





Abstract
Congenital isolated TSH deficiency (i-TSHD) is a rare form of congenital hypothyroidism. Five genes (IGSF1, IRS4, TBL1X, TRHR, and TSHB) responsible for the disease have been identified, although their relative frequencies and hypothalamic/pituitary unit phenotypes have remained to be clarified.
To define the relative frequencies and hypothalamic/pituitary unit phenotypes of congenital i-TSHD resulting from single gene mutations.
Thirteen Japanese patients (11 boys and 2 girls) with congenital i-TSHD were enrolled. IGSF1, IRS4, TBL1X, TRHR, and TSHB were sequenced. For a TBL1X mutation (p.Asn382del), its pathogenicity was verified in vitro. For a literature review, published clinical data derived from 74 patients with congenital i-TSHD resulting from single-gene mutations were retrieved and analyzed.
Genetic screening of the 13 study subjects revealed six mutation-carrying patients (46%), including five hemizygous IGSF1 mutation carriers and one hemizygous TBL1X mutation carrier. Among the six mutation carriers, one had intellectual disability and the other one had obesity, but the remaining four did not show nonendocrine phenotypes. Loss of function of the TBL1X mutation (p.Asn382del) was confirmed in vitro. The literature review demonstrated etiology-specific relationship between serum prolactin (PRL) levels and TRH-stimulated TSH levels with some degree of overlap.
The mutation screening study covering the five causative genes of congenital i-TSHD was performed, showing that the IGSF1 defect was the leading genetic cause of the disease. Assessing relationships between serum PRL levels and TRH-stimulated TSH levels would contribute to predict the etiologies of congenital i-TSHD.
Congenital defects involving the hypothalamus-pituitary-thyroid axis cause hypothyroidism from the birth, which is collectively referred to as congenital hypothyroidism (CH). About 85% of CH is due to dysfunction of the thyroid itself (primary CH), whereas the remaining 15% is CH of central origin (central CH) (1). Central CH is clinically classified into two categories, including congenital isolated TSH deficiency (i-TSHD) and congenital combined pituitary hormone deficiency, in which TSH and other pituitary hormone(s) are deficient. In addition to the pure form of congenital i-TSHD, combined TSH/prolactin (PRL) deficiency is considered a variant type of the disease, because TRH receptor signaling affects not only TSH secretion but also PRL secretion in both physiological and pathological conditions. Congenital i-TSHD accounts for ∼40% of central CH cases (2) with an estimated frequency of 1 in 40,000 newborns (1, 2).
Early studies have identified two genetic defects that are reasonably associated with congenital i-TSHD: mutations in TSHB (encoding the β subunit of TSH) (3) and TRHR (encoding the receptor for TRH) (4). Molecular genetic studies conducted so far have revealed that the two defects are extremely rare, except for a founder effect TSHB mutation, “313 delta T,” exclusively reported from Germany (5, 6). Introduction of the next-generation sequencing technique into research of congenital i-TSHD led the identification of additional causative genes: IGSF1 encoding a transmembrane glycoprotein expressed in the pituitary gland (7) and TBL1X encoding a component of the nuclear corepressor (NCoR) complex (8). IGSF1 mutations have been reported in multiple ethnicities including Europeans and Asians, implying the relatively high prevalence of the defect (7, 9). Very recently, mutations of IRS4 in X-linked congenital i-TSHD was reported (10), although the molecular mechanisms remain to be clarified.
Previous genetic studies on congenital i-TSHD have been conducted in a focused manner. In fact, no study has performed mutational screening of the five causative genes in a defined patient cohort. Here, we report the genetic screening study covering the five known causative genes (IGSF1, IRS4, TBL1X, TRHR, and TSHB) in a Japanese patient cohort with congenital i-TSHD. We also performed a literature review to identify the etiology-specific characteristics of the hypothalamic/pituitary unit.
Patients and Methods
Patients
This study, conducted in accordance with the Helsinki Declaration, was approved by the Ethics Committee of Keio University School of Medicine. Written informed consent for participating in the study was obtained from the study subjects and/or their parents.
Thyroid function tests of the study subjects were performed in each institution, with a total of four distinct kits including ARCHITECT (Abbott Japan, Tokyo, Japan), ECLusys (Roche Diagnostic Japan, Tokyo, Japan), Immulyze (Siemens Healthcare Diagnostics, Tokyo, Japan), and Lumipulse (Fujirebio, Tokyo, Japan). Each free T4 value was standardized with conversion formulas, and shown as ARCHITECT free T4 value (reference range, 0.81 to 1.38 ng/dL). In this study, congenital i-TSHD was defined with following three criteria: (i) low serum free T4 level (≤0.80 ng/dL) with no compensatory TSH rise (serum TSH level <10.0 mU/L), (ii) no deficiency of other pituitary hormone(s) except for PRL, and (iii) diagnosis as having i-TSHD at age younger than 12 years. To exclude patients with transient abnormalities (e.g., euthyroid sick syndrome, malnutrition), patients that showed spontaneous recovery of free T4 levels were excluded. Patients that were born preterm (gestational age <37 weeks) and/or as a low birth weight infant (<2500 g) were excluded. We also excluded patients who were treated with levothyroxine but did not require physiological replacement dose (1.0 µg/kg/d or more) in the follow-up period. The eligible 13 patients were followed in 12 institutions across Japan. The median age of diagnosis was 0.2 years (range, 0.0 to 10 years), and the median age of genetic testing was 7 years (range, 2 to 19 years).
