





Abstract
Central precocious puberty (CPP) may be the first presentation of nonclassical congenital adrenal hyperplasia (NCCAH) in girls. Data on the prevalence and the clinical phenotype of CPP associated with NCCAH are sparse.
To study the clinical and laboratory characteristics that could differentiate idiopathic CPP from CPP associated with NCCAH and to determine the prevalence of NCCAH among girls with CPP.
Case-control study.
Tertiary pediatric endocrinology institute.
From 2008 to 2017, 147 girls who had undergone stimulation tests with gonadotropin-releasing hormone and ACTH were diagnosed with CPP; of these, seven (4.8%) were eventually diagnosed with NCCAH. These seven patients together with 30 girls who presented with CPP during 1984 to 2008 and were later diagnosed with NCCAH comprised the NCCAH group. Demographic, anthropometric, clinical, and laboratory data were compared between the NCCAH group and the 140 girls with idiopathic CPP (ICPP group).
No between-group differences were found in height, weight, body mass index, bone age, and Tanner stage. Mean basal levels of androstenedione, dehydroepiandrosterone-sulphate, and 17-hydroxyprogesterone were significantly higher in the NCCAH group, although ranges overlapped between the groups, and stimulated cortisol level was higher in the ICPP group.
NCCAH was found in 4.8% of girls presenting with true CPP over 10 years, and no single parameter could differentiate between the diagnoses. Thus, in girls with true CPP from populations in which NCCAH is prevalent, assessment of adrenal androgens is required, and ACTH test should be considered.
Precocious puberty (PP) in girls is usually caused by premature activation of the hypothalamic GnRH pulse generator [termed central PP (CPP)], and in most cases it is idiopathic (1). True CPP may be secondary to the classic form of congenital adrenal hyperplasia (CAH), due to 21-hydroxylase deficiency (2). CPP in CAH may be associated with elevated sex-steroid levels (3), advanced bone age (BA), and possibly a decline in negative sex-steroid feedback after treatment initiation. CPP may also be secondary to nonclassical congenital adrenal hyperplasia (NCCAH), with reported prevalence rates of up to 28% (4–6). Researchers have suggested that every child with signs of excess androgen activity or early puberty should be evaluated for possible NCCAH (1).
The standard assessment for PP includes a GnRH stimulation test (1, 7). At our institute, the workup for suspected CPP also includes an ACTH stimulation test to rule out NCCAH. However, the routine performance of basal androgen and/or ACTH tests as part of the workup for CPP is controversial, and most centers apply them only when adrenarche is prominent (1, 8). Our policy is based on previous reports by our group and others (4, 6, 9) showing that CPP may be a presenting sign of NCCAH. In these cases, like for CAH, CPP is considered to be secondary to the excessive adrenal steroid secretion and advanced skeletal maturation associated with NCCAH. The presumed mechanism is a reduction in the sensitivity of the GnRH pulse generator to sex-steroid inhibition due to the excess pubertal androgens (10).
The prevalence of NCCAH in the Israeli population is 10 times higher than in the general world population (∼1:100 compared with 1:1000) (11, 12), making it more likely that girls diagnosed with CPP at our institute have NCCAH. Missing the diagnosis of NCCAH poses a risk of long-term sequelae, such as short stature (5, 13), subfertility (14), and other features of hyperandrogenism. Moreover, the prevalence of signs of androgen excess among women with NCCAH increases with age, suggesting the disease is progressive (15). Thus, early diagnosis and treatment with glucocorticoids or at least close follow-up are important. Screening for NCCAH is not performed routinely in most clinics; thus, clinical markers are needed that can differentiate idiopathic CPP from NCCAH-associated CPP. To our knowledge, data on the clinical characteristics of CPP in girls with NCCAH are sparse.
The aim of this study was to identify clinical and laboratory characteristics that could help clinicians distinguish between idiopathic CPP and CPP associated with NCCAH, and to determine the prevalence of NCCAH among girls with CPP.
Patients and Methods
A retrospective comparative cohort study design was used. Search of the database of a tertiary endocrinology institute yielded 147 girls who presented with CPP during 2008 to 2017. Inclusion criteria were breast budding before 8 years of age and referral before 9 years of age. This sample was divided into two groups: girls with idiopathic CPP (ICPP; the ICPP group; n = 140) and girls eventually diagnosed with NCCAH (n = 7), and prevalence rates were calculated. The latter group was then combined with an additional 30 girls who presented with CPP and were diagnosed with NCCAH during 1984 to 2008 (the NCCAH group). The NCCAH group was compared with the ICPP group for demographic, anthropometric, clinical, and laboratory data.
