





Abstract
Inactivating mutations within the AR gene are present in only ~40% of individuals with clinically and hormonally diagnosed androgen insensitivity syndrome (AIS). Previous studies revealed the existence of an AR gene mutation–negative group of patients with AIS who have compromised androgen receptor (AR) function (AIS type II).
To investigate whether AIS type II can be due to epigenetic repression of AR transcription.
Quantification of AR mRNA and AR proximal promoter CpG methylation levels in genital skin–derived fibroblasts (GFs) derived from patients with AIS type II and control individuals.
University hospital endocrine research laboratory.
GFs from control individuals (n = 11) and patients with AIS type II (n = 14).
Measurement of AR mRNA and AR promoter CpG methylation as well as activity of AR proximal promoter in vitro.
Fifty-seven percent of individuals with AIS type II (n = 8) showed a reduced AR mRNA expression in their GFs. A significant inverse correlation was shown between AR mRNA abundance and methylation at two consecutive CpGs within the proximal AR promoter. Methylation of a 158-bp-long region containing these CpGs was sufficient to severely reduce reporter gene expression. This region was bound by the runt related transcription factor 1 (RUNX1). Ectopic expression of RUNX1 in HEK293T cells was able to inhibit reporter gene expression through this region.
Aberrant CpGs methylation within the proximal AR promoter plays an important role in the control of AR gene expression and may result in AIS type II. We suggest that transcriptional modifiers, such as RUNX1, could play roles therein offering new perspectives for understanding androgen-mediated endocrine diseases.
Androgen insensitivity syndrome (AIS) is classically defined as a defect in androgen action caused by mutations in the X-chromosomal androgen receptor (AR) gene. Phenotypically, this results in incomplete male external genital development in 46,XY individuals, ranging from completely female external genitalia in individuals with complete androgen insensitivity syndrome (CAIS), various degrees of undervirilization in individuals with partial androgen insensitivity syndrome to infertility and gynecomastia in individuals with mild androgen insensitivity syndrome (1). Although in >90% of individuals with clinically diagnosed CAIS a disruptive mutation in the AR gene can be found, this holds true for <40% of individuals clinically diagnosed as having partial androgen insensitivity syndrome, indicating that factors outside the androgen receptor (AR) might lead to a similar phenotype (2). We previously set up an assay to assess androgen-dependent AR function in human genital skin fibroblasts (GFs) by measuring the DHT-mediated induction of the endogenous AR target gene apolipoprotein D (APOD) (3, 4). Applying this assay to GFs from individuals with the clinical diagnosis of AIS but no mutation in the AR gene revealed reduced AR function, manifested as reduced DHT-mediated upregulation of APOD mRNA, in about one third of cases. This functional subgroup of AIS, since then named AIS type II (3), suggests that coregulators of AR activity can be involved in the etiology of AIS when disrupted.
Cofactors can influence AR activity at the transcriptional as well as posttranscriptional level. Concerning the latter, we could show that a mutation in the 5′ untranslated region of the AR is able to severely interfere with normal AR protein translation (5). Once translated, the AR has been shown to undergo context-dependent posttranslational modifications (6) and to interact with many cofactors to activate transcription of its target genes (7, 8). It is conceivable that changes in these cofactors can impair AR function, but no cofactor mutation leading to AIS type II has been identified so far. Surprisingly little is known about the tissue-specific transcriptional control of the AR. Among other proteins, SP1, MYC, and ZEB1 (zinc finger E-box–binding homeobox 1) have been shown to activate AR transcription by binding to the AR proximal promoter, whereas AR itself can both activate and repress its own transcription (depending on the cellular context) through binding to androgen response elements located within the promoter as well as the gene body (9).
One important mechanism of transcriptional gene silencing is DNA methylation at cytosine bases in CpG-rich promoters where high methylation levels often correlate with low transcription of the corresponding gene (10). This is usually accompanied by binding of methyl-CpG–binding proteins as well as histone tail modifiers that confer repressive marks to the histone tails. The resulting chromatin compaction can impede activating transcription factors to bind. Transcriptional regulation through DNA methylation plays important roles in mammalian development and establishment of cellular identity and is a dynamic process depending on the cellular context (10).
