





Abstract
Patients born small for gestational age (SGA) who present with persistent short stature could have an underlying genetic etiology that will account for prenatal and postnatal growth impairment. We applied a unique massive parallel sequencing approach in cohort of patients with exclusively nonsyndromic SGA to simultaneously interrogate for clinically substantial genetic variants.
To perform a genetic investigation of children with isolated short stature born SGA.
Screening by exome (n = 16) or targeted gene panel (n = 39) sequencing.
Tertiary referral center for growth disorders.
We selected 55 patients born SGA with persistent short stature without an identified cause of short stature.
Frequency of pathogenic findings.
We identified heterozygous pathogenic or likely pathogenic genetic variants in 8 of 55 patients, all in genes already associated with growth disorders. Four of the genes are associated with growth plate development, IHH (n = 2), NPR2 (n = 2), SHOX (n = 1), and ACAN (n = 1), and two are involved in the RAS/MAPK pathway, PTPN11 (n = 1) and NF1 (n = 1). None of these patients had clinical findings that allowed for a clinical diagnosis. Seven patients were SGA only for length and one was SGA for both length and weight.
These genomic approaches identified pathogenic or likely pathogenic genetic variants in 8 of 55 patients (15%). Six of the eight patients carried variants in genes associated with growth plate development, indicating that mild forms of skeletal dysplasia could be a cause of growth disorders in this group of patients.
Children born small for gestational age (SGA) for length and/or weight with persistent short stature during childhood have a high probability of having a height shorter than the normal range in adulthood (1). Usually, after an extensive medical evaluation, the cause of this growth disorder will not be identified, and therapy with recombinant human growth hormone (rhGH) might be indicated to promote improvement in adult height (1–3).
The regulation of prenatal growth and the lack of catch-up growth in SGA children are not fully understood. The causes of SGA are multifactorial, including maternal/uterine-placental factors, fetal epigenetics, and genetic abnormalities (2). Numerous genes have been associated with the regulation of human height and implicated in growth disorders (4, 5). Several case reports (6, 7) and candidate gene studies (8–10) have demonstrated that a percentage of children born SGA will have a monogenic condition that explains their short stature (2).
Knowledge of the genetic basis for the growth disorders in these children has important consequences for their treatment and follow-up. For example, a broad genetic test will allow for early recognition of mild forms of genetic conditions that can increase the risk of cancer, such as Bloom syndrome, for which rhGH therapy is contraindicated (7). Children with IGF1R haploinsufficiency have a variable phenotype that hinders their diagnosis using on clinical characteristics (11, 12). Nevertheless, the recognition of this genetic defect explains the elevated IGF-1 level associated with a poor response to rhGH therapy (13). Finally, identification of a heterozygous defect in ACAN will allow for the prediction of accelerated bone maturation and a short growth spurt (9). Additionally, positive results for all these genetic tests have substantial implications for genetic counseling and early recognition of additional cases in the affected families (14).
The advent of new genomic technologies, especially massive parallel sequencing, has provided a genetic diagnosis for many children with short stature of unknown cause, especially among patients with syndromic conditions (15–18). To the best of our knowledge, however, no study has thus far focused on children born SGA with isolated short stature. Consequently, the present study evaluated the diagnostic yield of massive parallel sequencing approaches of the whole exome sequencing (WES) and/or targeted panel sequencing in a cohort of 55 nonsyndromic children born SGA without catch-up growth.
Patients and Methods
Patients
We enrolled children born SGA [birth length or weight SD score (SDS) ≤ −2] (19) referred for evaluation of short stature. Patients underwent a clinical assessment. The inclusion criterion was persistent short stature (height SDS ≤ −2) after the second year of life without any known cause. We excluded patients with characteristics in addition to short stature, such as several dysmorphic features, major malformations, microcephaly, neurodevelopmental delay, intellectual disabilities, or skeletal dysplasia. We did not exclude patients with short stature and minor malformations, defined as an unusual morphologic feature found in the general population and causing no serious medical or cosmetic significance to the affected individual (20). Children born at late preterm were also included (gestational age >34 weeks but <37 weeks). Thus, 55 patients were selected from an initial cohort of 297 children born SGA without catch-up growth (Fig. 1). The cases of two of these patients had been previously reported in studies that did not involve the evaluation of children born SGA (21, 22).
