





Abstract
Familial hypoparathyroidism has a heterogeneous presentation where patients usually have low parathyroid hormone (PTH) levels due to impaired production or secretion. This contrasts with pseudohypoparathyroidism, in which PTH resistance is usually associated with an elevated serum PTH. High levels of circulating PTH can also be due to bioinactive PTH, which is difficult to distinguish from pseudohypoparathyroidism on biochemical grounds.
We report on 2 sisters from consanguineous parents who presented with tetany at birth and were diagnosed with congenital hypocalcemia. Serum PTH levels were normal for many years, but progressively increased in midadulthood to greater than 100x the upper limit of normal on multiple assays. Homozygosity mapping was performed on 1 sister that demonstrated loss of heterozygosity (LOH) around PTH. Sequencing revealed a previously unreported variant, c.94T>C, predicting a codon change of p.Ser32Pro that is biologically inactive.
This case report shows a previously unreported unusual biochemical phenotype of a rising PTH in the context of a novel PTH mutation. This expands the evolving genotypes associated with hypoparathyroidism without established gene mutations.
Congenital hypoparathyroidism is a heterogeneous disorder characterized by hypocalcemia secondary to impaired production, action, or secretion of parathyroid hormone (PTH). Presentation can vary from early-onset tetany through to asymptomatic discovery in adulthood. Chronic complications of hypoparathyroidism most commonly include symptomatic hypocalcemia, renal complications, and—rarely—extrapyramidal disorders or variable skeletal abnormalities. Congenital hypoparathyroidism can be isolated or syndromic. Genetic causes of isolated hypoparathyroidism include inactivation of PTH or GCM2, X-linked recessive SOX3 mutations, or activation of the calcium-sensing receptor CASR or its coupled G-protein GNA11 (1–3). Syndromic hypoparathyroidism is associated with pathogenic variants in GATA3 (Barakat syndrome) (4), TBCE (Kenny-Caffey syndrome), 22q11 deletion encompassing the TBX1 mutation (vertebral anomalies, variable endocrine and T-cell dysfunction), CHD7 mutations (CHARGE syndrome) (5), and various mitochondrial deletions (Kearns Sayre syndrome) (1).
Human PTH contains 3 exons and is found on chromosome 11. Exon 1 is noncoding; exons 2 and 3 encode preparathyroid hormone (pre-ProPTH), which is the initial translation product. This undergoes 2 proteolytic cleavages to ProPTH and then to mature PTH (1–84). Few case reports harbouring pre-ProPTH and ProPTH mutations have been described (6–10) and there is only a single case in mature PTH (11).
Herein we describe 2 sisters with a novel homozygous c.94T>C, p.(Ser32Pro) mutation in the first amino acid residue of mature PTH. This is the first case of rising and circulating—but bioinactive—PTH in the context of familial hypoparathyroidism.
Case Report
Subject 1 (proband)
The proband was diagnosed at birth with hypocalcemia and was commenced on calcium supplements (20 mL of calcium oil), and calcitriol was added when she was a teenager (due to production availability). At age 7 she began having seizures, which has continued through her early adult life and was attributed to idiopathic epilepsy. Her last seizure was >30 years ago and she remains on carbamazepine. Growth and development was normal, and menarche occurred at age 13. Throughout her youth and middle age, she was intermittently symptomatic with tetany and occasional paraesthesiae, which resolved with additional calcium treatment. She presented to our department as an adult, and the earliest PTH we have recorded at age 34 years is 8.8 pmol/L, then 6 months later 2.5 pmol/L (NR 1.6–6.9 pmol/L), see Fig. 1. She has had 3 uncomplicated episodes of renal colic secondary to nephrocalcinosis, the first at age 30. Initial presentation was with flank pain, and renal ultrasound demonstrated 5 stones in her left kidney, and 4 in her right. Two subsequent episodes were less severe and none required intervention. Cerebral MRIs performed for epilepsy management have been unremarkable and show no evidence of basal ganglia calcification. Her postmenopausal bone density showed mild osteopenia in the hip (see Table 1 for T-scores) and spine, with a trabecular bone score of 1.51 at the lumbar spine. Neck ultrasound demonstrated enlarged parathyroid glands.