In this study we provide evidence that aberrantly high cytosine methylation in a defined proximal AR promoter region in individuals with AIS type II is accompanied by low AR mRNA expression. Chromatin modifiers as well as the transcription factor runt related transcription factor 1 (RUNX1) are able to bind to this AR proximal promoter. In vitro reporter gene expression can be inhibited both by methylation of the AR proximal promoter region and by overexpression of RUNX1. We suggest that this methylation-dependent repression of AR mRNA expression may contribute to an incomplete male genital development in a subset of individuals with AIS type II.
Material and Methods
The study was performed in agreement with the vote of the Ethical Committee of the Medical Faculty of the Christian Albrechts University, Kiel, Germany (AZ: D415/11). GFs received from collaborating partners were included in this study according to the recommendations of the local ethical committees. Control GFs were obtained from males with fully developed male external genitalia (i.e., neither hypospadias nor micropenis) during orchidopexy. All GFs included in this study were double encrypted. Primary culturing of genital skin biopsy specimens and further methods are described in the Supplemental Material and Methods.
Results
To see whether reduced AR mRNA expression could be a cause of AIS type II, we compared AR mRNA accumulation levels between scrotum control and labioscrotal AIS type II–derived GFs. The age range in both cohorts was 0 to 16 years (Supplemental Table 1). In all samples, the coding region including intron-exon boundaries of the AR gene was sequenced to exclude disruptive AR gene mutations. Furthermore, APOD analysis was performed to measure AR activity. Eight of 14 AIS type II–derived samples had AR mRNA expression levels outside the lower SD of the control group (<0. 05) (Fig. 1a; and Supplemental Table 1). Western blot analysis of the eight samples with lower AR mRNA also showed a reduced AR protein expression as compared with control GFs (Fig. 1b).
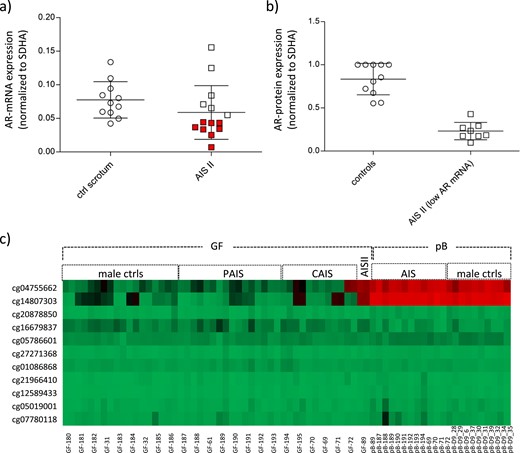
AR expression and AR promoter DNA methylation levels in GFs from controls and individuals with AIS. (a) AR mRNA expression in control GFs (n = 11) and GFs from patients with AIS type II (n = 14) as measured by quantitative PCR. AR mRNA levels were normalized to the housekeeping gene SDHA and are shown as mean of three independent measurements. AR mRNA levels below the SD are indicated in red. (b) AR protein measurement in control GFs and GFs from patients with AIS type II with reduced AR mRNA expression. Shown are all controls and patients with AIS type II indicated in red in panel (a) (GF-40, GF-89, GF-107, GF-109, GF-174, GF-176, GF-177, GF-179). (c) Heatmap of CpG methylation in the promoter region of the AR as present on the Human-Methylation27 DNA Analysis BeadChip. Depicted are DNA methylation levels from 9 control GF, 14 AR mutation–positive AIS II–derived GFs, and 1 AIS type II–derived GF, as well as from peripheral blood (pB) of a selection of patients with AR mutation–positive AIS and of independent male controls. All samples were run in duplicates. For pB samples, mean values of the duplicates are shown. Red indicates high DNA methylation; green indicates low DNA methylation. ctrls, controls; PAIS, partial androgen insensitivity syndrome.
We hypothesized that low AR mRNA expression levels could be due to high CpG methylation within the proximal AR promoter. Therefore, we analyzed AR promoter methylation on a Human-Methylation27 BeadChip (Illumina, San Diego, CA), choosing the AIS type II–derived GFs with the lowest AR mRNA levels (GF-89). DNA methylation data resulting from this chip were published in part before (11). The patient presented at birth with ambiguous external genitalia and a male 46,XY karyotype. Around mini-puberty, serum testosterone ranged from 4.85 to 9.43 nmol/L (normal male range, 3.1 to 8.8 nmol/L), serum DHT was 3.23 nmol/L (normal male range, 0.2 to 4.5 nmol/L), and LH was 3.9 mIU/mL. Ultrasonography did not reveal Müllerian structures, which together with the hormone levels supports the clinical diagnosis of AIS (12).