As part of the workup, all patients underwent clinical, biochemical, and BA evaluation. Pubertal stage was determined according to Marshall and Tanner (16). Height was measured using a Harpenden stadiometer (Holtain Ltd, Pembrokeshire, United Kingdom) and calculated as a height-SD score (SDS) using the Centers for Disease Control and Prevention growth charts (17). Body weight was expressed as body mass index (calculated as weight in kilograms divided by height in meters squared), and the body mass index SDS was calculated according to the method of Rosner et al. (18). BA was assessed and BA-SDS was calculated according to Greulich and Pyle (19).
GnRH and ACTH stimulation tests for the diagnosis of CPP were performed on the day of referral, according to our departmental policy. Basal and GnRH-stimulated levels of LH and FSH were measured using a solid-phase, two-site chemiluminescent immunometric assay (Immulite 2000; DPC, Los Angeles, CA). The GnRH stimulation test was performed with 100 μg of GnRH, given as an IV bolus; serum LH and FSH concentrations were measured at 0, 30, and 60 minutes. Peak LH levels >5.0 IU/L were considered diagnostic for CPP (20). For the ACTH stimulation test, levels of 17-hydroxyprogesterone (17-OHP) and cortisol were measured before and 60 minutes after IV injection of 0.25 mg of ACTH (Synacthen, Novartis, New York, NY). Both tests in all patients were carried out between 8:00 am and 10:00 am after a heparin-locked cannula was placed in a forearm vein. The hormonal analyses were performed in the endocrine laboratory of our hospital with commercial kits: double antibody estradiol procedure (DPC) and androstenedione radioimmunoassay (Diagnostic Systems Laboratories, Webster, TX). Serum cortisol, 17-OHP, testosterone, and dehydroepiandrosterone (DHEAS) levels were measured using the Coat-A-Count RIA (DPC).
The clinical diagnosis of CPP was based on the appearance of breast buds before 8 years of age accompanied by one or more of the following findings: menses, pubic hair, accelerated growth velocity, or BA >2 SD above chronological age. Cases that were equivocal on referral were diagnosed after a 6-month follow-up on the basis of clinical judgment by an experienced clinician.
Other forms of PP were ruled out by further laboratory tests (i.e., prolactin, liver functions, α-fetoprotein, β-human chorionic gonadotropin) and imaging studies (i.e., central nervous system magnetic resonance imaging, abdominal ultrasound), according to clinical judgment.
The diagnosis of NCCAH was based on a stimulated 17-OHP serum level of ≥45 nmol/L with or without molecular confirmation or >30 nmol/L if confirmed by molecular analysis, as previously described (6). Girls with chronic disease, bone dysplasia, organic brain disease, McCune-Albright syndrome, or other endocrinological abnormalities were excluded, as were girls who had received radiation therapy and/or chemotherapy, and girls with known exposure to exogenous androgens or estrogens. The study was approved by the institutional review board of Rabin Medical Center, Israel.
Statistical analysis
Data analyses were carried out using BMDP statistical software (21). Data were expressed as means and SDs for normally distributed variables, and as medians and interquartile ranges for variables with a skewed distribution.
Continuous variables were compared between groups using ANOVA, and discrete variables, using Pearson χ2 test or Fisher exact test, as appropriate. For variables that did not have a Gaussian distribution, and because of the extremely small numbers, the Mann-Whitney U test was used. The variables found to be significant on univariate analysis were entered into a stepwise logistic regression to identify those significantly associated with a diagnosis of NCCAH. P < 0.05 was considered significant.
Results
The girls diagnosed with NCCAH accounted for 4.8% of the 147 girls who presented with CPP at our institute during 2008 to 2017. The clinical characteristics of the ICPP group (n = 140) and the whole NCCAH group (n = 37) are presented in Table 1. There were no statistically significant differences between the groups in age at onset of puberty or anthropometric characteristics and extent of BA advancement. The ICPP group had a higher percentage of girls without signs of pubarche (either pubic and/or axillary hair) at presentation (27.3%) than did the NCCAH group (5.6%) (P = 0.03). However, the percentage of those in Tanner stage 1 or 2 compared with those in Tanner stage 3 or 4 was similar in the two groups (14.2% and 22.2%, respectively; P = 0.3).