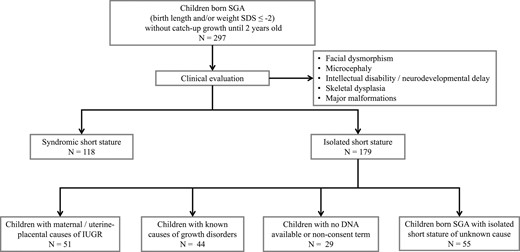
Strategy used to select children born SGA with isolated short stature of unknown cause included in the present study. IUGR, intrauterine growth restriction.
The clinical assessment was performed at the same period of the day and included measurements of weight (measured using an electronic scale) and standing and sitting height (mean of three measurements using a calibrated stadiometer to the nearest 0.1 cm). The body mass index was calculated as the weight in kilograms divided by the height in square meters). These results were converted to the SDS using age- and sex-specific norms (23, 24). The height of the parents was obtained during the patients’ visit using the same method for 85% of the patients. The anthropometric data from 14 fathers was obtained from the medical records reported by the family. We do not have access to the height of three fathers. The target height of the child was calculated as [(father’s height + mother’s height ± 13 cm)/2] and expressed as the SDS. Determination of bone age using left hand and wrist x-rays was assessed by the method of Greulich and Pyle (25).
The ethics committee for analysis of research projects of the School of Medicine, University of Sao Paulo, approved the present study (approval no. 1645329). All procedures were in accordance with the ethical standards of our ethics committee and the Declaration of Helsinki. All the patients and their parents provided written informed consent after receiving a full explanation of the purpose and nature of all procedures used.
Massive parallel sequencing
Targeted panel and exome sequencing
Genomic DNA was isolated from peripheral blood leukocytes from all 55 patients and from 36 mothers and 25 fathers using standard procedures. Target panel sequencing (n = 39) or WES (n = 16) was performed according to availability. Target panel sequencing was performed only for the index patients, and WES was also performed for five of these patients. Of the other 11 patients who had undergone WES, 4 had had a trio analysis. An autosomal dominant short stature was suspected in the other seven patients; therefore, for these families, WES was performed for the index case, affected parent, and additional relatives.
We developed customized target panel sequencing (Agilent SureSelect XT assay; Agilent Technologies, Santa Clara, CA) that included 388 genes (26). The selected genes were those associated with short stature [growth-related genes derived from the Online Mendelian Inheritance in Man (OMIM) and MedGen databases], genes involved in GH-IGF1 pathway regulation, and candidate genes thought to be associated with growth disorders from our previous results (27) and unpublished data. An exome library was prepared according to the Sure Select Human All Exon, version 5 or 6 (Agilent Technologies).
Sequencing was performed using an Illumina NextSeq 500 (Illumina, San Diego, CA) platform for the targeted gene panel and a HiSeq 2500 (Illumina) platform for the exome, both in paired-end mode. In-house bioinformatic analysis was performed as previously reported (28). The sequences were aligned with the human reference assembly (GRCh37/hg19).
Variant assessment
The exome and the targeted panel sequencing data were screened for rare variants (minor allele frequency <0.1%) in public international and national (Genome Aggregation Database; available at: http://gnomad.broadinstitute.org/; and ABraOM; available at: http://abraom.ib.usp.br/) and in-house databases, located in exonic regions and consensus splice site sequences. Subsequently, our variant filtration prioritized genes according to their potential to be pathogenic, including loss-of-function variants and variants predicted to be pathogenic by multiple in silico programs (n = 7) (29). For variants identified by WES, we selected those based on different modes of inheritance (autosomal dominant de novo, autosomal recessive, and X-linked) according to the possible model of inheritance of the family. The sequencing reads carrying candidate variants were inspected visually using the Integrative Genomics Viewer to reduce false-positive findings. The assessment of gene function was performed using OMIM and the PubMed databases. Sanger sequencing and segregation were performed to validate the candidate variant identified. All variants were classified in accordance with the American College of Medical Genetics and Genomics/Association for Molecular Pathology variant pathogenicity guidelines (30).
Results
Cohort description
We analyzed the data from 55 patients born SGA without catch-up growth. The clinical characteristics of the studied group are presented in Table 1. The age of the children at the first evaluation ranged from 2 to 16.3 years. The cohort was characterized by a male predominance (64%) and a family history of short stature (69%). For seven patients, we did not have access to their length at birth (13%). For the other 48 patients, 25% were SGA for both weight and length, 55% were SGA just for length, and 7% were SGA only for weight. Nine patients (16%) had a disproportionate short stature defined by a sitting height/height ratio >2 SDS from the mean (31). All these patients had undergone a skeletal survey with only normal and nonspecific findings. Among these disproportionate short stature children, six were SGA just for length and three for length and weight.