Current laboratory results for both subjects, P1NP procollagen type 1 N-terminal propeptide, CTX C-terminal collagen type 1 telopeptide, L1-4 lumbar spine 1–4
| Subject 1 (Proband) | Subject 2 | Reference Range | |
|---|---|---|---|
| Serum | |||
| PTH, pmol/L | 185 | 150 | 1.6-6.9 |
| Calcium, mmol/L | 2.25 | 2.35 | 2.2-2.7 |
| Phosphate, mmol/L | 1.50 | 1.40 | 0.8-1.3 |
| 25 hydroxyvitamin D, nmol/L | 91 | 79 | >50 |
| eGFR, ml/min | 80 | >90 | >90 |
| Bone turnover markers | |||
| P1NP, µg/L | 60 | 32 | 15-70 |
| CTX, ng/L | 388 | 157 | 150-800 |
| Urine | |||
| Calcium, mmol/day | 5.2 | 5.4 | <7.5 mmol/day |
| Fractional excretion of calcium | 15.8% | 14.3% | – |
| Bone density; T-score | |||
| L1–4 | -0.8 | 1.8 | – |
| Femoral neck | -1.3 | 0.9 | – |
| Calciotropic medications | |||
| Calcium carbonate, mg | – | 600 | – |
| Calcitriol, mcg | 1.25 | 1.25 | – |
| Magnesium, mg | 4500 | 500 | – |
| Calcium citrate, mg | 250 | – | – |
| Subject 1 (Proband) | Subject 2 | Reference Range | |
|---|---|---|---|
| Serum | |||
| PTH, pmol/L | 185 | 150 | 1.6-6.9 |
| Calcium, mmol/L | 2.25 | 2.35 | 2.2-2.7 |
| Phosphate, mmol/L | 1.50 | 1.40 | 0.8-1.3 |
| 25 hydroxyvitamin D, nmol/L | 91 | 79 | >50 |
| eGFR, ml/min | 80 | >90 | >90 |
| Bone turnover markers | |||
| P1NP, µg/L | 60 | 32 | 15-70 |
| CTX, ng/L | 388 | 157 | 150-800 |
| Urine | |||
| Calcium, mmol/day | 5.2 | 5.4 | <7.5 mmol/day |
| Fractional excretion of calcium | 15.8% | 14.3% | – |
| Bone density; T-score | |||
| L1–4 | -0.8 | 1.8 | – |
| Femoral neck | -1.3 | 0.9 | – |
| Calciotropic medications | |||
| Calcium carbonate, mg | – | 600 | – |
| Calcitriol, mcg | 1.25 | 1.25 | – |
| Magnesium, mg | 4500 | 500 | – |
| Calcium citrate, mg | 250 | – | – |
Current laboratory results for both subjects, P1NP procollagen type 1 N-terminal propeptide, CTX C-terminal collagen type 1 telopeptide, L1-4 lumbar spine 1–4
| Subject 1 (Proband) | Subject 2 | Reference Range | |
|---|---|---|---|
| Serum | |||
| PTH, pmol/L | 185 | 150 | 1.6-6.9 |
| Calcium, mmol/L | 2.25 | 2.35 | 2.2-2.7 |
| Phosphate, mmol/L | 1.50 | 1.40 | 0.8-1.3 |
| 25 hydroxyvitamin D, nmol/L | 91 | 79 | >50 |
| eGFR, ml/min | 80 | >90 | >90 |
| Bone turnover markers | |||
| P1NP, µg/L | 60 | 32 | 15-70 |
| CTX, ng/L | 388 | 157 | 150-800 |
| Urine | |||
| Calcium, mmol/day | 5.2 | 5.4 | <7.5 mmol/day |