Characteristics at Diagnosis of Girls With ICPP or NCCAH-Associated CPP
| Characteristic | ICPP Group, n = 140 | NCCAH Group, n = 37 | |||
|---|---|---|---|---|---|
| Mean ± SD | Range | Mean ± SD | Range | P | |
| Clinical feature | |||||
| Age at onset, y | 6.96 ± 1.14 | 1.5–8.8 | 7.2 ± 0.8 | 4.5–8.3 | 0.34 |
| Age at diagnosis, y | 7.54 ± 1.1 | 3.3–9.5 | 7.6 ± 1.1 | 4.6–9 | 0.85 |
| Pubarche, %a | 27.3, 59, 9.4, 4.3, 0 | 5.6, 72.2, 22.2, 0, 0 | 0.007 | ||
| Thelarche, %a | 0, 54.3, 41.4, 4.3, 0 | 0, 83.8, 16.2, 0, 0 | 0.004 | ||
| Axillary hair, %a | 59.3, 37.1, 2.9, 0, 0 | 61.1, 30.6, 8.3, 0, 0 | 0.43 | ||
| Anthropometric measurement | |||||
| Height SDS | 0.69 ± 1.09 | −2.6 to 4.37 | 0.52 ± 0.82 | −1.16 to 2.22 | 0.37 |
| Weight SDS | 0.93 ± 0.90 | −2to 2.81 | 0.68 ± 1.06 | −1.7 to 2.3 | 0.16 |
| BMI SDS | 0.87 ± 0.86 | −2.14 to 2.43 | 0.55 ± 1.19 | −2.75 to 2.32 | 0.07 |
| BA, y | 9.03 ± 1.53 | 4.5–12.5 | 9.28 ± 1.71 | 3.5–11.5 | 0.39 |
| BA − chronological age, y | 1.48 ± 1.07 | −0.93 to 5.1 | 1.7 ± 1.04 | −1.17 to 3.83 | 0.29 |
| Hormonal profile | |||||
| Estradiol, pmol/L | 49.16 ± 55.54 | 3–212.5 | 54.35 ± 35.88 | 2.4–116.6 | 0.78 |
| DHEAS, μmol/L | 1.5 ± 1.23 | 0.3–6.7 | 3.6 ± 2.23 | 0.6–10.26 | <0.001 |
| Androstenedione, nmol/L | 2.15 ± 2.26 | 0.1–22 | 5.01 ± 4.18 | 0.04–23.6 | <0.001 |
| Basal LH, IU/L | 0.62 ± 0.77 | 0.08–4.4 | 0.52 ± 0.59 | 0.08–1.9 | 0.49 |
| Stimulated LH, IU/L | 13.23 ± 13.55 | 0.3–68.4 | 4.77 ± 3.7 | 0.08–15.7 | <0.002 |
| Basal FSH, IU/L | 3.23 ± 1.98 | 0.13–9.4 | 2.57 ± 2.51 | 0.9–10.9 | 0.1 |
| Stimulated FSH, IU/L | 10.40 ± 4.54 | 1.09–30.7 | 7.92 ± 3.26 | 2.3–13.4 | 0.008 |
| Basal 17-OHP, nmol/L | 1.86 ± 1.291 | 0.3–9.6 | 32.62 ± 26.66 | 2.2–96 | <0.001 |
| Stimulated 17-OHP, nmol/L | 7.2 ± 3.2 | 2–22.7 | 123.6 ± 47.1 | 28.5–203 | <0.001 |
| Basal cortisol, nmol/L | 262.9 ± 125.4 | 68–629 | 306.8 ± 153.5 | 114–646 | 0.08 |
| Stimulated cortisol, nmol/L | 697.6 ± 97 | 419–1046 | 550.7 ± 140.9 | 340–899 | <0.001 |