CpG methylation analysis of GF-89, together with control GFs, as well as samples from patients with AIS with a mutation in the AR gene (11) (Supplemental Table 2) revealed high methylation levels in GF-89 compared with the controls at cg04755662 (chrX:66,763,359; CpG7) and cg14807303 (chrX:66,763,445; CpG8) (Fig. 1c and Fig. 2a). GF-72 (AR-mutation positive CAIS) also showed high methylation at cg04755662 (chrX:66,763,359). Methylation levels in blood samples from the same patients with AIS as well as independent male control blood samples were analyzed in parallel. Interestingly, both cg04755662 and cg14807303 were highly methylated in peripheral blood in all samples, underlining a tissue-specific AR promoter methylation pattern (Fig. 1c). In line with this, AR mRNA expression has been shown to be significantly lower in peripheral blood than in genital skin (13).
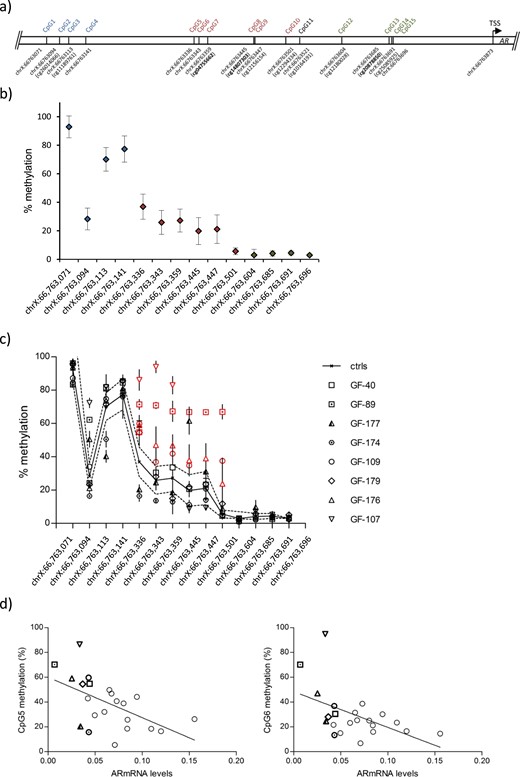
CpG methylation analysis within the proximal AR promoter in GFs from controls and individuals with AIS type II. (a) Schematic representation of the analyzed AR promoter region covering the CpGs (hg19). Only cg10164191 at chrX:66763521 (CpG11) was excluded from the analysis. CpGs present on the Human-Methylation27 DNA Analysis BeadChip are shown in bold. A color code distinguishes three regions CpG1-4 (blue), CpG5-10 (red), and CpG12-15 (green). (b) Mean methylation levels (percentages) at all individual CpGs shown in scrotal control GFs (n = 9). The analysis was performed in triplicate. Error bars indicate the SD. The color code reflects the position indicated in panel (a). (c) Methylation levels at the same CpGs in GFs from controls (black line; SDs are dotted lines) and patients with AIS type II (mean and SD) with relative AR mRNA expression <0.05 (GF-40, GF-89, GF-177, GF-174, GF-109, GF-179, GF-176, GF-107). Significantly higher methylation levels from chrX:66,763,336-66,763,501 are highlighted in red. Individual methylation values are shown in Supplemental Table 3. (d) Linear regression of the correlation between AR mRNA expression and methylation at CpG5 and CpG6. The correlation calculation included all controls and AIS type II samples analyzed by pyrosequencing. The correlation coefficient for CpG5 was −0.55 (P = 0.008); that for CpG6 was −0.5 (P = 0.01). GF-40, GF-89, GF-177, GF-174, GF-109, GF-179, GF-176, and GF-107 are highlighted by using the same symbols as in panel (c).