Characteristics of 55 Patients Born SGA With Isolated Short Stature
| Characteristic | Patients Born SGA |
|---|---|
| Consanguinity | 3 (5.5) |
| Familial short staturea | 38 (69.1) |
| Sex | |
| Male | 35 |
| Female | 20 |
| Gestational age, wk | 39.1 ± 1.1 |
| Prematureb | 6 (10.9) |
| Birth weight SDS | −1.7 ± 0.7 |
| Birth length SDS | −3,0 ± 0.2 |
| Age at first evaluation, y | 9.7 ± 3.5 |
| Height SDS | −2.7 ± 0.5 |
| Sitting height/total height ratio SDS | 0.8 ± 1.0 |
| BMI SDS | −0.7 ± 1.0 |
| Characteristic | Patients Born SGA |
|---|---|
| Consanguinity | 3 (5.5) |
| Familial short staturea | 38 (69.1) |
| Sex | |
| Male | 35 |
| Female | 20 |
| Gestational age, wk | 39.1 ± 1.1 |
| Prematureb | 6 (10.9) |
| Birth weight SDS | −1.7 ± 0.7 |
| Birth length SDS | −3,0 ± 0.2 |
| Age at first evaluation, y | 9.7 ± 3.5 |
| Height SDS | −2.7 ± 0.5 |
| Sitting height/total height ratio SDS | 0.8 ± 1.0 |
| BMI SDS | −0.7 ± 1.0 |
Data presented as mean ± SD for continuous variables and as absolute number (percentage) for numerical variables.
Abbreviation: BMI, body mass index.
First-degree family members with height SDS < −2.
Birth before 37th week of pregnancy.
Characteristics of 55 Patients Born SGA With Isolated Short Stature
| Characteristic | Patients Born SGA |
|---|---|
| Consanguinity | 3 (5.5) |
| Familial short staturea | 38 (69.1) |
| Sex | |
| Male | 35 |
| Female | 20 |
| Gestational age, wk | 39.1 ± 1.1 |
| Prematureb | 6 (10.9) |
| Birth weight SDS | −1.7 ± 0.7 |
| Birth length SDS | −3,0 ± 0.2 |
| Age at first evaluation, y | 9.7 ± 3.5 |
| Height SDS | −2.7 ± 0.5 |
| Sitting height/total height ratio SDS | 0.8 ± 1.0 |
| BMI SDS | −0.7 ± 1.0 |
| Characteristic | Patients Born SGA |
|---|---|
| Consanguinity | 3 (5.5) |
| Familial short staturea | 38 (69.1) |
| Sex | |
| Male | 35 |
| Female | 20 |
| Gestational age, wk | 39.1 ± 1.1 |
| Prematureb | 6 (10.9) |
| Birth weight SDS | −1.7 ± 0.7 |
| Birth length SDS | −3,0 ± 0.2 |
| Age at first evaluation, y | 9.7 ± 3.5 |
| Height SDS | −2.7 ± 0.5 |
| Sitting height/total height ratio SDS | 0.8 ± 1.0 |
| BMI SDS | −0.7 ± 1.0 |
Data presented as mean ± SD for continuous variables and as absolute number (percentage) for numerical variables.
Abbreviation: BMI, body mass index.
First-degree family members with height SDS < −2.
Birth before 37th week of pregnancy.
Variants identified by massive parallel sequencing
We identified eight heterozygous pathogenic (n = 3) or likely pathogenic (n = 5) variants in 8 of 55 patients (15%). All these variants were identified in genes already associated with growth disorders (Table 2) and included Indian hedgehog (IHH) in 2, natriuretic peptide receptor 2 (NPR2) in 2, short stature homeobox (SHOX) in 1, aggrecan (ACAN) in 1, neurofibromin 1 (NF1) in 1, and protein-tyrosine phosphatase nonreceptor-type 11 (PTPN11) in 1. Four variants were found using targeted panel sequencing and four using WES. All variants were absent or extremely rare in the local and public databases. Five variants were inherited from affected parents. One of the variants was in a consensus splice site, and all the others were missense and predicted to be pathogenic by multiple in silico tools.