| Characteristic | ICPP Group, n = 140 | NCCAH Group, n = 37 | |||
|---|---|---|---|---|---|
| Mean ± SD | Range | Mean ± SD | Range | P | |
| Clinical feature | |||||
| Age at onset, y | 6.96 ± 1.14 | 1.5–8.8 | 7.2 ± 0.8 | 4.5–8.3 | 0.34 |
| Age at diagnosis, y | 7.54 ± 1.1 | 3.3–9.5 | 7.6 ± 1.1 | 4.6–9 | 0.85 |
| Pubarche, %a | 27.3, 59, 9.4, 4.3, 0 | 5.6, 72.2, 22.2, 0, 0 | 0.007 | ||
| Thelarche, %a | 0, 54.3, 41.4, 4.3, 0 | 0, 83.8, 16.2, 0, 0 | 0.004 | ||
| Axillary hair, %a | 59.3, 37.1, 2.9, 0, 0 | 61.1, 30.6, 8.3, 0, 0 | 0.43 | ||
| Anthropometric measurement | |||||
| Height SDS | 0.69 ± 1.09 | −2.6 to 4.37 | 0.52 ± 0.82 | −1.16 to 2.22 | 0.37 |
| Weight SDS | 0.93 ± 0.90 | −2to 2.81 | 0.68 ± 1.06 | −1.7 to 2.3 | 0.16 |
| BMI SDS | 0.87 ± 0.86 | −2.14 to 2.43 | 0.55 ± 1.19 | −2.75 to 2.32 | 0.07 |
| BA, y | 9.03 ± 1.53 | 4.5–12.5 | 9.28 ± 1.71 | 3.5–11.5 | 0.39 |
| BA − chronological age, y | 1.48 ± 1.07 | −0.93 to 5.1 | 1.7 ± 1.04 | −1.17 to 3.83 | 0.29 |
| Hormonal profile | |||||
| Estradiol, pmol/L | 49.16 ± 55.54 | 3–212.5 | 54.35 ± 35.88 | 2.4–116.6 | 0.78 |
| DHEAS, μmol/L | 1.5 ± 1.23 | 0.3–6.7 | 3.6 ± 2.23 | 0.6–10.26 | <0.001 |
| Androstenedione, nmol/L | 2.15 ± 2.26 | 0.1–22 | 5.01 ± 4.18 | 0.04–23.6 | <0.001 |
| Basal LH, IU/L | 0.62 ± 0.77 | 0.08–4.4 | 0.52 ± 0.59 | 0.08–1.9 | 0.49 |
| Stimulated LH, IU/L | 13.23 ± 13.55 | 0.3–68.4 | 4.77 ± 3.7 | 0.08–15.7 | <0.002 |
| Basal FSH, IU/L | 3.23 ± 1.98 | 0.13–9.4 | 2.57 ± 2.51 | 0.9–10.9 | 0.1 |
| Stimulated FSH, IU/L | 10.40 ± 4.54 | 1.09–30.7 | 7.92 ± 3.26 | 2.3–13.4 | 0.008 |
| Basal 17-OHP, nmol/L | 1.86 ± 1.291 | 0.3–9.6 | 32.62 ± 26.66 | 2.2–96 | <0.001 |
| Stimulated 17-OHP, nmol/L | 7.2 ± 3.2 | 2–22.7 | 123.6 ± 47.1 | 28.5–203 | <0.001 |
| Basal cortisol, nmol/L | 262.9 ± 125.4 | 68–629 | 306.8 ± 153.5 | 114–646 | 0.08 |
| Stimulated cortisol, nmol/L | 697.6 ± 97 | 419–1046 | 550.7 ± 140.9 | 340–899 | <0.001 |
Abbreviation: BMI, body mass index.
Tanner stages 1–5.