Because the array only contained a selection of CpGs within the proximal AR promoter, we designed a bisulfite-pyrosequencing assay including cg04755662 and cg14807303 as well as directly surrounding CpGs. Figure 2a shows all CpGs in this area that were covered by the assay, apart from cg10164191 (CpG11) because the sequence surrounding this CpG did not permit an appropriate primer design for bisulfite-treated DNA. We first determined CpG methylation levels in the selected AR promoter region in GFs from the control group. This revealed high methylation levels (mean ± SD, 67% ± 23.9%) farthest upstream of the transcriptional start site (CpG1-4) with a continuous decrease in methylation levels in the central region covering CpG5-10 (mean ± SD, 23% ± 9%) and lowest methylation levels when approaching the transcriptional start site (CpG12-15; mean ± SD, 4% ± 1%) (Fig. 2b).
Next, we tested methylation levels in AIS type II–derived GFs with AR mRNA expression below the SD of the control group. Interestingly, several AIS II GFs with low AR mRNA levels showed significantly higher methylation levels, especially in the central region (chrX:66,763,336-66,763,501; CpG5-10), with GF-89, GF-176, and GF-109 showing high methylation throughout this region (P < 0.001) and GF-107 showing very high methylation levels at position chrX:66,763,336-66,763,359 (P < 0.001) (Fig. 2c). CpG methylation levels in all samples dropped from position chrX:66,763,604 onward. As a further control, we included AIS type II samples showing AR mRNA levels similar to those in the control group, which revealed no increase in methylation along the analyzed region (Supplemental Fig. 1a). Mean values and SDs of every single CpG analyzed are summarized in Supplemental Table 3. The control group only contained more recently obtained samples, the AIS type II group instead a mixture of more recently and formerly obtained samples that were kept for longer time periods in liquid nitrogen. Therefore, we compared AR mRNA levels and CpG methylation levels in the AR promoter in both older and more recently obtained samples. No significant differences were seen (Supplemental Fig. 1b and 1c), excluding a strong bias due to longer storage in liquid nitrogen.
Next, we wanted to investigate any possible correlation between methylation levels of the analyzed cytosines and AR mRNA expression levels. To achieve this aim, we calculated the correlation coefficient between AR mRNA expression and every single CpG analyzed in duplicates by our pyrosequencing assay in all samples. A significant negative correlation (P ≤ 0.01) was found at positions chrX:66,763,336 (CpG5) and chrX:66,763,343 (CpG6) after multiple testing against both duplicates and each single duplicate, indicating that high methylation at these CpGs is negatively associated with AR mRNA expression (Fig. 2d). Sequencing of the AR promoter region revealed no mutation in the analyzed AIS type II samples with low AR mRNA expression, suggesting that the effect was of an epigenetic rather than a genetic nature.
To validate this correlation, we experimentally cloned the central region from position chrX:66,763,278 to 66,763,597 (CpG5-11) in a CpG-free β-galactosidase reporter vector, replacing the cytomegalovirus enhancer. An enhancer free vector served as negative control. A schematic representation of all AR promoter fragments cloned in this reporter vector is shown in Fig. 3a. After transfection into HEK293T cells, plasmid AR CpG5-11 showed higher β-galactosidase activity compared with the negative control plasmid, indicating that this AR promoter region might enhance transcription (Fig. 3b). In vitro CpG methylation of both plasmids before transfection caused a dramatic decrease in reporter gene expression in AR CpG5-11–transfected cells. No reduction was seen in the enhancer free control vector–transfected cells (Fig. 3b). Hence, CpG methylation of this AR region strongly interferes with reporter gene expression.
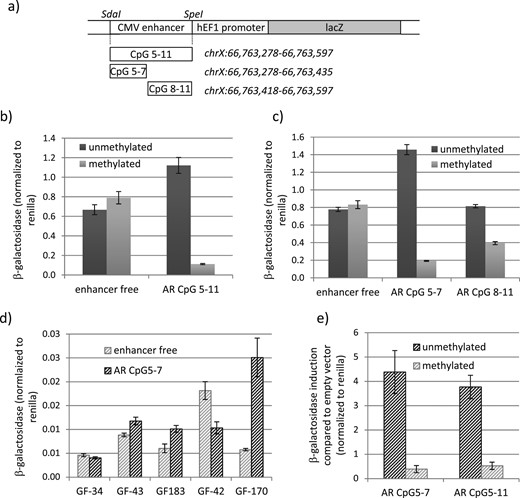
CpG methylation–dependent silencing of the AR proximal promoter region. (a) Schematic representation of the AR promoter constructs used for the β-galactosidase reporter assay. (b) β-Galactosidase activity in HEK293T cells transfected with the enhancer free and the AR CpG5-11 reporter vector in the methylated and unmethylated forms. β-Galactosidase activity significantly differed between the unmethylated and the methylated AR CpG 5-11 (P < 0.001). (c) β-Galactosidase activity in HEK293T cells transfected with the enhancer free, AR CpG5-7, and AR CpG8-11 reporter vectors in the methylated and unmethylated forms. β-Galactosidase activity significantly differed between both unmethylated and methylated AR CpG5-7 and AR CpG8-11 (P < 0.001). (d) β-Galactosidase activity in control GF transfected with the enhancer free and the AR CpG5-7 reporter vectors in the unmethylated form. (e) β-Galactosidase activity in GF170 transfected with the AR CpG5-7 and AR CpG5-11 reporter vectors in the methylated and unmethylated forms relative to enhancer free transfected cells. β-Galactosidase activity significantly differed between unmethylated and methylated AR CpG5-7 and AR CpG8-11 (P < 0.005). All β-galactosidase measurements were normalized to Renilla measurements. Error bars indicate the SD. CMV, cytomegalovirus.