Mutation detection
Genomic DNA samples, which were collected between January 2012 and June 2018, were prepared from peripheral leukocytes of each patient. All coding exons of IGSF1, IRS4, TBL1X, TRHR, and TSHB were captured with the SureSelect protocol (Agilent Technologies, Santa Clara, CA) as previously described (11) (for IGSF1, TBL1X, TRHR, and TSHB), or the Nextera XT protocol (Illumina, San Diego, CA) with long-range PCR (for IRS4). Next-generation sequencing, read mapping, variant calling, and annotation were performed as previously described (11). Detected variants were searched in public variant databases, including gnomAD (www.gnomad.broadinstitute.org), 1000 genomes (www.internationalgenome.org/), HGVD (www.hgvd.genome.med.kyoto-u.ac.jp/), and dbSNP150 (www.ncbi.nlm.nih.gov/snp). Variants with allele frequency of 0.001 or more (in any databases) were not classified as disease-causing mutations. Nonsense variants, frameshift variants, splice site variants, and genomic deletions were classified as disease-causing mutations. The pathogenicity of missense variants was assessed with 10 in silico programs, including CADD (12), DANN (13), FATHMM (14), M-CAP (15), MutationAssessor (16), MutationTaster (17), PolyPhen-2 (18), PROVEAN (19), SIFT (20), and VEST-4 (21). A variant was classified as disease-causing if seven or more programs judged it as deleterious. Parental genotyping was performed in five of the six mutation-carrying families. Descriptions of IGSF1 mutations and a TBL1X mutation were based on NM_001170961.1 and NM_005647, respectively.
Functional analyses of Asn382del-TBL1X
Embryonic mouse hypothalamus cell line N-1 was purchased from CELLutions Biosystems (Westbury, NY). African green monkey kidney fibroblast cell line CV-1 was provided by Dr. Shigekazu Sasaki. These cells were cultured in DMEM supplemented with 10% fetal bovine serum, ampicillin (100 U/mL), and streptomycin (100 µg/mL). Expression vectors for human thyroid hormone receptor β1 (NM_000461.4), mouse thyroid hormone receptor β2 (NM_009380.3), mouse NCoR (NM_011308.3), mouse GATA2 (NM_008090.5), mouse pituitary-specific positive transcription factor 1 (NM_008849.5), and human TBL1X (NM_005647) were described previously (22). Luciferase reporter vectors driven by the mouse Trh promoter (−256/+83)(Trh-luc) and the human TSHB promoter (−1192/+38)(TSHB-luc) were also described previously (22). The Asn382del TBL1X mutation was introduced with a standard site-directed mutagenesis technique.
For Western blotting, N-1 cells were transfected with each TBL1X-expressing vector [wild-type (WT) or Asn382del] using Lipofectamine 2000 reagent (Thermo Fisher Scientific, Waltham, MA). Whole cell lysates prepared from the transfected cells were subject to SDS-PAGE, and Western blotting was performed as described previously (22). For luciferase assays, N-1 cells (for Trh-luc) or CV-1 cells (for TSHB-luc) were seeded onto six-well plates and were cultured in DMEM supplemented with 10% resin-charcoal double-stripped fetal bovine serum. Three microgram of each luciferase reporter and 150 ng of expression vectors were cotransfected with a calcium phosphate precipitation method. Total amounts of transfected plasmids were adjusted by adding empty vectors. From 24 hours after transfection, the cells were incubated with 10 nM T3 (Sigma Aldrich, St. Louis, MO), or vehicle, for 24 hours. Then, luciferase assays were performed as previously reported (23).
Literature review
A review of the articles published before October 1, 2018 was conducted by two authors (C.S. and S.N.). PubMed and Google Scholar search engines were used to identify relevant articles using keywords “central congenital hypothyroidism,” “mutation,” “TSHB,” “TRHR,” “IGSF1,” and “TBL1X.” We also searched the HGMD Professional database (www.hgmd.cf.ac.uk/ac/index.php) to find previously reported mutation-carrying patients. To characterize the hypothalamic/pituitary unit phenotypes, we collected data of serum PRL levels and TRH-stimulated serum TSH levels of mutation-carrying patients. Considering the physiologically high PRL levels in early infancy, we omitted PRL data measured at age younger than 3 months.