| Fractional excretion of calcium | 15.8% | 14.3% | – |
| Bone density; T-score | |||
| L1–4 | -0.8 | 1.8 | – |
| Femoral neck | -1.3 | 0.9 | – |
| Calciotropic medications | |||
| Calcium carbonate, mg | – | 600 | – |
| Calcitriol, mcg | 1.25 | 1.25 | – |
| Magnesium, mg | 4500 | 500 | – |
| Calcium citrate, mg | 250 | – | – |
| Subject 1 (Proband) | Subject 2 | Reference Range | |
|---|---|---|---|
| Serum | |||
| PTH, pmol/L | 185 | 150 | 1.6-6.9 |
| Calcium, mmol/L | 2.25 | 2.35 | 2.2-2.7 |
| Phosphate, mmol/L | 1.50 | 1.40 | 0.8-1.3 |
| 25 hydroxyvitamin D, nmol/L | 91 | 79 | >50 |
| eGFR, ml/min | 80 | >90 | >90 |
| Bone turnover markers | |||
| P1NP, µg/L | 60 | 32 | 15-70 |
| CTX, ng/L | 388 | 157 | 150-800 |
| Urine | |||
| Calcium, mmol/day | 5.2 | 5.4 | <7.5 mmol/day |
| Fractional excretion of calcium | 15.8% | 14.3% | – |
| Bone density; T-score | |||
| L1–4 | -0.8 | 1.8 | – |
| Femoral neck | -1.3 | 0.9 | – |
| Calciotropic medications | |||
| Calcium carbonate, mg | – | 600 | – |
| Calcitriol, mcg | 1.25 | 1.25 | – |
| Magnesium, mg | 4500 | 500 | – |
| Calcium citrate, mg | 250 | – | – |
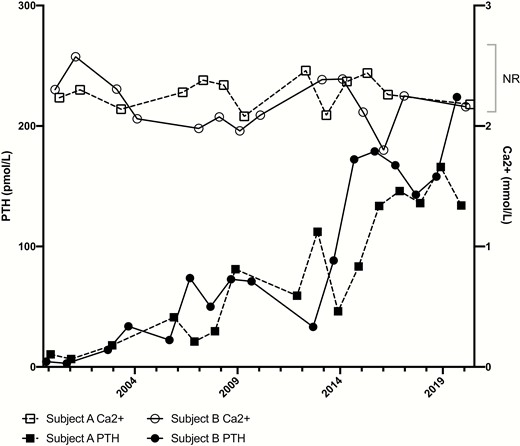
Trajectory of calcium and PTH levels in both subjects. Mean levels of PTH and calcium have been recorded for each year. Subject A was 33 years old and Subject B was 42 years old when PTH levels began to rise substantially. Abbreviations: Ca2+, calcium; PTH, parathyroid hormone; NR, normal range.
Subject 2
The younger sister was also diagnosed at birth with hypocalcemia and has a similar phenotype. She also recalls starting on calcium supplements (20 ml oil) and calcitriol when it became available. At birth, she was diagnosed with a congential heart malformation (Ebstein’s anomaly), resulting in tricuspid regurgitation. Menarche was slightly delayed at 14 years, but she achieved a normal height (163 cm). She has a history of clear cell renal carcinoma (diagnosed at age 41), with resultant nephrectomy and remains in remission. She has also suffered recurrent untriggered pulmonary emboli with no cause identified and remains on long-term anticoagulation. She reports no history of renal calculi. She suffers from intermittent headaches, unlike her sister. MRI to investigate headaches in midadulthood showed basal ganglia calcification. She is premenopausal with normal bone mineral density and trabecular bone score (see Table 1).