Next we divided the promoter region in a fragment containing CpG5-7 and a fragment containing CpG8-11 and tested their activity in the β-galactosidase reporter assay after 20 hours and 24 hours of transfection. As seen in Fig. 3c, only AR CpG5-7 had an activating function on transcription after 20 hours of transfection, restricting the regulatory region to chrX:66,763,278-66,763,435. The enhancing activity was lost 24 hours after transfection but notably could be restored upon mutating of CpG5 and CpG6, indicating that these two CpGs are important regulators of repressive functions in this chromosomal region (Supplemental Fig. 2). In line with this, in vitro CpG-methylation of CpG5-7 had a much stronger inhibitory effect on reporter gene activity than in vitro CpG-methylation of CpG8-11 (Fig. 3c).
To analyze this regulatory region in a more natural context, we transfected five different male control GFs and tested their ability to regulate the AR promoter region harboring CpG5-7. This revealed a variable regulation in different GFs from enhancement to repression (Fig. 3d). Nevertheless, transfection of the methylated plasmid in GF-170 showed a strong reduction in reporter gene activation, validating the methylation-dependent repression of this chromosomal region (Fig. 3e).
Aiming to identify putative factors binding to the methylated fragments, we performed a DNA pulldown using a fragment spanning the chromosomal region chrX:66,763,324-66,763,383 (containing CpG5-7) as bait. This fragment was chosen because the two first cytosines in this region showed a significant negative correlation between DNA methylation and AR mRNA expression in GFs and because it showed a strong repression on reporter gene expression when methylated. Unmethylated and methylated baits were incubated with nuclear extracts derived from control GFs to identify proteins binding preferentially to one or the other. Bound proteins were determined through mass spectrometry. As shown in Fig. 4a, the unmethylated oligonucleotide was preferentially bound by transcription factors (FOSL2, MAX) and proteins involved in DNA repair (RECQL, HMGB2). In contrast, the methylated fragment was bound by DNA methyl-binding proteins [methyl-CpG binding domain protein 2 (MBD2), methyl-CpG binding protein 2 (MECP2), UHRF1], histone-modifying enzymes (HDAC1, HDAC2, MTA2), RNA-binding proteins (RBBP4, RBM4, RBM14, TIAL1), and RUNX1. RUNX1 was also successfully pulled down in an experiment using extracts from a different GF sample (Supplemental Fig. 3).