Characteristics at Diagnosis of Girls With ICPP or NCCAH-Associated CPP
| Characteristic | ICPP Group, n = 140 | NCCAH Group, n = 37 | |||
|---|---|---|---|---|---|
| Mean ± SD | Range | Mean ± SD | Range | P | |
| Clinical feature | |||||
| Age at onset, y | 6.96 ± 1.14 | 1.5–8.8 | 7.2 ± 0.8 | 4.5–8.3 | 0.34 |
| Age at diagnosis, y | 7.54 ± 1.1 | 3.3–9.5 | 7.6 ± 1.1 | 4.6–9 | 0.85 |
| Pubarche, %a | 27.3, 59, 9.4, 4.3, 0 | 5.6, 72.2, 22.2, 0, 0 | 0.007 | ||
| Thelarche, %a | 0, 54.3, 41.4, 4.3, 0 | 0, 83.8, 16.2, 0, 0 | 0.004 | ||
| Axillary hair, %a | 59.3, 37.1, 2.9, 0, 0 | 61.1, 30.6, 8.3, 0, 0 | 0.43 | ||
| Anthropometric measurement | |||||
| Height SDS | 0.69 ± 1.09 | −2.6 to 4.37 | 0.52 ± 0.82 | −1.16 to 2.22 | 0.37 |
| Weight SDS | 0.93 ± 0.90 | −2to 2.81 | 0.68 ± 1.06 | −1.7 to 2.3 | 0.16 |
| BMI SDS | 0.87 ± 0.86 | −2.14 to 2.43 | 0.55 ± 1.19 | −2.75 to 2.32 | 0.07 |
| BA, y | 9.03 ± 1.53 | 4.5–12.5 | 9.28 ± 1.71 | 3.5–11.5 | 0.39 |
| BA − chronological age, y | 1.48 ± 1.07 | −0.93 to 5.1 | 1.7 ± 1.04 | −1.17 to 3.83 | 0.29 |
| Hormonal profile | |||||
| Estradiol, pmol/L | 49.16 ± 55.54 | 3–212.5 | 54.35 ± 35.88 | 2.4–116.6 | 0.78 |
| DHEAS, μmol/L | 1.5 ± 1.23 | 0.3–6.7 | 3.6 ± 2.23 | 0.6–10.26 | <0.001 |
| Androstenedione, nmol/L | 2.15 ± 2.26 | 0.1–22 | 5.01 ± 4.18 | 0.04–23.6 | <0.001 |
| Basal LH, IU/L | 0.62 ± 0.77 | 0.08–4.4 | 0.52 ± 0.59 | 0.08–1.9 | 0.49 |
| Stimulated LH, IU/L | 13.23 ± 13.55 | 0.3–68.4 | 4.77 ± 3.7 | 0.08–15.7 | <0.002 |
| Basal FSH, IU/L | 3.23 ± 1.98 | 0.13–9.4 | 2.57 ± 2.51 | 0.9–10.9 | 0.1 |
| Stimulated FSH, IU/L | 10.40 ± 4.54 | 1.09–30.7 | 7.92 ± 3.26 | 2.3–13.4 | 0.008 |
| Basal 17-OHP, nmol/L | 1.86 ± 1.291 | 0.3–9.6 | 32.62 ± 26.66 | 2.2–96 | <0.001 |
| Stimulated 17-OHP, nmol/L | 7.2 ± 3.2 | 2–22.7 | 123.6 ± 47.1 | 28.5–203 | <0.001 |
| Basal cortisol, nmol/L | 262.9 ± 125.4 | 68–629 | 306.8 ± 153.5 | 114–646 | 0.08 |
| Stimulated cortisol, nmol/L | 697.6 ± 97 | 419–1046 | 550.7 ± 140.9 | 340–899 | <0.001 |
| Characteristic | ICPP Group, n = 140 | NCCAH Group, n = 37 | |||
|---|---|---|---|---|---|
| Mean ± SD | Range | Mean ± SD | Range | P | |
| Clinical feature | |||||
| Age at onset, y | 6.96 ± 1.14 | 1.5–8.8 | 7.2 ± 0.8 | 4.5–8.3 | 0.34 |
| Age at diagnosis, y | 7.54 ± 1.1 | 3.3–9.5 | 7.6 ± 1.1 | 4.6–9 | 0.85 |
| Pubarche, %a | 27.3, 59, 9.4, 4.3, 0 | 5.6, 72.2, 22.2, 0, 0 | 0.007 | ||
| Thelarche, %a | 0, 54.3, 41.4, 4.3, 0 | 0, 83.8, 16.2, 0, 0 | 0.004 | ||
| Axillary hair, %a | 59.3, 37.1, 2.9, 0, 0 | 61.1, 30.6, 8.3, 0, 0 | 0.43 | ||
