.7 (-2.9) | 12.6 (-4.14) |
| Triangular face | Yes | Yes | Yes | Yes | Yes | |
| Further symptoms | Type 2 diabetes mellitus since age of 30 y | Café au lait spots, genu valgum, teeth abnormalities | Delayed teething, deep set eyes, clinodactyly V, small nose and ears, craniofacial disproportion, narrow palate | Clinodactyly V, midface hypoplasia, small nose and ears, craniofacial disproportion, narrow palate |
Abbreviations: BMI, body mass index; DOB, date of birth; NA, not available; NR, not reported; SDS, standard deviation score.
Overview on the Main Clinical Features in the 5 Patients
| P1 | Father of P1 | P2 | P3 | Brother of P3 | ||
|---|---|---|---|---|---|---|
| Small for gestational age | Gestational weeks | 39 + 6 | 42 | 39 | 39 | 38 |
| DOB length in cm (SDS) | 46 (-2.82) | 45 (-3.5) | 47 (-2.13) | 36 (-6.86) | NA | |
| DOB weight in g (SDS) | 2365 (-2.79) | 2280 (-3.27) | 2360 (-2.6) | 1400 (-4.65) | 1050 (-5.18) | |
| Postnatal growth retardation | Age at investigation (year) | 2 | 32 | 3 8/12 | 3 | 1 1/12 |
| Length at investigation in cm (SDS) | 67 (-6.21) | 160 (-2.77) | 88 (-3.24) | 70 (-6.73) | 57 (-6.19) | |
| Weight at investigation in g (SDS) | 6990 (-4.57) | NA | 10000 (-3.93) | 6200 (-7.09) | 4100 (-6.76) | |
| Relative macrocephaly at birth | Head circumference at birth (SDS) | 33 (-1.97) | 34.5 (-1.15) | 31 (-3.31) | NA | NA |
| Protruding forehead | Yes | Yes | Yes | Yes | Yes | |
| Body asymmetry | No | No | No | Uncertain | Uncertain | |
| Feeding difficulties | BMI at investigation (SDS) | 15.6 (-0.33) | NR | 12.9 (-2.47) | 12.7 (-2.9) | 12.6 (-4.14) |
| Triangular face | Yes | Yes | Yes | Yes | Yes | |
| Further symptoms | Type 2 diabetes mellitus since age of 30 y | Café au lait spots, genu valgum, teeth abnormalities | Delayed teething, deep set eyes, clinodactyly V, small nose and ears, craniofacial disproportion, narrow palate | Clinodactyly V, midface hypoplasia, small nose and ears, craniofacial disproportion, narrow palate |
| P1 | Father of P1 | P2 | P3 | Brother of P3 | ||
|---|---|---|---|---|---|---|
| Small for gestational age | Gestational weeks | 39 + 6 | 42 | 39 | 39 | 38 |
| DOB length in cm (SDS) | 46 (-2.82) | 45 (-3.5) | 47 (-2.13) | 36 (-6.86) | NA | |
| DOB weight in g (SDS) | 2365 (-2.79) | 2280 (-3.27) | 2360 (-2.6) | 1400 (-4.65) | 1050 (-5.18) | |
| Postnatal growth retardation | Age at investigation (year) | 2 | 32 | 3 8/12 | 3 | 1 1/12 |
| Length at investigation in cm (SDS) | 67 (-6.21) | 160 (-2.77) | 88 (-3.24) | 70 (-6.73) | 57 (-6.19) | |
| Weight at investigation in g (SDS) | 6990 (-4.57) | NA | 10000 (-3.93) | 6200 (-7.09) | 4100 (-6.76) | |
| Relative macrocephaly at birth | Head circumference at birth (SDS) | 33 (-1.97) | 34.5 (-1.15) | 31 (-3.31) | NA | NA |
| Protruding forehead | Yes | Yes | Yes | Yes | Yes | |
| Body asymmetry | No | No | No | Uncertain | Uncertain | |
| Feeding difficulties | BMI at investigation (SDS) | 15.6 (-0.33) | NR | 12.9 (-2.47) | 12.7 (-2.9) | 12.6 (-4.14) |
| Triangular face | Yes | Yes | Yes | Yes | Yes | |
| Further symptoms | Type 2 diabetes mellitus since age of 30 y | Café au lait spots, genu valgum, teeth abnormalities | Delayed teething, deep set eyes, clinodactyly V, small nose and ears, craniofacial disproportion, narrow palate | Clinodactyly V, midface hypoplasia, small nose and ears, craniofacial disproportion, narrow palate |
Abbreviations: BMI, body mass index; DOB, date of birth; NA, not available; NR, not reported; SDS, standard deviation score.