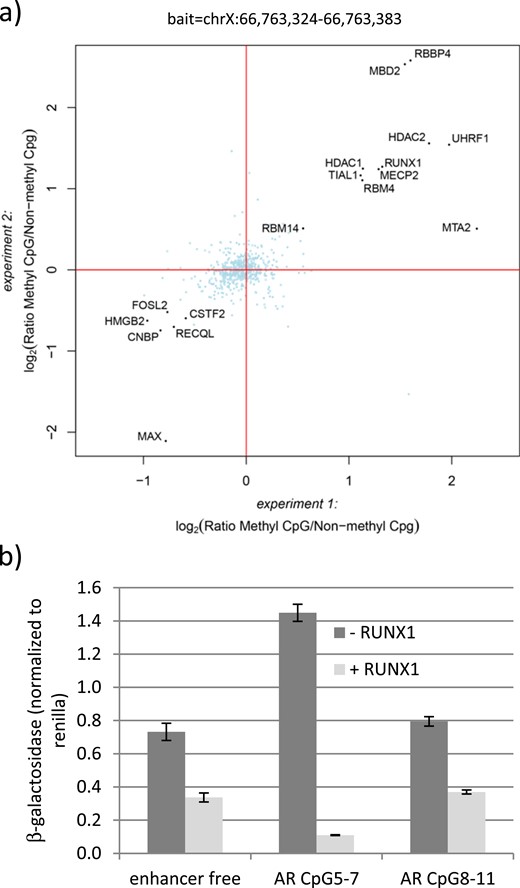
RUNX1 binding and silencing of the chromosomal region chrX:66,763,324-66,763,383. (a) DNA pulldown with oligonucleotides spanning the chromosomal region chrX:66,763,324-66,763,383. Pulldowns using the same methylated or unmethylated oligonucleotides and nuclear extract from GFs were performed in duplicate for each oligonucleotide and were analyzed separately by liquid chromatography/tandem mass spectrometry. Log2 ratios of all identified and quantified proteins in both experiments were plotted against each other. Proteins binding equally well to both oligonucleotides center around log2(ratio) = 0 and are marked in light blue. Proteins binding significantly better to either the methylated or the unmethylated oligonucleotide were determined by intensity-based outlier statistics, requiring a significance of B < 0.001 in both experiments. These proteins are marked with black dots. (b) β-Galactosidase activity in HEK293T cells transfected with the enhancer free, AR CpG5-7, and AR CpG8-11 reporter vectors. All vectors were transfected in unmethylated form and cotransfected with a RUNX1 expression vector (+RUNX1) or the mock vector as control (−RUNX1). Error bars indicate the SD.
To test a possible role of RUNX1 in transcriptional regulation, we cotransfected in HEK293T-cells a RUNX1 expression vector together with either the enhancer free reporter vector or the same vector carrying the CpG5-7 or the CpG8-11 regions. As a control we used cells transfected with an empty vector containing only the backbone of the RUNX1 expression vector (mock). Successful RUNX1 overexpression was verified by Western blot (Supplemental Fig. 4). As seen in Fig. 4b, RUNX1 revealed a repressive effect already on the enhancer free vector, possibly by acting on the hEF1 promoter present in the reporter plasmid. Nevertheless, RUNX1 was able to reduce the activating function of AR CpG5-7 on reporter gene expression further three- to fourfold compared with the control vector and 13-fold compared with the co–mock-transfected AR CpG5-7. AR CpG8-11 behaved like the empty vector control, suggesting that RUNX1 acts mainly on AR CpG5-7. Of note, RUNX1 did not bind to the chromosomal region chrX:66,763,422-66,763,481 covering CpG8 and CpG9, underlining a specific role for RUNX1 at region chrX:66,763,324-66,763,383 (Supplemental Fig. 5).
Usually, DNA methylation alone is not sufficient to repress transcription. In line with this, we identified the methyl-CpG binding protein MBD2, MECP2, and other proteins involved in chromatin compaction bound to the AR promoter fragment (Fig. 4a). We therefore sought to analyzed the chromatin state in the highly methylated endogenous AR promoter region of the AIS type II–derived fibroblast GF-89. To achieve this aim, we performed chromatin analysis discriminating between open and closed regions by using nuclease accessibility followed by quantitative RT-PCR. The PCR primer spanned the chromosomal region chrX:66,763,273-66,763,602 covering CpG5-11. As shown in Fig. 5, nuclease accessibility in the analyzed region was strongly reduced in GF-89 compared with the controls and GF derived from patients with classic (AR mutation–positive) CAIS. Of note, the CAIS-derived GF-72 showed a high methylation at cg04755662 (Fig. 1c) but low methylation in the surrounding CpGs (Supplemental Fig. 6), indicating that methylation solely at this side is not sufficient to induce chromatin compaction.