| Anthropometric measurement | |||||
| Height SDS | 0.69 ± 1.09 | −2.6 to 4.37 | 0.52 ± 0.82 | −1.16 to 2.22 | 0.37 |
| Weight SDS | 0.93 ± 0.90 | −2to 2.81 | 0.68 ± 1.06 | −1.7 to 2.3 | 0.16 |
| BMI SDS | 0.87 ± 0.86 | −2.14 to 2.43 | 0.55 ± 1.19 | −2.75 to 2.32 | 0.07 |
| BA, y | 9.03 ± 1.53 | 4.5–12.5 | 9.28 ± 1.71 | 3.5–11.5 | 0.39 |
| BA − chronological age, y | 1.48 ± 1.07 | −0.93 to 5.1 | 1.7 ± 1.04 | −1.17 to 3.83 | 0.29 |
| Hormonal profile | |||||
| Estradiol, pmol/L | 49.16 ± 55.54 | 3–212.5 | 54.35 ± 35.88 | 2.4–116.6 | 0.78 |
| DHEAS, μmol/L | 1.5 ± 1.23 | 0.3–6.7 | 3.6 ± 2.23 | 0.6–10.26 | <0.001 |
| Androstenedione, nmol/L | 2.15 ± 2.26 | 0.1–22 | 5.01 ± 4.18 | 0.04–23.6 | <0.001 |
| Basal LH, IU/L | 0.62 ± 0.77 | 0.08–4.4 | 0.52 ± 0.59 | 0.08–1.9 | 0.49 |
| Stimulated LH, IU/L | 13.23 ± 13.55 | 0.3–68.4 | 4.77 ± 3.7 | 0.08–15.7 | <0.002 |
| Basal FSH, IU/L | 3.23 ± 1.98 | 0.13–9.4 | 2.57 ± 2.51 | 0.9–10.9 | 0.1 |
| Stimulated FSH, IU/L | 10.40 ± 4.54 | 1.09–30.7 | 7.92 ± 3.26 | 2.3–13.4 | 0.008 |
| Basal 17-OHP, nmol/L | 1.86 ± 1.291 | 0.3–9.6 | 32.62 ± 26.66 | 2.2–96 | <0.001 |
| Stimulated 17-OHP, nmol/L | 7.2 ± 3.2 | 2–22.7 | 123.6 ± 47.1 | 28.5–203 | <0.001 |
| Basal cortisol, nmol/L | 262.9 ± 125.4 | 68–629 | 306.8 ± 153.5 | 114–646 | 0.08 |
| Stimulated cortisol, nmol/L | 697.6 ± 97 | 419–1046 | 550.7 ± 140.9 | 340–899 | <0.001 |
Abbreviation: BMI, body mass index.
Tanner stages 1–5.
Thelarche tended to be more advanced in the ICPP group, with 45.7% of the girls presenting in Tanner stage 3 or 4 compared with 16.3% of the girls in the NCCAH group (P = 0.001). The remaining girls in each group presented in Tanner stage 2; there were no cases of Tanner stage 5.
The ICPP and NCCAH groups had similar mean levels of basal LH (0.62 and 0.52 IU/L, respectively; P = 0.49) and basal FSH (3.23 and 2.57 IU/L, respectively; P=0.1), but stimulated levels of both hormones were significantly higher in the ICPP group (LH: 13.22 vs 4.77 IU/L, P=0.002; FSH: 10.40 vs 7.92 IU/L; P = 0.008).
Mean basal androgen levels were significantly higher in the NCCAH than the ICPP group: androstenedione, 5.01 vs 2.15 nmol/L (P < 0.001); basal 17-OHP, 32.62 vs 1.86 nmol/L (P < 0.001); and DHEAS, 30.6 vs 1.5 nmol/L (P < 0.001); however, the ranges overlapped (Fig. 1). In addition, as expected, mean stimulated cortisol levels were lower in the NCCAH group (550.7 vs 697.6 nmol/L; P < 0.001). Testosterone was below detection level (0.7 nmol/L) in 93.7% of the ICPP group and 66.7% of the NCCAH group (P < 0.001).