Results
Patient characteristics
In Japan, most regional newborn screening (NBS) programs use TSH-based NBS. Simultaneous measurements of TSH and T4 (TSH/T4-based NBS), in which TSH deficiency is detectable, are conducted in ∼12% of all newborns. In this study, 13 Japanese patients (11 boys and 2 girls) with congenital i-TSHD were enrolled (Table 1). Ten of the 13 subjects were born in areas with TSH/T4-based NBS, and seven of them were diagnosed in the frame of NBS. The remaining three patients (patients 4, 10, and 13; Table 1), and another three patients born in areas with TSH-based NBS (patients 7, 11, and 12), presented hypothyroidism-associated symptoms (e.g., poor growth, developmental delay) and came to medical attention at a median age of 0.9 years. Family history of i-TSHD was noted in one patient (a cousin was affected). Medians of serum free T4 and TSH at diagnosis were 0.69 ng/dL and 2.9 mU/L, respectively. PRL deficiency, defined as peak TRH-stimulated serum PRL level <25 ng/mL, was noted in two of the seven evaluated patients. One patient (patient 4) had macroorchidism. None had hearing impairment. At the last visit, two patients had short stature, four patients had intellectual disability, and one patient had obesity.
Characteristics of the Study Subjects
| ID | Sex | Mutation | Family History | Result of T4-Based NBS | TFT at the Diagnosis | Basal PRL, ng/mL | Basal/Peak TSH by TRH Test, mU/L | Clinical Information at the Last Visit | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age, y | TSH, mU/L | Free T4, ng/dL | Age, y | Height SD Score | Intellectual Disability | L-T4 Dose, μg/kg/d | |||||||
| 1 | M | IGSF1 p.Leu680Pro | No | Positive | 0.1 | 4.4 | 0.71 | 33.2 | NE | 3 | −0.2 | No | 4.0 |
| 2 | M | IGSF1 p.Tyr856* | No | Positive | 0.1 | 2.6 | 0.44 | NE | NE | 6 | +1.0 | No | 1.7 |
| 3 | M | IGSF1 p.Arg1189* | No | Positive | 0.1 | 3.0 | 0.63 | 7.4 | 3.0/5.8 | 9 | +0.1 | Present | 2.3 |
| 4 | M | IGSF1 c.2056+1G>A | Present | Negative | 10 | 7.1 | 0.65 | 1.0 | 7.1/6.0 | 23 | −1.0 | No | 1.4 |
| 5 | M | IGSF1 4.8-kb deletion | No | Positive | 0.1 | 2.9 | 0.45 | 11.7 | 2.9/5.8 | 14 | −1.4 | No | 1.0 |
| 6 | M | TBL1X p.Asn382del | No | Positive | 0.2 | 2.7 | 0.53 | 34.0 | 2.7/21.4 | 6 | −1.5 | No | Not treated |
| 7 | M | No | NAa | 0.0 | 2.0 | 0.73 | NE | NE | 10 | −4.3 | Present | 3.3 | |
| 8 | M | No | Positive | 0.1 | 2.5 | 0.55 | NE | 2.5/9.0 | 6 | −2.0 | No | 1.4 | |
| 9 | M | No | Positive | 0.2 | 6.4 | 0.73 | 174.3 | 6.4/29.3 | 9 | +0.4 | No | 1.7 | |
| 10 | F | No | Negative | 0.9 | 3.2 | 0.68 | 6.6 | NE | 5 | −1.1 | No | 2.0 | |
| 11 | M | No | NAa | 0.9 | 1.2 | 0.79 | 5.0 | 1.2/11.9 | 5 | −0.7 | No | 2.1 | |
| 12 | M | No | NAa | 1.1 | 5.9 | 0.79 | 6.4 | NE | 8 | −2.5 | Present | 3.9 | |
| 13 | F | No | Negative | 4 | 2.1 | 0.70 | 7.3 | 2.1/42.0 | 20 | +1.8 | Present | 1.5 | |
| Ref | 1.7-9.1b | 0.9-2.3b | M, 4.3–13.7 | ||||||||||
| F, 4.9–29.3 | |||||||||||||
| ID | Sex | Mutation | Family History | Result of T4-Based NBS | TFT at the Diagnosis | Basal PRL, ng/mL | Basal/Peak TSH by TRH Test, mU/L | Clinical Information at the Last Visit | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age, y | TSH, mU/L | Free T4, ng/dL | Age, y | Height SD Score | Intellectual Disability | L-T4 Dose, μg/kg/d | |||||||
| 1 | M | IGSF1 p.Leu680Pro | No | Positive | 0.1 | 4.4 | 0.71 | 33.2 | NE | 3 | −0.2 | No | 4.0 |
| 2 | M | IGSF1 p.Tyr856* | No | Positive | 0.1 | 2.6 | 0.44 | NE | NE | 6 | +1.0 | No | 1.7 |