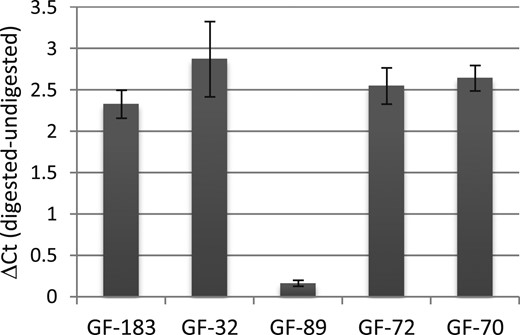
Heterochromatin state of the chromosomal region chrX:66,763,273-66,763,602. The difference between digested and undigested DNA was calculated on the cycle thresholds (Ct) of the quantitative PCR run and reflects the level of nuclease accessibility. GF-183 and GF-32 are control GFs, GF-72 and GF-70 are AR mutation–positive GFs from patients with CAIS, and GF-89 is an AIS type II–derived GF. All experiments were performed in triplicate. Error bars indicate the SD.
Discussion
We hypothesized that AIS type II could be a consequence of low AR expression throughout male sexual development. Measuring AR mRNA levels in AIS type II GFs showed significantly lower AR mRNA levels in 57% of the cases, indicating that a reduction in AR mRNA is a frequent event in AIS type II. Because CpG methylation within promoter regions can repress transcription of the corresponding gene (14), we compared CpG methylation levels of the proximal AR promoter region in GFs derived from controls as well as patients with AIS type II. We here show that methylation levels of two CpGs (chrX:66,763,336; CpG5 and chrX:66,763,343; CpG6) within the proximal AR promoter region inversely correlate with AR mRNA expression levels and that this correlation is significant. Four of eight AIS type II cases with low AR mRNA expression show high methylation of these two cytosines, two cases only at the first cytosine. This is important because GF-72 (AR mutation–positive CAIS) showed a high methylation at one single CpG (cg04755662; CpG7), indicating that in GFs harboring a mutation in the AR cytosine methylation can also occur in the analyzed region. Nevertheless, it turns out that only methylation over at least one of the first two consecutive CpGs might be a feature specific for AIS type II.
A correlation between CpG methylation within the AR promoter and loss of AR transcription has been analyzed before in human prostate cancer cell lines (15, 16). However, that study concentrated on the CpG island overlapping the transcriptional start site, whereas we concentrated on the CpG shore upstream of the island. Another study associated high methylation within the CpG island of the AR promoter with a lower AR mRNA expression in foreskin tissue from children with hypospadias (17). Nevertheless, a negative correlation between AR mRNA expression and AR methylation at two specific CpGs outside the CpG island in GFs has not been shown before.
We show that the chromosomal region chrX:66,763,278-66,763,435 covering CpG5-7 can act as a cis-acting element when unmethylated. In vitro methylation of CpG5-7 has a strong negative effect on reporter gene expression. This corroborates the hypothesis that methylation of CpG5 and CpG6 interferes with AR expression in GFs from patients with AIS type II with low AR mRNA levels. To our knowledge, this region has not been described as a regulatory region of the AR before. Currently we do not know which cellular conditions lead to an enhancing function of this chromosomal region. In HEK293T cells, the activating function is only transient and in GF this region can act as both an activator or silencer. By performing DNA pulldown experiments, we identified several proteins in vitro preferentially binding to the methylated or unmethylated promoter fragment. Although not all proteins could be reproducibly pulled down from different GF samples, one can speculate that proteins such as FosL2, a subunit of the AP-1 transcription factor complex, or MAX (maybe in complex with MYC) could be involved in positive regulation of the AR gene when the promoter is unmethylated. MYC activates transcription of the AR through binding to a consensus E box in exon 4 of the AR (18), and a noncanonical MYC-MAX binding site (CACGAG) (19) covers CpG5.
The only transcription factor identified as binding reproducibly in vitro to the methylated bait covering chrX:66,763,324-66,763,383 in the DNA pulldown was RUNX1. Looking for a RUNX1 consensus binding site within chrX:66,763,324-66,763,383 showed only a partial core consensus (ccacg instead of ccaca) (20). Data mining of chromatin immunoprecipitation sequencing experiments in prostate cancer and breast cancer cell lines (21–23) revealed a minimal overlapping RUNX1 binding from position chrX:66,763,331-66,763,349 covering CpG5 and CpG6, suggesting a potential noncanonical RUNX1 binding at this site. RUNX1 is necessary for normal hematopoiesis, and loss-of-function mutations are associated with acute myeloic leukemia and familial platelet disorder in humans (24). Also in the mouse, Runx1 is indispensable for the emergence of hematopoietic stem cells. Interestingly, mouse Runx1 also plays a role in gonadal differentiation; Runx1 is expressed in the bipotent gonad and is further activated in the developing ovary but not in the developing testis (25). This activation depends on the Wnt-4 signaling cascade (26). Recent studies on human gonadal gene expression also showed a slightly but significantly higher expression of RUNX1 in the developing ovary as compared with testis (27).