Examination of both subjects revealed no dysmorphic features, such as brachydactyly or short stature. There was no radiological evidence of Albrights hereditary osteodystrophy. Biochemical data from 2019 (at ages 54 and 47) is presented in Table 1. The sisters have another unaffected elder sister and had 2 brothers who both died at 10 weeks and 8 months, respectively. The cause of death is not known to the family. The extended pedigree reveals no other family members affected with calcium or PTH disorders. Both parents are alive and well with normal PTH and calcium levels.
Although both subjects had low-normal serum PTH for many years, over the past 2 decades, serum PTH concentrations have increased markedly in both despite remaining normocalcemic (Fig. 1). This was first observed in Subject 1 in 2000 (age 36 years) when her PTH started to rise with a slow gradient, which accelerated from age 43 years until her current age of 55 years, where it now remains >150 pmol/L (Fig. 1). Subject 2 had a steeper gradient of PTH drift in 2007 (age 34 years) and up to 263.3 pmol/L in 2016 (age 43 years), where it has largely remained. Assay interference was excluded by confirming elevated PTH levels on multiple sandwich assays. The proband’s PTH levels were 81.2 (10x ULN) on Roche in 2009, 46 (8xULN) on Centeur in 2010, and 29.9 (4x ULN) on Immulite in 2008. In addition, PTH diluted in parallel and there was no evidence of heterophile antibodies.
Homozygosity mapping performed using the proband’s DNA showed several large regions of homozygosity, encompassing >1.7% of the genome. There was no significant copy number change detected. Regions of LOH were examined and cross-referenced to PTH, GNAS, and other associated genes involved in calcium pathways. One region with LOH was identified in chromosome 11, which encompassed the PTH locus. Direct sequencing of PTH identified a homozygous missense variant c.94T>C, p.(Ser32Pro) in exon 3 (Fig. 2).
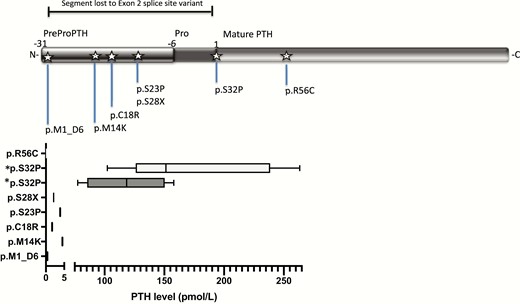
Identified mutations in PTH with corresponding hormone levels reported in the literature. *Shows p.R56C mutation, where PTH measured with (1–84) antibody identified high levels of the mutated peptide, while using the ntPTH assay, PTH measured below normal, as reviewed in Lee et al (11). The novel mutation described in this report, p.S32P, is shown here for both subjects.
This variant has not been reported in ClinVar and is not present in population databases gnomAD- nor 1000 genomes. To assess the functional impact of this amino acid substitution, we performed in silico analysis with Provean, SIFT, and Polyphen-2, which produced predictions consistent with damaging effects on protein function (Table 2).
Results from in silico analysis for newly identified variant p.Ser32Pro
| Tool | Score | Prediction |
|---|---|---|
| Polyphen | 0.771 | Possibly damaging |
| PROVEAN | -3.06 | Deleterious |
| SIFT | 0.025 | Damaging |
| Tool | Score | Prediction |
|---|---|---|
| Polyphen | 0.771 | Possibly damaging |
| PROVEAN | -3.06 | Deleterious |
| SIFT | 0.025 | Damaging |
Results from in silico analysis for newly identified variant p.Ser32Pro
| Tool | Score | Prediction |
|---|---|---|
| Polyphen | 0.771 | Possibly damaging |
| PROVEAN | -3.06 | Deleterious |
| SIFT | 0.025 | Damaging |
| Tool | Score | Prediction |
|---|---|---|
| Polyphen | 0.771 | Possibly damaging |
| PROVEAN | -3.06 | Deleterious |
| SIFT | 0.025 | Damaging |
Discussion
This trajectory of serum PTH levels over the past 20 years is remarkable and not previously reported in the literature associated with congenital hypoparathyroidism. Typically, serum PTH remains low throughout life in familial isolated hypoparathyroidism (1). Conversely, high PTH levels seen in pseudohypoparathyroidism type 1a (PHP1a) and 1b are consequent to PTH resistance. A rising PTH level has been reported in PHP1a, where in a retrospective study all patients with inactivating GNAS mutations had PTH levels, which increased over time, reflecting the gradual silencing of the paternal GS⍺ in the renal proximal tubule (12). In that series, none of the 20 patients ever had low levels of PTH. The novel biochemical phenotype in our 2 subjects that began with a low PTH at birth to young adulthood, followed by high levels of PTH in midadulthood has not been previously described. In the diabetes literature, a similar scientific narrative is observed when insulin gene mutations cause hyperinsulinemia (13). This culminates in reduced biological activity, receptor binding, and causes diabetes mellitus. To our knowledge there has been no report on increasing insulin levels over time in patients harbouring the mutations.