| 3 | M | IGSF1 p.Arg1189* | No | Positive | 0.1 | 3.0 | 0.63 | 7.4 | 3.0/5.8 | 9 | +0.1 | Present | 2.3 |
| 4 | M | IGSF1 c.2056+1G>A | Present | Negative | 10 | 7.1 | 0.65 | 1.0 | 7.1/6.0 | 23 | −1.0 | No | 1.4 |
| 5 | M | IGSF1 4.8-kb deletion | No | Positive | 0.1 | 2.9 | 0.45 | 11.7 | 2.9/5.8 | 14 | −1.4 | No | 1.0 |
| 6 | M | TBL1X p.Asn382del | No | Positive | 0.2 | 2.7 | 0.53 | 34.0 | 2.7/21.4 | 6 | −1.5 | No | Not treated |
| 7 | M | No | NAa | 0.0 | 2.0 | 0.73 | NE | NE | 10 | −4.3 | Present | 3.3 | |
| 8 | M | No | Positive | 0.1 | 2.5 | 0.55 | NE | 2.5/9.0 | 6 | −2.0 | No | 1.4 | |
| 9 | M | No | Positive | 0.2 | 6.4 | 0.73 | 174.3 | 6.4/29.3 | 9 | +0.4 | No | 1.7 | |
| 10 | F | No | Negative | 0.9 | 3.2 | 0.68 | 6.6 | NE | 5 | −1.1 | No | 2.0 | |
| 11 | M | No | NAa | 0.9 | 1.2 | 0.79 | 5.0 | 1.2/11.9 | 5 | −0.7 | No | 2.1 | |
| 12 | M | No | NAa | 1.1 | 5.9 | 0.79 | 6.4 | NE | 8 | −2.5 | Present | 3.9 | |
| 13 | F | No | Negative | 4 | 2.1 | 0.70 | 7.3 | 2.1/42.0 | 20 | +1.8 | Present | 1.5 | |
| Ref | 1.7-9.1b | 0.9-2.3b | M, 4.3–13.7 | ||||||||||
| F, 4.9–29.3 | |||||||||||||
Abbreviations: F, female; L-T4, levothyroxine; M, male; NA, not applicable; NE, not evaluated; TFT, thyroid function tests.
These patients received TSH-based NBS.
Reference range for neonates.
Characteristics of the Study Subjects
| ID | Sex | Mutation | Family History | Result of T4-Based NBS | TFT at the Diagnosis | Basal PRL, ng/mL | Basal/Peak TSH by TRH Test, mU/L | Clinical Information at the Last Visit | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age, y | TSH, mU/L | Free T4, ng/dL | Age, y | Height SD Score | Intellectual Disability | L-T4 Dose, μg/kg/d | |||||||
| 1 | M | IGSF1 p.Leu680Pro | No | Positive | 0.1 | 4.4 | 0.71 | 33.2 | NE | 3 | −0.2 | No | 4.0 |
| 2 | M | IGSF1 p.Tyr856* | No | Positive | 0.1 | 2.6 | 0.44 | NE | NE | 6 | +1.0 | No | 1.7 |
| 3 | M | IGSF1 p.Arg1189* | No | Positive | 0.1 | 3.0 | 0.63 | 7.4 | 3.0/5.8 | 9 | +0.1 | Present | 2.3 |
| 4 | M | IGSF1 c.2056+1G>A | Present | Negative | 10 | 7.1 | 0.65 | 1.0 | 7.1/6.0 | 23 | −1.0 | No | 1.4 |
| 5 | M | IGSF1 4.8-kb deletion | No | Positive | 0.1 | 2.9 | 0.45 | 11.7 | 2.9/5.8 | 14 | −1.4 | No | 1.0 |
| 6 | M | TBL1X p.Asn382del | No | Positive | 0.2 | 2.7 | 0.53 | 34.0 | 2.7/21.4 | 6 | −1.5 | No | Not treated |
| 7 | M | No | NAa | 0.0 | 2.0 | 0.73 | NE | NE | 10 | −4.3 | Present | 3.3 | |
| 8 | M | No | Positive | 0.1 | 2.5 | 0.55 | NE | 2.5/9.0 | 6 | −2.0 | No | 1.4 | |
| 9 | M | No | Positive | 0.2 | 6.4 | 0.73 | 174.3 | 6.4/29.3 | 9 | +0.4 | No | 1.7 | |
| 10 | F | No | Negative | 0.9 | 3.2 | 0.68 | 6.6 | NE | 5 | −1.1 | No | 2.0 | |
| 11 | M | No | NAa | 0.9 | 1.2 | 0.79 | 5.0 | 1.2/11.9 | 5 | −0.7 | No | 2.1 | |
| 12 | M | No | NAa | 1.1 | 5.9 | 0.79 | 6.4 | NE | 8 | −2.5 | Present | 3.9 | |
| 13 | F | No | Negative | 4 | 2.1 | 0.70 | 7.3 | 2.1/42.0 | 20 | +1.8 | Present | 1.5 | |
| Ref | 1.7-9.1b | 0.9-2.3b | M, 4.3–13.7 | ||||||||||
| F, 4.9–29.3 | |||||||||||||
| ID | Sex | Mutation | Family History | Result of T4-Based NBS | TFT at the Diagnosis | Basal PRL, ng/mL | Basal/Peak TSH by TRH Test, mU/L | Clinical Information at the Last Visit | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age, y | TSH, mU/L | Free T4, ng/dL | Age, y | Height SD Score | Intellectual Disability | L-T4 Dose, μg/kg/d | |||||||
| 1 | M | IGSF1 p.Leu680Pro | No | Positive | 0.1 | 4.4 | 0.71 | 33.2 | NE | 3 | −0.2 | No | 4.0 |
| 2 | M | IGSF1 p.Tyr856* | No | Positive | 0.1 | 2.6 | 0.44 | NE | NE | 6 | +1.0 | No | 1.7 |