Pathogenic and Likely Pathogenic Variants Identified by WES and Targeted Panel Sequencing for 55 Patients Born SGA With Isolated Short Stature
| Pt. No. | Gene | Variant | Frequency (MAF)a | Functional Annotation | Inheritance Pattern | ACMG/AMP |
|---|---|---|---|---|---|---|
| 1 | IHH | c.446G>A:p.Arg149Hisb | 0 | Missense | Inherited from affected mother | Likely pathogenicf |
| 2 | IHH | c.172G>A:p.Glu58Lysb | 0 | Missense | Inherited from affected father | Likely pathogenicf |
| 3 | NPR2 | c.1249C>G:p.Gln417Gluc | 0.000004 | Missense | Unavailabled | Pathogenicf |
| 4 | NPR2 | c.94C>A:p.Pro32Thrc | 0 | Missense | Inherited from affected mother | Likely pathogenic |
| 5 | PTPN11 | c.794G>A:p.Arg265Glnc | 0.00003 | Missense | Unavailabled | Pathogenicf |
| 6 | SHOX | c.503G>A:p.Arg168Glnc | 0 | Missense | Inherited from affected mothere | Likely pathogenic |
| 7 | ACAN | c.532A>T:p.Asn178Tyrb | 0 | Missense | Inherited from affected mother | Likely pathogenicf |
| 8 | NF1 | c.1261-1G>Cb | 0 | Splice site acceptor | Unavailablec | Pathogenic |
| Pt. No. | Gene | Variant | Frequency (MAF)a | Functional Annotation | Inheritance Pattern | ACMG/AMP |
|---|---|---|---|---|---|---|
| 1 | IHH | c.446G>A:p.Arg149Hisb | 0 | Missense | Inherited from affected mother | Likely pathogenicf |
| 2 | IHH | c.172G>A:p.Glu58Lysb | 0 | Missense | Inherited from affected father | Likely pathogenicf |
| 3 | NPR2 | c.1249C>G:p.Gln417Gluc | 0.000004 | Missense | Unavailabled | Pathogenicf |
| 4 | NPR2 | c.94C>A:p.Pro32Thrc | 0 | Missense | Inherited from affected mother | Likely pathogenic |
| 5 | PTPN11 | c.794G>A:p.Arg265Glnc | 0.00003 | Missense | Unavailabled | Pathogenicf |
| 6 | SHOX | c.503G>A:p.Arg168Glnc | 0 | Missense | Inherited from affected mothere | Likely pathogenic |
| 7 | ACAN | c.532A>T:p.Asn178Tyrb | 0 | Missense | Inherited from affected mother | Likely pathogenicf |
| 8 | NF1 | c.1261-1G>Cb | 0 | Splice site acceptor | Unavailablec | Pathogenic |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; AMP, Association for Molecular Pathology; MAF, minor allele frequency; Pt. No., patient number.
MAF based on the Genome Aggregation Database.
Result obtained from exome sequencing.
Result obtained from targeted panel sequencing.
DNA from one of the parents was not available.
Heterozygous from mosaic mother.
Variants previously reported to be associated with short stature.
Pathogenic and Likely Pathogenic Variants Identified by WES and Targeted Panel Sequencing for 55 Patients Born SGA With Isolated Short Stature
| Pt. No. | Gene | Variant | Frequency (MAF)a | Functional Annotation | Inheritance Pattern | ACMG/AMP |
|---|---|---|---|---|---|---|
| 1 | IHH | c.446G>A:p.Arg149Hisb | 0 | Missense | Inherited from affected mother | Likely pathogenicf |
| 2 | IHH | c.172G>A:p.Glu58Lysb | 0 | Missense | Inherited from affected father | Likely pathogenicf |
| 3 | NPR2 | c.1249C>G:p.Gln417Gluc | 0.000004 | Missense | Unavailabled | Pathogenicf |
| 4 | NPR2 | c.94C>A:p.Pro32Thrc | 0 | Missense | Inherited from affected mother | Likely pathogenic |
| 5 | PTPN11 | c.794G>A:p.Arg265Glnc | 0.00003 | Missense | Unavailabled | Pathogenicf |
| 6 | SHOX | c.503G>A:p.Arg168Glnc | 0 | Missense | Inherited from affected mothere | Likely pathogenic |
| 7 | ACAN | c.532A>T:p.Asn178Tyrb | 0 | Missense | Inherited from affected mother | Likely pathogenicf |
| 8 | NF1 | c.1261-1G>Cb | 0 | Splice site acceptor | Unavailablec | Pathogenic |
| Pt. No. | Gene | Variant | Frequency (MAF)a | Functional Annotation | Inheritance Pattern | ACMG/AMP |
|---|---|---|---|---|---|---|
| 1 | IHH | c.446G>A:p.Arg149Hisb | 0 | Missense | Inherited from affected mother | Likely pathogenicf |
| 2 | IHH | c.172G>A:p.Glu58Lysb | 0 | Missense | Inherited from affected father | Likely pathogenicf |
| 3 | NPR2 | c.1249C>G:p.Gln417Gluc | 0.000004 | Missense | Unavailabled | Pathogenicf |
| 4 | NPR2 | c.94C>A:p.Pro32Thrc | 0 | Missense | Inherited from affected mother | Likely pathogenic |
| 5 | PTPN11 | c.794G>A:p.Arg265Glnc | 0.00003 | Missense | Unavailabled | Pathogenicf |
| 6 | SHOX | c.503G>A:p.Arg168Glnc | 0 | Missense | Inherited from affected mothere | Likely pathogenic |
| 7 | ACAN | c.532A>T:p.Asn178Tyrb | 0 | Missense | Inherited from affected mother | Likely pathogenicf |
| 8 | NF1 | c.1261-1G>Cb | 0 | Splice site acceptor | Unavailablec | Pathogenic |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; AMP, Association for Molecular Pathology; MAF, minor allele frequency; Pt. No., patient number.