RUNX1 can act as both a transcriptional activator and repressor in a context-specific manner (28–30). It complexes with a variety of epigenetic modifiers and interacts with HDAC1 in repressive complexes (31). In our DNA pulldown, we identified several chromatin modifiers binding to CpG methylated DNA. Although the binding of many of these proteins needs to be confirmed, most of them are known to be able to control gene transcription by controlling the level of chromatin compaction. The methyl CpG binding proteins MBD2 and MECP2 both can confer transcriptional repression. MTA2 is a component of a nucleosome remodeling deacetylase complex and mediates transcriptional repression by facilitating the association of repressor molecules to the chromatin (32, 33). Of note, in rat Sertoli cells, Mta2 is activated by the androgen-activated AR to confer a negative feedback loop to FSH signaling and thereby AR expression (34). RNA-binding proteins (RBM4 and RBM14) are also interesting candidates because they could use sequence specificity conferred by RNA molecules to induce transcriptional gene silencing to the genomic target (10). In line with this, AR promoter targeted transcriptional gene silencing has been performed in a mammary gland cancer cell line and is accompanied by heterochromatin formation involving a noncoding RNA spanning the AR proximal promoter (35). Taken together, the AR proximal promoter region can recruit, when methylated, several chromatin modifiers involved in chromatin compaction and thereby confer transcriptional repression. This observation is corroborated by the fact that this region is protected from nuclease digestion in GF-89 but not in the control GFs.
The reporter assays indicate that RUNX1 binding to the unmethylated AR promoter fragment (chrX:66,763,278-66,763,435; CpG5-7) leads to transcriptional silencing. It could be that both promoter methylation and RUNX1 are required for full repression but are limiting in the tested cells. The exact mechanism by which RUNX1 may confer transcriptional repression awaits further research.
In conclusion, we provide evidence for epigenetic control of AR mRNA expression in human GFs. This epigenetic regulation of AR expression might be set up during embryonic development and maintained after differentiation to ensure proper cellular identity. When disturbed, this misregulation might be propagated throughout male genital development by interfering with AR expression, thereby leading to AIS type II. The identification of upstream factors responsible for this epigenetic AR mRNA repression is the next planned step. Our results not only give a molecular diagnosis to a considerable number of individuals with AIS type II but also reveal a previously unrecognized AR regulatory region that could be relevant for other AR-dependent conditions, such as prostate cancer.
Abbreviations:
- AIS
androgen insensitivity syndrome
- APOD
apolipoprotein D
- AR
androgen receptor
- CAIS
complete androgen insensitivity syndrome
- GF
genital skin–derived fibroblast
- MBD2
methyl-CpG binding domain protein 2
- MECP2
methyl-CpG binding protein 2
- RUNX1
runt related transcription factor 1
Acknowledgments
We thank Brigitte Karwelies, Tanja Stampe, and Gila Hohmann for their excellent laboratory support and Ingram Iaccarino for critical reading of the manuscript. We thank the KinderKrebsInitiative Buchholz/Holm-Seppensen for providing infrastructure.
Financial Support: The study has been funded by the Medical Faculty of the Christian-Albrechts-University, CAU, Kiel, Germany (Forschungsförderung 2015–Anschub to N.C. Hornig) and the German Research Council (Deutsche Forschungsgemeinschaft) (Ho 2073/7-1/7-3 to P.-M.H. and Am 343/2-1/2-3 to O.A.) and SFB1074(B9) to R.S.
Author Contributions: Designing research studies: N.C. Hornig, P.-M.H., O.A.; conducting experiments: N.C. Hornig, P.R., N.C. Hubner, M.W.; acquiring data: N.C. Hornig, P.R., N.C. Hubner; analyzing data: N.C. Hornig, S.B., O.A., R.S., A.E.K., P.-M.H.; providing reagents: H.D., H.-U.S., O.H., R.W., S.G., A.K.E., M.C., A.V.-S., H.G.S; writing the manuscript: N.C. Hornig.
Disclosure Summary: The authors have nothing to disclose.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}