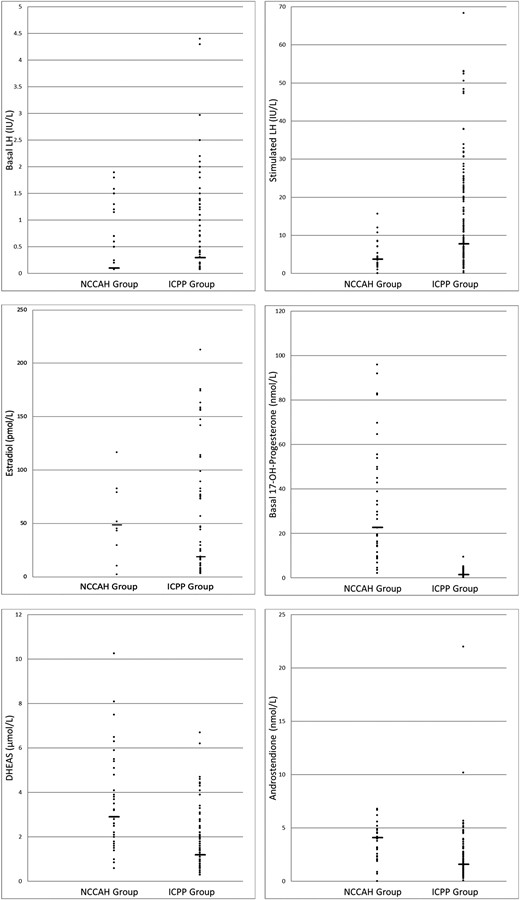
Individual hormonal values of girls with ICPP or NCCAH and CPP. The horizontal lines indicate the median for each group.
On stepwise logistic regression analysis of the hormones with significantly different basal levels in the two study groups (i.e., 17-OHP, androstenedione, DHEAS), only 17-OHP was predictive of a diagnosis of NCCAH (OR, 2.17; 95% CI, 1.48 to 3.16). The other two variables did not enter into the equation. A 17-OHP cutoff value of 6.0 nmol/L had 97.1% sensitivity and 97.2% specificity.
No between-group differences were found for birth weight or gestational age (Table 2). As expected, girls with ICPP were more likely to have another family member with CPP, and girls with NCCAH were more likely to have family history of NCCAH. Interestingly, 8.1% of the girls with NCCAH had a family history of CPP. Maternal age at menarche was similar in the two groups.
Background Characteristics of Patients With ICPP or CPP Associated With NCCAH
| Characteristic | ICPP, n = 140 | CPP Associated With NCCAH, n = 37 | |||||
|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | Range | Mean ± SD | Median | Range | P | |
| Birth weight, g | 2959 ± 622 | 2880 | 800–4300 | 2931 ± 619 | 2880 | 980–4220 | 0.82 |
| Gestational age, > 37, 34–37, <34 wk, % | 88.1, 7.6, 4.2 | 90.9, 3.0, 6.1 | 0.6 | ||||
| Maternal age at menarche, y | 12.19 ± 1.4 | 12 | 8–16.5 | 12.27 ± 1.19 | 12.5 | 9–16 | 0.78 |
| Family history of precocious puberty, % | 40.4 | 8.1 | <0.001 | ||||
| Family history of NCCAH, % | 0 | 16.7 | <0.001 | ||||
| Characteristic | ICPP, n = 140 | CPP Associated With NCCAH, n = 37 | |||||
|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | Range | Mean ± SD | Median | Range | P | |
| Birth weight, g | 2959 ± 622 | 2880 | 800–4300 | 2931 ± 619 | 2880 | 980–4220 | 0.82 |
| Gestational age, > 37, 34–37, <34 wk, % | 88.1, 7.6, 4.2 | 90.9, 3.0, 6.1 | 0.6 | ||||
| Maternal age at menarche, y | 12.19 ± 1.4 | 12 | 8–16.5 | 12.27 ± 1.19 | 12.5 | 9–16 | 0.78 |
| Family history of precocious puberty, % | 40.4 | 8.1 | <0.001 | ||||
| Family history of NCCAH, % | 0 | 16.7 | <0.001 | ||||
Background Characteristics of Patients With ICPP or CPP Associated With NCCAH
| Characteristic | ICPP, n = 140 | CPP Associated With NCCAH, n = 37 | |||||
|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | Range | Mean ± SD | Median | Range | P | |
| Birth weight, g | 2959 ± 622 | 2880 | 800–4300 | 2931 ± 619 | 2880 | 980–4220 | 0.82 |
| Gestational age, > 37, 34–37, <34 wk, % | 88.1, 7.6, 4.2 | 90.9, 3.0, 6.1 | 0.6 | ||||
| Maternal age at menarche, y | 12.19 ± 1.4 | 12 | 8–16.5 | 12.27 ± 1.19 | 12.5 | 9–16 | 0.78 |
| Family history of precocious puberty, % | 40.4 | 8.1 | <0.001 | ||||
| Family history of NCCAH, % | 0 | 16.7 | <0.001 | ||||
| Characteristic | ICPP, n = 140 | CPP Associated With NCCAH, n = 37 | |||||
|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | Range | Mean ± SD | Median | Range | P | |
| Birth weight, g | 2959 ± 622 | 2880 | 800–4300 | 2931 ± 619 | 2880 | 980–4220 | 0.82 |
| Gestational age, > 37, 34–37, <34 wk, % | 88.1, 7.6, 4.2 | 90.9, 3.0, 6.1 | 0.6 | ||||
| Maternal age at menarche, y | 12.19 ± 1.4 | 12 | 8–16.5 | 12.27 ± 1.19 | 12.5 | 9–16 | 0.78 |
| Family history of precocious puberty, % | 40.4 | 8.1 | <0.001 | ||||
| Family history of NCCAH, % | 0 | 16.7 | <0.001 | ||||
Discussion