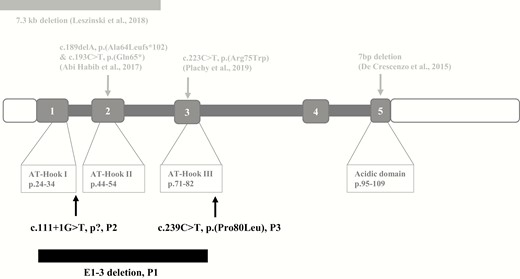
Structure of the HMGA2 gene and its protein, and localization of the reported (in gray, upper panel) and the new pathogenic variants (black, lower panel). (From Leszinski GS, Warncke K, Hoefele J, Wagner M. A case report and review of the literature indicate that HMGA2 should be added as a disease gene for Silver-Russell syndrome. Gene 2018;663:110-114). (not to scale)
Familial deletion of exons 1 to 3 in HMGA2—family 1
Heterozygosity for a deletion of exons 1 to 3 of the HMGA2 gene (NM_003483.4) was detected in a 2-year-old German patient and his likewise affected father (Fig. 2A-D). Intrauterine growth retardation was reported, and the boy was born at 39 + 6 gestational weeks with reduced weight (standard deviation score [SDS], -2.82) and length (SDS, -2.79), and a head circumference of 33 cm (SDS, -1.97). At the age of 24 months, growth was severely restricted (SDS, -6.21). The patient exhibited a triangular face and a protruding forehead. He developed feeding difficulties, but feeding with nasogastric tube was not required. Asymmetry was not reported. The deletion was inherited from his father, who presented similar symptoms (Table 2), such as small stature and facial dysmorphisms. Final height of the father was 160 cm (SDS, -2.77), without treatment with recombinant growth hormone (rGH). At the age of 30 years, the father developed type 2 diabetes mellitus (T2D). Clinical scoring according to the NH-CSS (2) revealed a score of 4 of 6 and 4 of 5 in the patient and his father, respectively. Parents of the father were 172 cm (grandfather) and taller than 160 cm (grandmother), respectively. His sister and his brother were taller than 170 cm.
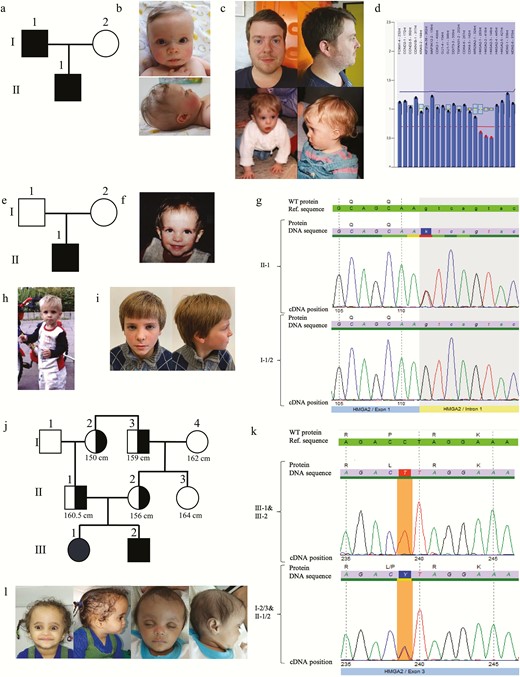
Phenotypes and genetic testing results in the 3 families. Pedigree of (A) family 1, (B) father of patient 1 as child (age 1 year) and adult (32 years), (C) patient 1 (age 9 months), and (D) multiplex ligation-dependent probe amplification. Pedigree of the (F) family, (E) patient 2 (age 3.8 years), (G) electropherogram, (H) patient 2 (age 4 years), and (I) patient 2 (age 8 years). Pedigree of (J) family 3, (K) electropherogram of patient 3 and heterozygous affected relatives (I-2, I-3, II-1, II-2), and (L) patient 3 and her brother.