MAF based on the Genome Aggregation Database.
Result obtained from exome sequencing.
Result obtained from targeted panel sequencing.
DNA from one of the parents was not available.
Heterozygous from mosaic mother.
Variants previously reported to be associated with short stature.
Phenotype of patients with pathogenic or likely pathogenic variants associated with short stature
All eight patients with pathogenic variants had been born at full term. Seven patients were SGA only for length and one was SGA for both length and weight. Six of the patients with pathogenic variants had a family history of short stature; four had inherited the pathogenic variant from an affected parent. Two patients had minor malformations (Table 3). Six of eight patients had pathogenic variants in genes directly involved in growth plate development. Of these patients, two had body disproportion (IHH and ACAN). The skeletal survey depicted shortening of the middle phalanx of the second and fifth fingers with cone-shaped epiphyses in both patients with IHH variants. Additionally, two patients had a slightly advanced bone age at prepubertal age (bone age SDS of 0.6 and 0.7 for one patient each with NPR2 and ACAN, respectively), and three patients had a remarkably delayed bone age (bone age SDS of −4.4, −4.8, and −5.8 for one patient each with SHOX, PTPN11, and NF1, respectively; Table 3).
Clinical Characteristics at First Evaluation of Patients With Pathogenic or Likely Pathogenic Variants Identified by Exome or Targeted Panel Sequencing
| Pt. No. | Gene | Sex | Birth Weight,g (SDS) | Birth Length, cm (SDS) | Age, y | Bone Age, y | Height SDS | SH/H SDS | BMI SDS | Mother Height SDS | Father Height SDS | Additional Features |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | IHH | F | 2785 (−0.9) | 44 (−3.2) | 8.0 | 6.8 | −2.0 | +3.4 | −0.8 | −2.6a | 2.3 | |
| 2 | IHH | M | 2965 (−1.0) | 46 (−2.5) | 5.6 | NA | −3.0 | +0.6 | +0.3 | −1.6 | −4.3a | |
| 3 | NPR2 | F | 2600 (−1.8) | 46 (−2.5) | 10.6 | 11.1 | −2.0 | +1.3 | +1.1 | −1.5 | −0.7 | |
| 4 | NPR2 | F | 3310 (−0.2) | 46 (−2.5) | 2.2 | NA | −4.3 | −1.0 | −1.7 | −2.9a | −0.6 | Triangular face, frontal bossing |
| 5 | SHOX | M | 3200 (−0.4) | 46 (−2.5) | 7.5 | 4.0 | −2.7 | NA | +0.2 | −1.3b | −1.9 | |
| 6 | ACAN | M | 3080 (−0.7) | 46 (−2.5) | 11.8 | 13.0 | −2.0 | +3.7 | +1.0 | −3.7a | 0.9 | |
| 7 | PTPN11 | M | 2680 (−1.6) | 44 (−3.6) | 12.3 | 8.0 | −3.1 | NA | +0.3 | −2.4 | −1.4 | |
| 8 | NF1 | M | 2480 (−2.1) | 47 (−2.0) | 7.3 | 2.8 | −3.0 | +0.8 | −0.8 | −2.0 | NA | Two café au lait spots |
| Pt. No. | Gene | Sex | Birth Weight,g (SDS) | Birth Length, cm (SDS) | Age, y | Bone Age, y | Height SDS | SH/H SDS | BMI SDS | Mother Height SDS | Father Height SDS | Additional Features |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | IHH | F | 2785 (−0.9) | 44 (−3.2) | 8.0 | 6.8 | −2.0 | +3.4 | −0.8 | −2.6a | 2.3 | |
| 2 | IHH | M | 2965 (−1.0) | 46 (−2.5) | 5.6 | NA | −3.0 | +0.6 | +0.3 | −1.6 | −4.3a | |
| 3 | NPR2 | F | 2600 (−1.8) | 46 (−2.5) | 10.6 | 11.1 | −2.0 | +1.3 | +1.1 | −1.5 | −0.7 | |