| 3 | M | IGSF1 p.Arg1189* | No | Positive | 0.1 | 3.0 | 0.63 | 7.4 | 3.0/5.8 | 9 | +0.1 | Present | 2.3 |
| 4 | M | IGSF1 c.2056+1G>A | Present | Negative | 10 | 7.1 | 0.65 | 1.0 | 7.1/6.0 | 23 | −1.0 | No | 1.4 |
| 5 | M | IGSF1 4.8-kb deletion | No | Positive | 0.1 | 2.9 | 0.45 | 11.7 | 2.9/5.8 | 14 | −1.4 | No | 1.0 |
| 6 | M | TBL1X p.Asn382del | No | Positive | 0.2 | 2.7 | 0.53 | 34.0 | 2.7/21.4 | 6 | −1.5 | No | Not treated |
| 7 | M | No | NAa | 0.0 | 2.0 | 0.73 | NE | NE | 10 | −4.3 | Present | 3.3 | |
| 8 | M | No | Positive | 0.1 | 2.5 | 0.55 | NE | 2.5/9.0 | 6 | −2.0 | No | 1.4 | |
| 9 | M | No | Positive | 0.2 | 6.4 | 0.73 | 174.3 | 6.4/29.3 | 9 | +0.4 | No | 1.7 | |
| 10 | F | No | Negative | 0.9 | 3.2 | 0.68 | 6.6 | NE | 5 | −1.1 | No | 2.0 | |
| 11 | M | No | NAa | 0.9 | 1.2 | 0.79 | 5.0 | 1.2/11.9 | 5 | −0.7 | No | 2.1 | |
| 12 | M | No | NAa | 1.1 | 5.9 | 0.79 | 6.4 | NE | 8 | −2.5 | Present | 3.9 | |
| 13 | F | No | Negative | 4 | 2.1 | 0.70 | 7.3 | 2.1/42.0 | 20 | +1.8 | Present | 1.5 | |
| Ref | 1.7-9.1b | 0.9-2.3b | M, 4.3–13.7 | ||||||||||
| F, 4.9–29.3 | |||||||||||||
Abbreviations: F, female; L-T4, levothyroxine; M, male; NA, not applicable; NE, not evaluated; TFT, thyroid function tests.
These patients received TSH-based NBS.
Reference range for neonates.
Mutation detection
Mutational screening of the five genes (IGSF1, IRS4, TBL1X, TRHR, and TSHB) led us to identify six mutation-carrying male patients. Five patients had hemizygous IGSF1 mutations, including a missense mutation (p.Leu680Pro), nonsense mutations (p.Tyr856* and p.Arg1189*), a splice site mutation (c.2056+1G>A), and a 4.8-kb microdeletion (hg19:130,405,658- 130,410,468)(Fig. 1A and 1B). We also found one male patient with a hemizygous single amino-acid deletion TBL1X mutation (p.Asn382del) (Fig. 1A and 1B). None of the mutations were registered in any variant databases. None had a mutation in IRS4, TRHR, and TSHB.
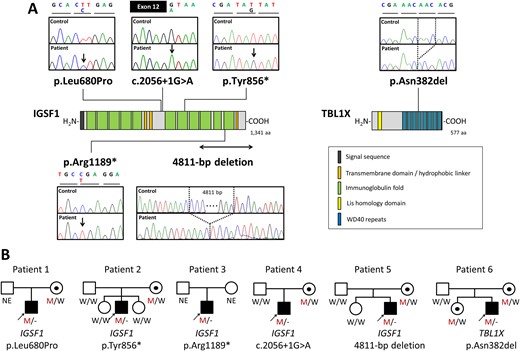
Identification of the five IGSF1 mutations and one TBL1X mutation. (A) Schematic diagrams of the IGSF1 protein (left) and the TBL1X protein (right) are shown along with electropherograms of each mutation-carrying patient. The mutated nucleotides are shown by arrows. For deletions, deleted nucleotides are shown by dashed lines. (B) Pedigrees of the six mutation-carrying patients. Black symbols indicate individuals affected with congenital i-TSHD. Abbreviations: M, mutated; NE, not evaluated; W, wild-type.
Clinical phenotypes of the mutation carrying patients
IGSF1 mutation carriers
Four of the five IGSF1 mutation-carrying patients were diagnosed as having i-TSHD in the frame of NBS. One patient had a negative result in TSH/T4-based NBS, and was subsequently diagnosed as i-TSHD at age 10 years. The presenting symptom was short stature (height SD score, −2.4), and he was successfully treated with normal-range adult height SD score (−1.0). Weight SD score at birth and at the last visits (age 3 to 23 years) were 1.4 ± 0.4 (mean ± SD) and 1.4 ± 1.3, respectively. One patient had obesity [body mass index (BMI) 26.1 kg/m2 at age 23 years]. Testicular volume of the three prepubertal patients (patients 1 to 3) were normal (1to 2 mL; reference 1 to 3 mL). Patient 4 had macroorchidism at age 18 years (testicular volume, 28 mL; reference, 12.5 to 20 mL), whereas testicular growth of patient 5 seemed to be delayed (testicular volume, 3 mL at age 14 years; age-matched reference, 8 to 20 mL). PRL deficiency was noted in two of two patients evaluated with the TRH test.