De novo splice-site variant c.111 + 1G>T in HMGA2—patient 2
Next-generation sequencing-based analysis revealed a de novo HMGA2 splice-site variant in a 4-year-old boy from Lithuania (NM_003483.4:c.111 + 1G>T; Chr12(GRCh38):g.65825382G>T) (Fig. 2E-I). The patient was born small for gestational age (39 gestational weeks; SDS, -2.6), postnatal growth restriction persisted (SDS, -3.24 at the age of 3.8 years) (Table 2). Head circumference was 31 cm (SDS, -3.31). Facial features suggestive for SRS included a protruding forehead and a triangular face. Feeding difficulties were reported as well, but asymmetry was not present. In total, an NH-CSS of 4 of 6 could be delineated. The patient had received recombinant rGH therapy since the age of 3.8 years with a positive response at the age of 6.9 years (from SDS -3.24 to -1.16). The variant could not be detected in the healthy parents and gnomAD and ExAC databases and was therefore classified as de novo.
Familial missense variant c.239C>T; p.(Pro80Leu) in HMGA2—family 3
In a consanguineous (first cousin) Egyptian family, we identified a homozygous missense variant in exon 3 of HMGA2 (NM_003483.4:c.239C>T; p.(Pro80Leu); Chr12(GRCh37):g.66232339C>T) in 2 siblings with severe short stature (SDS, -6.73; and SDS, -6.19 at the age of 3 and 1.1 years, respectively; Table 2) (Fig. 2J-L). They were born small for gestational age and exhibited a pronounced dysmorphic facial gestalt of the typical SRS features such as a protruding forehead, a triangular face, a high hairline, and clinodactyly of the fifth finger. NH-CSS calculation resulted in a score of 4/4 in both children. Head circumference at birth and estimation of body asymmetry were not available but the mother reported head circumference at birth as relatively large. The parents were heterozygous for the variant and presented smaller than their relatives not carrying the variant (father [II-1], 160.5 cm; mother [II-2], 156 cm)(Fig. 2J). Segregation analysis of the variant in the maternal family revealed a correlation between the variant and height. The paternal grandmother of the patients (I-2) and the paternal grandfather (I-3) were of small stature (150 cm and 159 cm, respectively) and carried the mutation, whereas the maternal grandmother (I-4) was relatively tall (162 cm) and did not show the mutation. A noncarrier sister of the mother (II-3) was relatively tall as well (164 cm) (Fig. 2H). The variant is neither listed in gnomAD, ExAC, nor the 1000G database.
Discussion
HMGA2 is 1 of the 4 members of the high-motility group AT-hook family, a protein family of architectural transcription factors. The protein contains 3 AT-hooks that interact with DNA in AT-rich domains. Expression of HMGA2 is repressed in normal adult cells, but it is high in cells during embryogenesis (11). The function as an upstream regulator of IGF2 has been suggested. This hypothesis is additionally corroborated by functional assays showing a decrease of IGF2 expression when HMGA2 is silenced (5). IGF2 is a central factor in human growth regulation, and in SRS its downregulation by LOM of the IC1 in 11p15.5 and by pathogenic variants cause growth restriction (1, 3, 12, 13). Other evidence for the functional role of HMGA2 in growth and early development is the “pigmy” phenotype in Hmga2 homozygous and heterozygous mice (14, 15) with a decreased body size of 25% and 60%, respectively (16).
Up to now, 6 patients with pathogenic variants in HMGA2 (5-8) and about 15 additional growth-retarded patients with microdeletions in chromosome 12p14 affecting the HMGA2 gene have been reported (for review, see (17)). All 6 patients with HMGA2 variants reported so far presented with short stature and features reminiscent to SRS, such as a protruding forehead and feeding difficulties (for review, see (6)). In the 5 patients presented in this report at least 4 NH-CSS criteria were fulfilled as well, and at least 1 of them also had a relative macrocephaly (father of patient 1).
With the identification of 3 new variants in HMGA2, its role as an SRS-causing gene is further corroborated as all 3 molecular changes can be regarded as pathogenic.