| 4 | NPR2 | F | 3310 (−0.2) | 46 (−2.5) | 2.2 | NA | −4.3 | −1.0 | −1.7 | −2.9a | −0.6 | Triangular face, frontal bossing |
| 5 | SHOX | M | 3200 (−0.4) | 46 (−2.5) | 7.5 | 4.0 | −2.7 | NA | +0.2 | −1.3b | −1.9 | |
| 6 | ACAN | M | 3080 (−0.7) | 46 (−2.5) | 11.8 | 13.0 | −2.0 | +3.7 | +1.0 | −3.7a | 0.9 | |
| 7 | PTPN11 | M | 2680 (−1.6) | 44 (−3.6) | 12.3 | 8.0 | −3.1 | NA | +0.3 | −2.4 | −1.4 | |
| 8 | NF1 | M | 2480 (−2.1) | 47 (−2.0) | 7.3 | 2.8 | −3.0 | +0.8 | −0.8 | −2.0 | NA | Two café au lait spots |
Abbreviations: BMI, body mass index; F, female; M, male; NA, not available; Pt. No., patient number; SH/H, sitting height/height (ratio).
Presence of the mutation in heterozygous state.
Presence of mutation in mosaicism.
Clinical Characteristics at First Evaluation of Patients With Pathogenic or Likely Pathogenic Variants Identified by Exome or Targeted Panel Sequencing
| Pt. No. | Gene | Sex | Birth Weight,g (SDS) | Birth Length, cm (SDS) | Age, y | Bone Age, y | Height SDS | SH/H SDS | BMI SDS | Mother Height SDS | Father Height SDS | Additional Features |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | IHH | F | 2785 (−0.9) | 44 (−3.2) | 8.0 | 6.8 | −2.0 | +3.4 | −0.8 | −2.6a | 2.3 | |
| 2 | IHH | M | 2965 (−1.0) | 46 (−2.5) | 5.6 | NA | −3.0 | +0.6 | +0.3 | −1.6 | −4.3a | |
| 3 | NPR2 | F | 2600 (−1.8) | 46 (−2.5) | 10.6 | 11.1 | −2.0 | +1.3 | +1.1 | −1.5 | −0.7 | |
| 4 | NPR2 | F | 3310 (−0.2) | 46 (−2.5) | 2.2 | NA | −4.3 | −1.0 | −1.7 | −2.9a | −0.6 | Triangular face, frontal bossing |
| 5 | SHOX | M | 3200 (−0.4) | 46 (−2.5) | 7.5 | 4.0 | −2.7 | NA | +0.2 | −1.3b | −1.9 | |
| 6 | ACAN | M | 3080 (−0.7) | 46 (−2.5) | 11.8 | 13.0 | −2.0 | +3.7 | +1.0 | −3.7a | 0.9 | |
| 7 | PTPN11 | M | 2680 (−1.6) | 44 (−3.6) | 12.3 | 8.0 | −3.1 | NA | +0.3 | −2.4 | −1.4 | |
| 8 | NF1 | M | 2480 (−2.1) | 47 (−2.0) | 7.3 | 2.8 | −3.0 | +0.8 | −0.8 | −2.0 | NA | Two café au lait spots |
| Pt. No. | Gene | Sex | Birth Weight,g (SDS) | Birth Length, cm (SDS) | Age, y | Bone Age, y | Height SDS | SH/H SDS | BMI SDS | Mother Height SDS | Father Height SDS | Additional Features |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | IHH | F | 2785 (−0.9) | 44 (−3.2) | 8.0 | 6.8 | −2.0 | +3.4 | −0.8 | −2.6a | 2.3 | |
| 2 | IHH | M | 2965 (−1.0) | 46 (−2.5) | 5.6 | NA | −3.0 | +0.6 | +0.3 | −1.6 | −4.3a | |
| 3 | NPR2 | F | 2600 (−1.8) | 46 (−2.5) | 10.6 | 11.1 | −2.0 | +1.3 | +1.1 | −1.5 | −0.7 | |
| 4 | NPR2 | F | 3310 (−0.2) | 46 (−2.5) | 2.2 | NA | −4.3 | −1.0 | −1.7 | −2.9a | −0.6 | Triangular face, frontal bossing |
| 5 | SHOX | M | 3200 (−0.4) | 46 (−2.5) | 7.5 | 4.0 | −2.7 | NA | +0.2 | −1.3b | −1.9 | |
| 6 | ACAN | M | 3080 (−0.7) | 46 (−2.5) | 11.8 | 13.0 | −2.0 | +3.7 | +1.0 | −3.7a | 0.9 | |
| 7 | PTPN11 | M | 2680 (−1.6) | 44 (−3.6) | 12.3 | 8.0 | −3.1 | NA | +0.3 | −2.4 | −1.4 | |
| 8 | NF1 | M | 2480 (−2.1) | 47 (−2.0) | 7.3 | 2.8 | −3.0 | +0.8 | −0.8 | −2.0 | NA | Two café au lait spots |
Abbreviations: BMI, body mass index; F, female; M, male; NA, not available; Pt. No., patient number; SH/H, sitting height/height (ratio).