The cousin of patient 4 was also affected by i-TSHD, and he also carried the identical IGSF1 mutation (c.2056+1G>A). He was diagnosed in the frame of TSH/T4-based NBS, and has been treated with levothyroxine. He had obesity (BMI, 25.9 kg/m2) and macroorchidism (testicular volume, 35 mL).
We identified four female heterozygous IGSF1 mutation carriers; detailed clinical evaluation was performed in three (mothers of patients 1, 2, and 4). They had normal serum free T4 levels ranged 0.91 to 1.21 ng/dL. The mother of patient 2 had hypogalactia that was accompanied by a low serum PRL level (0.5 ng/mL). The mother of patient 4 had severe obesity (BMI, 35.0 kg/m2).
TBL1X mutation carrier
The TBL1X mutation-carrying patient, a 6-year-old boy, was the second child of healthy unrelated parents. He was born at gestational age 41 weeks, 1 day, with birth length 50.5 cm (0.3 SD) and weight 3188 g (−0.5 SD). He had a low blood-spot free T4 level at NBS (0.68 ng/dL; cutoff level, 0.70), and was referred to us at age 58 days. Blood tests revealed a low free T4 level (0.53 ng/dL; reference, 0.9 to 2.3), a low free T3 level (3.1 pg/mL; reference, 3.3 to 4.9) and a normal serum TSH level (2.7 mU/L; reference, 1.7 to 9.1). Thyroid ultrasonography showed a hypoplastic gland. A TRH test performed at age 69 days showed a normal TSH response (peak TSH, 21.4 mU/L). The patient was carefully followed without therapy, because he lacked hypothyroidism-related manifestations and had normal TSH response to exogenous TRH. TRH tests were repeated at age 1.5 and 3 years, showing consistently normal TSH responses (peak TSH levels, 12.9 mU/L and 17.6 mU/L). Serum PRL levels were also measured at the second TRH test, showing high levels (basal, 34.0 ng/mL; peak, 102 ng/mL). Despite the consistently low thyroid hormone levels (free T4, 0.55 ± 0.07 ng/dL), he showed normal-range growth (height SD score −1.5 and weight SD score 0.0 at age 4 years) and satisfactory development until the last visit. Hearing loss was not noted throughout the course.
Functional analyses of Asn382del-TBL1X
The pathogenicity of the Asn382del TBL1X mutation was verified in vitro. Western blotting of WT-TBL1X and Asn382del-TBL1X showed comparable protein expression levels (Fig. 2A). In the absence of T3, expression of WT-TBL1X along with TRβ1 and NCoR resulted in approximately fivefold activation of Trh-luc compared with empty vector alone (Fig. 2B). The activation was abolished in the presence of T3 [i.e., ligand-independent activation (LIA)]. As for Asn382del-TBL1X, LIA on Trh-luc was significantly reduced, although the mutant retains the ability to repress Trh-luc in the presence of T3 (Fig. 2B). We additionally characterized Asn382del-TBL1X using the TSHB-luc reporter, obtaining similar results: Asn382del-TBL1X showed loss of LIA (Fig. 2C).
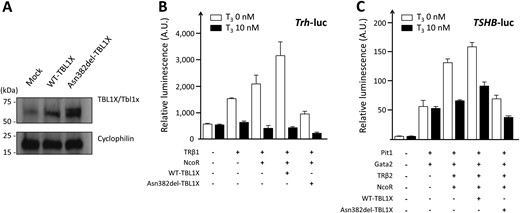
Functional characterization of the TBL1X p.Asn382del mutation. Western blotting, performed with use of N-1 cells, showed similar protein expression levels between WT and (A) the mutant. Cyclophilin was used as a loading control. Note that N-1 cells express endogenous mouse Tbl1x protein with a relatively low level. (B) In the luciferase assay using the Trh-luc reporter, LIA of Trh-luc was observed with coexpression of TRβ1 and NCoR. The LIA was further augmented with addition of WT-TBL1X, but was abrogated with addition of Asn382-del-TBL1X. (C) Similar results were obtained with the TSHB-luc reporter. The data of the luciferase assays are representative of three experiments showing similar results. Bars indicate SEM.