The deletion of exons 1 to 3 in patient 1 and his father leads to the loss of the first 84 amino acids of HMGA2 including the start-codon, probably resulting in the loss of HMGA2 expression. Furthermore, in vitro analysis showed a significantly reduced protein function if the first 3 exons of HMGA2 are mutated, thus introduction of a new start codon should lead to loss of protein function as well (18). Unfortunately, samples of the normal heighted grandparents and relatives were not available. According to the severity of the growth retardation phenotype, we assume the variant as de novo in the father (Fig. 2A: I-1).
The splicing variant c.111 + 1G>T in patient 2 can be assumed to encode for an elongated transcript as intron 1 is retained, which is predicted to introduce a premature stop codon and to cause nonsense RNA mediated decay. The resulting protein, if any, should not be functional.
In family 3, the missense variant c.239C>T in exon 3 leads to an exchange of proline to leucine at protein position 80 (p.(Pro80Leu)). Pathogenicity prediction tools (Mutation taster, SIFT) predicted this variant as disease causing and damaging. Because this residue is the last amino acid of the conserved DNA binding motif in the AT-hook III of HMGA2, it can be supposed to result in a dysfunction of AT-hook III. In fact, the relevance of the AT-Hook III in HMGA2 is not yet understood, but first studies have shown a correlation between the amount of functional AT hooks and the protein function in general (18). In HMGA1, a sister gene of HMGA2, the AT hook III has the lowest DNA-binding capacity in comparison to the other 2, whereas AT hook II is the most important one (19, 20). In summary, the presumably minor functional impact of the variant p.(Pro80Leu) in the third AT hook might explain the mild phenotype in the heterozygote members of family III with only slightly reduced growth (Fig. 2J: I-2, I-3, II-1, II-2) whereas the homozygous patients (Fig. 2J: III-1, III-2) exhibit a severe phenotype. Despite the lack of verification for the impact of this variant to HMGA2 protein function, the genotype–phenotype correlation in the family strongly indicates a functional consequence. Furthermore, no other pathogenic variants compatible with the phenotype and the segregation in the family were found by whole exome analysis. To the best of our knowledge, this is the first report on homozygosity for a genomic variant in HMGA2 associated with SRS.
Clinically, the precise identification of the molecular cause of growth retardation and SRS is important for prognosis and treatment. The catch-up growth in the course of rGH treatment in patient 2 suggests that rGH treatment might be appropriate in management of SRS patients with HMGA2 variants. However, further investigations and follow-up studies are required to confirm these effects. An interesting observation is the discrepancy in head circumference in HMGA2 patients even within the same family. Although the father of patient 1 and the siblings of family 3 are more macrocephalic, patients 1 and 2 are microcephalic. In fact, this is accordance with the literature (Table 1) (6). A possible explanation might be an incomplete penetrance of HMGA2 variants. Furthermore, patients with pathogenic variants in HMGA2 might be at risk to develop T2D because the father in family 1 developed T2D at the age of 30. Genome-wide association studies showed a correlation between HMGA2 single nucleotide polymorphisms and T2D (21, 22). In addition, variants in HMGA1 lead to T2D and insulin resistance in human and mice (23). We suggest that patients with an HMGA2-associated SRS should receive regular endocrinological follow-ups to recognize diabetes mellitus as early as possible and to avoid complications.
Conclusion
Based on literature data and the identification of 3 novel variants, we confirm that pathogenic variants in HMGA2 are associated with the characteristic SRS phenotype and can therefore be regarded as an SRS-causing gene. Furthermore, we show for the first time that pathogenic variants in HMGA2 lead to a phenotype, the severity of which depends on the localization of the variant in the gene and its impact on HMGA2 protein function.
Weblinks
https://gnomad.broadinstitute.org/
http://exac.broadinstitute.org/
http://www.mutationtaster.org/
https://sift.bii.a-star.edu.sg/
Abbreviations
- IC1
imprinting center 1
- LOM
loss of methylation
- NH-CSS
Netchine-Harbison clinical scoring system
- rGH
recombinant growth hormone
- SDS
standard deviation score
- SRS
Silver-Russell syndrome
- T2D
type 2 diabetes.
Acknowledgments
Financial Support: The group is funded by the Deutsche Forschungsgemeinschaft (DFG, EG110/15-1; 948/32-1 FUGG).
Additional Information
Disclosure Summary: None of the authors has a conflict of interest relevant to the subject matter or materials included in this work.
Data Availability: The datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
References

{kind=link}
{kind=link}