Presence of the mutation in heterozygous state.
Presence of mutation in mosaicism.
Two patients had pathogenic variants in genes involved in the RAS-MAPK pathway (PTPN11 and NF1). At the first evaluation, none of these patients had characteristics that allowed for the diagnosis of Noonan syndrome (OMIM no. 163950) or neurofibromatosis type 1 (OMIM no. 162200). The patient with the NF1 mutation had two small café au lait spots and normal ophthalmologic evaluation findings at 7 years old when he started attending our clinic. Ten years later, at 17 years of age, he had developed a plexiform neurofibroma, confirming the clinical diagnosis.
Discussion
The evaluation of children born SGA with persistent short stature and absence of syndromic features is a common challenge for both pediatricians and pediatric endocrinologists. Usually, the clinical evaluation, followed by traditional investigations using radiological and laboratory examinations, will be unable to determine a cause for the pre- and postnatal growth impairment. The frequent observation of short stature in other first-degree family members suggests a genetic component. However, the absence of specific clinical findings has the use of a candidate gene approach impractical. Thus, we used a multigene strategy with next generation sequencing and were able to provide a diagnosis for 15% of patients born SGA with persistent short stature without other characteristics.
To the best of our knowledge, a limited number of studies have applied the genomic approach to investigate children with short stature. Usually, these studies have included patients with short stature of unknown cause, regardless of birth weight or length and have also included syndromic conditions (16–18, 32). Pathogenic or likely pathogenic variants were observed in 2% to 46% of these children (16–18, 32, 33). This wide range of variability could be partially explained by the sequencing methods applied, the number of patients analyzed, and the inclusion and exclusion selection criteria of patients. A recent well-conducted study that evaluated 200 patients with growth disorders (134 classified as having isolated short stature) using WES identified pathogenic variants in known short-stature genes in 21% of syndromic cases and 14.2% of isolated short stature, with a marked preponderance of autosomal dominant inheritance (18). This result was similar to that observed in our study, which analyzed only nonsyndromic children born SGA.
Of the patients provided with a diagnosis in the present study, all were SGA for length, and the genes that participated in growth plate development were overrepresented (SHOX, ACAN, NPR2, and IHH). Our patients presented with a mild variation of a skeletal dysplasia phenotype. Four patients inherited the pathogenic variant from an affected, but previously undiagnosed, parent. The role of these growth plate-associated genes in explaining the persistence of short stature in children born SGA has not been fully investigated. Mutations in these genes have already been associated with distinct degrees of short stature with or without other mild features, such as a slightly disproportional short stature and a nonspecific skeletal abnormality (21, 31, 34–36). In many studies, the birth weight and length were not reported. Each of these genes explains a small fraction (1% to 4%) of the patients classified as having idiopathic short stature (21, 36–40), with a greater prevalence in familial cases (40). Only one study reported the specific prevalence of ACAN gene mutations in children born SGA. From a cohort of 290 SGA children, 29 with advanced bone age were selected for gene sequencing, and 4 patients (13.8%) were identified with ACAN mutations. Three of these patients were born SGA for length (9).