Review of hypothalamus pituitary phenotypes of mutation carriers
We found 27 papers describing 202 individuals carrying mutations in IGSF1, IRS4, TBL1X, TRHR, or TSHB. For rigorous comparison of the phenotypes, 102 individuals with heterozygous mutation carriers (monoallelic mutations in TSHB or TRHR; female mutation carriers in IGSF1, IRS4, or TBL1X) were excluded. Of the remaining 100 mutation carriers, thyroid function test data and/or serum PRL data were available in 74. The data of 80 mutation carriers (74 from the literature and six from the current study) were subject to the following analyses (IGSF1 defect: male 45, female 0; IRS4 defect: male 8, female 0; TBL1X defect: male 7, female 0); TRHR defect: male 4, female 2; TSHB defect: male 8, female 6). TRH-stimulated TSH levels [mU/L, median (interquartile range)] were low in the TSHB defect [0.8 (0.1, 3.2)] and TRHR defect [1.5 (0.8, 5.3)], whereas the values were mostly normal in the IGSF1 defect [7.2 (5.5, 8.5)], TBL1X defect [11.0 (9.9, 11.3)] and IRS4 defect [9.2 (6.8, 10.4)] (Fig. 3A). Basal serum PRL levels [ng/mL, median (interquartile range)] were slightly high in the TSHB defects [23.9 (16.0, 37.1)], were mostly normal in TBL1X defect [8.9 (7.9, 21.2)] and IRS4 defect [12.0 (5.8, 18.8)], and were low in the IGSF1 defect [2.0 (undetectable to 5.1)] and the TRHR defect (3.0, 6.2, and 6.8) (Fig. 3B). A scatter plot of the paired TRH-stimulated TSH and basal PRL data, which were available for 31 patients, showed etiology-specific distributions (Fig. 3C). The TSHB defect showed a nonoverlapping “TSH low/PRL high” distribution. This was contrasting to the IGSF1 defect, showing a “TSH normal/PRL low” distribution. The TBL1X defect and the IRS4 defect showed an indistinguishable “TSH normal/PRL normal” distribution, which was partially overlapped with that of the IGSF1 defect. One TRHR mutation carrier showed the “TSH low/PRL low” pattern, which seemed consistent with physiological knowledge that TRHR signaling regulates both TSH secretion and PRL secretion.
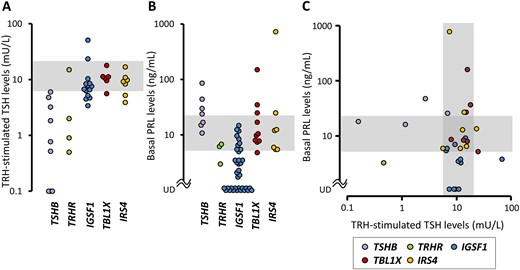
Hypothalamic/pituitary unit phenotypes of TSH deficiency resulting from single gene mutations. Data of (A) TRH-stimulated TSH levels and (B) basal PRL levels were obtained from the literature and were plotted according to the genotypes. (C) The data with paired PRL/TRH-stimulated TSH measurements were visualized as a scatter plot. The shaded areas indicate reference ranges. UD, undetectable.
Discussion
We described here the genetic screening targeting congenital i-TSHD, in which all five known causative genes were analyzed. We found that approximately one-half of the patients had any single gene mutations, and the most frequently mutated gene was IGSF1. To our knowledge, this is the first direct evidence showing the IGSF1 defect as the leading genetic cause of congenital i-TSHD. No ethnic-specific common mutation, as seen in the TSHB defect, was found. Most of IGSF1 mutations (including those found in the current study) were observed only within pedigrees (i.e., private mutations): 24 distinct mutations have been found in 26 unrelated families. This suggests that IGSF1 mutations have been randomly acquired in the populations, and that racial disparity of the frequency would be minimal. Congenital i-TSHD has been predominantly seen in boys: Zwaveling-Soonawala et al. (2) reported a male-to-female ratio of 4.7. We presume that the male predominance is at least partly from the high relative frequency of the IGSF1 defect, which follows X-linked inheritance.
Eight hemizygous TBL1X mutation carriers with congenital i-TSHD have been described so far (8). In this study, we reported the ninth mutation carrier that showed comparable clinical phenotypes with previous cases: mild i-TSHD with no prominent nonendocrine complications. TBL1X consists of an N-terminal Lis homology domain and a C-terminal WD40 repeats. Five single amino-acid mutations (Asn365Tyr, Ala366Thr, Trp369Arg, Asn382del, and Tyr458Cys) (8) identified so far have been exclusively found in the WD40 repeats, implying the correlation between the specific genotypes and i-TSHD. We have previously shown that two mutant TBL1X reported by Heinen et al. (8) (Asn365Tyr and Tyr458Cys) were characterized by loss of LIA on the Trh-luc and TSHB-luc (22). In the current study, we showed that Asp382del-TBL1X also had the similar molecular characteristics.