Parathyroid hormone binds to its receptor PTHR1 and G proteins mediate subsequent intracellular signaling, leading to increased production of cAMP. Patients who are heterozygous for PTH mutations are normocalcemic and have normal levels of PTH, suggesting that 1 functioning gene is sufficient to ensure signaling and maintains physiological levels of calcium and phosphate (11). Several PTH mutations have been described that variably affect cleavage from preproPTH (8–10) to proPTH or from proPTH to mature PTH, leading to inadequate PTH synthesis (14) or secretion (Fig. 2). If the mutation is the pro or prepro sequence, PTH is unable to be cleaved and mutant PTH is trapped intracellularly, which is toxic for cells affecting translocation and function (15). Measured PTH levels have been graphed by mutation in Fig. 2.
Lee and colleagues reported the first and only other mutation in mature PTH: c.166C>T (11). In that case, measurement of PTH was highly assay-dependent and was only raised when measured with a PTH (1–84) assay. In the cases described here, assays employed throughout the 20 years of records were all 2-site sandwich assays, which have epitopes directed against either end of the PTH molecule, ruling out assay interference as the cause for PTH increase.
In our novel mutation, proline is substituted for serine in position 32, or the first amino acid in mature PTH or “position 1.” In silico analysis of this variant predicts deleterious effects on protein function. Moreover, the effect of this variant on protein function was examined directly in vitro by Gardella and colleagues through COS cell expression with degenerate oligonucleotides followed by assays of PTH receptor-binding and cAMP stimulation in rat osteosarcoma ROS17/2.8 cells (16). Remarkably, these studies showed that PTH p.Ser32Pro had no detectable binding to PTH receptor nor postreceptor cAMP signaling. In their study, several other position 1 mutants besides proline (Tyr, Val, Asp) also showed inactivity, although alanine at this position was tolerated, reflecting the evolutionary observation that serine or alanine are the only residues seen at PTH codon 1. These functional studies, taken together with the clinical phenotype in our subjects, strongly support that this p.Ser32Pro variant is nonfunctional.
Homozygosity mapping is an effective mapping method to identify candidate genes in rare conditions associated with consanguinity and allowed us to quickly refine our search for a mutation in calciotropic pathways. In patients with “idiopathic” hypoparathyroidism, albeit rare, mutations in the PTH gene should be considered. We present the first report of familial hypoparathyroidism in which serum PTH was initially low normal but progressively increased in midadulthood, consistent with the overproduction of a nonfunctional PTH. The parathyroid enlargement may be the cause of the progressively increasing PTH levels that have developed over time. We speculate this reflects parathyroid hyperplasia, secondary to chronic mild undertreatment with calcium/calcitriol supplementation (Fig. 1) and/or chronic mild hyperphosphatemia (Table 1).
Additional Information
Disclosure Summary: The authors have nothing to disclose.
Data Availability: All data generated or analyzed during this study are included in this published article or in the data repositories listed in References.
References

{kind=link}
{kind=link}