In the present cohort of girls with CPP consecutively referred to our tertiary care center, almost 5% had NCCAH. Although the prevalence of NCCAH among children with prematur pubarche has been researched extensively (22–24), this study evaluates the prevalence of the disorder among girls presenting with true CPP.
The NCCAH prevalence found in our cohort is significantly higher than that reported in the general population (11) and supports previous data showing that NCCAH may trigger early activation of the hypothalamic-pituitary-gonadal axis (6, 9, 11). Thus, it seems that in girls with true CPP, from populations where the disorder is prevalent, an assessment of adrenal androgen levels is required, and an ACTH test should be considered. The need for an ACTH stimulation test to rule out NCCAH in children with premature pubarche is controversial (4, 22, 25). It has been suggested that using a cutoff basal 17-OHP level of 6.0 nmol/L may serve as a diagnostic screening tool for NCCAH. In our previous study of children aged 0 to 18 years with NCCAH, we showed that applying this cutoff may lead to misdiagnosis in 10.6% of cases (4). However, the present cohort had different characteristics, including a different age range. Thus, although the cutoff suggested may not be sensitive enough for adolescents and adults (16, 26), it may be useful in younger girls.
Neither basal estradiol level nor basal gonadotropin level could differentiate between the two groups. This was also true for basal androgen level. Although mean basal levels of 17-OHP, androstenedione, and DHEAS were significantly higher in the NCCAH group, there was a significant overlap of ranges, and none of these androgens served as an accurate diagnostic marker.
Not a single clinical characteristic was found to definitely differentiate between the two study groups. The presence of pubarche was more prevalent among the girls with NCCAH, but >70% of the girls with ICPP had pubarche presentation. Moreover, pubarche stage in the NCCAH group was as advanced as in the ICPP group. The rate of family history of PP in the ICPP group (40%) was even higher than the rate in our earlier study (∼27%) (27). However, owing to the retrospective nature of the current study, family history data could not be verified and could have been inaccurate.
The strength of this study lies in the relatively large cohort and the unbiased inclusion criteria, because GnRH and ACTH tests are routinely performed in all girls referred to our clinic for PP. In addition, all hormonal analyses were done in the same laboratory. One limitation of the study is the inclusion of girls in the NCCAH group who were diagnosed before 2008. Although the laboratory tests used have not changed over the years, possible secular trends might have influenced the results.
In conclusion, we describe the prevalence of NCCAH among girls presenting with true CPP. The high prevalence of NCCAH and the lack of even one parameter that was diagnostic for NCCAH suggest that the evaluation of girls presenting with ICPP should include basal androgens. The decision to perform an ACTH stimulation test should be based on clinical parameters, including family history and level of basal 17-OHP and possibly androstenedione.
Abbreviations:
- 17-OHP
17-hydroxyprogesterone
- BA
bone age
- CAH
congenital adrenal hyperplasia
- CPP
central precocious puberty
- ICPP
idiopathic central precocious puberty
- NCCAH
nonclassical congenital adrenal hyperplasia
- PP
precocious puberty
- SDS
SD score
Acknowledgments
We thank Pearl Lilos for the statistical analysis and Ruth Fradkin for editorial assistance.
Disclosure Summary: The authors have nothing to disclose.

{kind=link}