In our study, we also identified two patients with pathogenic variants affecting the RAS-MAPK pathway. In one of our patients, neurofibromatosis type 1 was diagnosed (OMIM no. 162200). Usually, the diagnosis of neurofibromatosis type 1 is made in children older than 8 years using the clinical features (41); however, it can be difficult to diagnose in young children. Our patient had an atypical presentation that delayed the clinical diagnosis until he was 17 years of age, when he developed a plexiform neurofibroma. In another patient, we identified a pathogenic variant in PTPN11 associated with Noonan syndrome (OMIM no. 163950). Usually, Noonan syndrome will be diagnosed by specific clinical criteria (42). The facial features and/or typical cardiac malformation frequently trigger the suspicion of the diagnosis. However, the patient analyzed in our study had a mild phenotype and did not meet the criteria for a clinical diagnosis. A recent functional study of the same variant found in this patient (PTPN11 p.Arg265Gln) demonstrated increased catalytic activity of the phosphatase and hyperactivation of MAPK pathway signaling when stimulated by a growth factor (epidermal growth factor). It was responsible for a mild phenotype without the typical characteristics that usually constitute the clinical features of Noonan syndrome (43). Other studies using the genomic approach in patients with isolated short stature also identified NF1 and PTPN11 mutations in patients without the typical clinical features (18, 32).
The early diagnosis provided by a genomic approach can have a substantial effect on familial genetic counseling, rhGH treatment decisions, and patient follow-up. In our study, all affected patients had an autosomal dominant form of growth disorders, with a recurrence risk of 50% for the patients’ offspring and for the siblings of the affected parents. The molecular diagnosis has therapeutic implications because rhGH treatment seems effective in improving the height of patients with SHOX, ACAN, and IHH gene defects (9, 21, 44) Additionally, children with neurofibromatosis type 1 should receive a multidisciplinary approach to optimize their follow-up and medical care, to facilitate early recognition of complications, and to promote general health (41).
Although we did not find differences between the rates of diagnosis using WES or target panel sequencing, the WES strategy permits the identification of new genes and allows for a hypothesis-free method to assess for genetic variations (45). However, all genes identified with pathogenic variants by WES were present in our target gene panel. Nevertheless, recent studies that prospectively compared the cost-effectiveness between clinical WES and the traditional investigation showed a greater rate of diagnosis and lower cost when WES was used (15). A recent systematic review has offered encouraging evidence of the cost-effectiveness of genomic sequencing compared with the usual care for pediatric patients (46). The use of a genomic approach significantly increased the diagnostic rate to 16% to 79% and lowered the cost by 11% to 64% compared with the standard diagnostic pathway (46).
In the present study, molecular investigation, using massive parallel sequencing, was effective in diagnosing one in seven patients with prenatal short stature of unknown etiology. All these patients had undergone extensive clinical investigations, and all were classified as children born SGA with isolated short stature. Without this genomic approach, these patients would have remained undiagnosed and probably would have had inadequate follow-up. Because a karyotype is indicated to investigate all girls with unexplained growth failure (47), we believe that with the greater availability and lower cost of genetic analyses, multigene molecular sequencing might be incorporated for the investigation of all children born SGA with persistent short stature. Our results should encourage prospective studies that include patient well-being and economic cost/benefit evaluations to guide future recommendations for genetic testing using a genomic approach for children born SGA.
Abbreviations:
- ACAN
aggrecan
- IHH
Indian hedgehog
- NF1
neurofibromin 1
- NPR2
natriuretic peptide receptor 2
- OMIM
Online Mendelian Inheritance in Man
- PTPN11
protein-tyrosine phosphatase nonreceptor-type 11
- rhGH
recombinant human growth hormone
- SDS
SD score
- SGA
small for gestational age
- SHOX
short stature homeobox
- WES
whole exome sequencing
Acknowledgments
We are grateful to the patients and families for their collaboration. We acknowledge the support of the São Paulo Research Foundation and the National Council for Scientific and Technological Development. We also thank the Laboratório de Sequenciamento em Larga Escala for their assistance with whole exome and targeted panel sequencing.
FinancialSupport: The present study was supported by the Laboratório de Sequenciamento em Larga Escala) from the São Paulo Research Foundation (grants 2013/03236-5 to A.A.L.J., 2015/26980-7 to T.K.H., and grant SELA 2014/50137-5); the National Council for Scientific and Technological Development (grant 304678/2012-0 to A.A.L.J.); and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–Brasil (finance code 001 to B.L.F.).
Disclosure Summary: The authors have nothing to disclose.
References
Author notes
B.L.F. and T.K.H. contributed equally to this study.

{kind=link}