Determining the site of abnormality in each i-TSHD case is not easy. TRH tests produce both blunted (“pituitary dysfunction”) or exaggerated (“hypothalamic dysfunction”) TSH responses, and thus interpretation of test results is often difficult. In several monogenic forms of i-TSHD, specific nonendocrine features have been reported, including macroorchidism and obesity in the IGSF1 defect (7), and hearing impairment in the TBL1X defect (8). However, in this study, we could hardly recognize such etiology-specific nonendocrine features in the six mutation-carrying patients. Through the literature review, we noticed that taking PRL levels into account in addition to TRH test results, would contribute to a better understanding of the etiology. For example, the TSHB defect is characterized by discordance between TSH levels and PRL levels. The high PRL levels suggest increased hypothalamic TRH secretion via the LIA mechanism (Fig. 4). Similar hypersecretion of TRH would occur in the TRHR defect, but in this case PRL secretion is defective because PRL-producing cells are insensitive to TRH (Fig. 4). As for the TBL1X defect, most patients showed normal TRH-stimulated TSH responses and normal PRL levels (8). This vague hypothalamic/pituitary unit phenotypes are probably associated with inappropriate negative feedback of both hypothalamus and pituitary (Fig. 4). Molecular pathogenesis of the remaining two forms (the IGSF1 defect and the IRS4 mutations) are less clear than the other three forms. Genetically engineered Igsf1-deficient mice have been analyzed, showing decreased levels of Thrh mRNA and Tshb mRNA in the pituitary (24). Although TRH resistance was hypothesized as a mechanism of TSH deficiency in the IGSF1 defect, this hypothesis does not fit well with distinct scatter plot distributions between the IGSF1 defect and the TRHR defect. From a practical point of view, we suppose the discordance between TRH-stimulated TSH levels (normal) and serum PRL levels (low) would be the most valuable clinical clue to suspect the IGSF1 defect. The mechanism(s) linking IRS4 mutations and i-TSHD is also unclear, though the defective leptin signaling is postulated (10). More clinical and experimental data will be required to delineate the mechanisms of the IRS4 defect.

Schematic diagrams showing the presumed hypothalamic/pituitary unit states of the TSHB defect, the TRHR defect, and the TBL1X defect. Hypothalamic TRH-producing cells are colored in gray, whereas pituitary TSH-producing cells and PRL-producing cells are colored in light blue and orange, respectively. TR and TBL1X are expressed in TRH-producing cells and TSH-producing cells, mediating negative feedback regulations. The TRHR is expressed in the surface of TSH-producing cells and PRL-producing cells. In the TSHB defect and the TRHR defect, low circulating thyroid hormones cause LIA of TRH, resulting in hypersecretion of TRH. In this condition, PRL secretion is increased in the TSHB defect, but it is not increased in the TRHR defect because of insensitivity to TRH. In the TBL1X defect, serum PRL levels are mostly normal, suggesting comparable TRH secretion levels between the defect and the physiological state. This indicates inappropriate negative feedback of the TRH-producing cells. Similarly, pituitary TSH secretion is inappropriately normal despite low circulating thyroid hormones, suggesting inappropriate negative feedback of the TSH-producing cells. Loss of LIA in the hypothalamus and pituitary agrees with those hypothalamic/pituitary unit phenotypes. TR, thyroid hormone receptor; TRHR, TRH receptor.
In our patient cohort of congenital i-TSHD, seven of the 13 patients (54%) were diagnosed in the frame of TSH/T4-based NBS. If all patients were diagnosed irrespective of the screening methods, the proportion should be ∼12% (the proportion of Japanese newborns receiving TSH/T4-based NBS), implying that a substantial number of congenital i-TSHD patients remain undiagnosed. The universal implementation of TSH/T4-based NBS has been debated in Japan.
In conclusion, we conducted the mutation screening study in a defined patient cohort of congenital i-TSHD, and found that a substantial proportion of the disease is due to monogenic causes. By reviewing previously reported mutation carriers, we demonstrated etiology-specific relationships between serum PRL levels and TRH-stimulated TSH levels that may aid differential diagnosis in patients with congenital i-TSHD.
Acknowledgments
The authors thank Dr. Rumi Hachiya (Tokyo Metropolitan Children’s Medical Center,), Dr. Nagisa Komatsu (Japanese Red Cross Hospital Kumamoto,), Dr. Masako Izawa (Aichi Children's Health and Medical Center), Dr. Yoichiro Oda (Chigasaki Municipal Hospital), Shigeyuki Ohtsu (Kitasato University School of Medicine), Masaki Takagi (Tokyo Metropolitan Children’s Medical Center) and Noriyuki Takubo (Kitasato University School of Medicine) for providing clinical materials and information and Dr. Shigekazu Sasaki (Hamamatsu Medical University,) for providing CV-1 cells.
Financial Support: This work was supported by JSPS KAKENHI Grant 15K09630 to S.N. and Grant 26461353 to T.S. and a grant from MHLW [Jitsuyoka (Nanbyo)-Ippan-014] to T.H.
Disclosure Summary: The authors have nothing to disclose.
Data Availability: All data generated or analyzed during this study are included in this published article or in the data repositories listed in References.
Abbreviations:
- BMI
body mass index
- CH
congenital hypothyroidism
- i-TSHD
isolated TSH deficiency
- LIA
ligand-independent activation
- NBS
newborn screening
- NCoR
nuclear corepressor
- PRL
prolactin
- WT
wild-type
References
Author notes
C.S., T.T., and K.A. contributed equally to this work.
M.Y. and S.N. jointly supervised this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}