





Abstract
The wheat Qu and Lin-fan yeast starter are two vital components of Huangjiu brewing, the microbiome composition of which can have a major impact on the end product's quality. This investigation aimed to investigate and compare the fungal diversity of eight starters, including four wheat Qus and four Lin-fan yeast starters collected from different manufacturers of Shaoxing Huangjiu using high-throughput sequencing (HTS) technology. Significant variation was observed in the fungal community composition among different starter samples. Fusarium, Epicoccum, Aspergillus and Alternaria were the most predominant fungal genera in wheat Qus. In contrast, Lin-fan yeast starters possessed relatively simple fungal communities and were predominated by Saccharomyces and Saccharomycopsis. LEfSe analysis showed that nine genus-level fungal taxa could be retrieved to differentiate the two starter types. Furthermore, the complicated association among the dominant fungi in traditional starters was revealed by utilising Spearman's correlation-based network analysis. A total of 42 positive and 28 negative correlations were identified respectively. Ultimately, this investigation described the fungal communities in wheat Qus and Lin-fan yeast starters and will aid in screening fermentation strains, which can remarkably enhance the quality of Shaoxing Huangjiu.
Introduction
The history of Huangjiu, commonly known as Chinese rice wine, dates back to >5000 years in China (Yu et al., 2012; Liu et al., 2015). It is known as the national banquet wine (Han & Xu, 2011; Yu et al., 2012) due to its unique aroma, low alcoholicity and rich nutritional ingredients, like oligosaccharides, vitamins, amino acids and peptides (Yongmei et al., 2007; Ren et al., 2019). The Yangtze River Delta, Shandong and Zhejiang produce most Huangjiu, including Hong Qu Glutinous Rice Wine, Shanghai rice wine, Ji Mo Lao Jiu and Shaoxing Huangjiu (Lv et al., 2013; Xu et al., 2015; Song et al., 2019). Of these, Shaoxing Huangjiu is the commonest Huangjiu with regional properties (Niu et al., 2008; Cao et al., 2010).
In most cases, wheat Qu and yeast are used as starting cultures in Shaoxing Huangjiu, which is then produced with steamed glutinous rice by brewing (Niu et al., 2008; Shen et al., 2012). Wheat Qu, comparable to the koji of Japanese sake, is traditionally produced from raw crushed wheat and inoculated naturally with moulds, yeast and bacteria (Que et al., 2006; Xu et al., 2010). Hydrolytic enzymes (including proteases, glucoamylases and amylases) are produced by the microorganisms and used for microbial metabolism and taste creation by breaking down the macromolecules in rice and wheat (Lai et al., 2019; Ren et al., 2019). Accordingly, wheat Qu is an essential ingredient in Huangjiu brewing, greatly enhancing the end-product. Another important component of the Huangjiu fermentation mechanism is the yeast starter, a bacterial and S. cerevisiae combination (Tian et al., 2022). Lin-fan yeast starter is a typical yeast starter type used for traditional Huangjiu production. S. cerevisiae, the predominant yeast in the representative Shaoxing Huangjiu fermentation pathway and originates mainly from the yeast starter, was estimated to be the main responsible for ethanol production (Zhang et al., 2012; Cai et al., 2018). Under appropriate temperature and humidity conditions, the yeast starter and wheat Qu are combined and then the fermentation process begins. Unfortunately, until now, the starters are still made under non-sterile environmental conditions using empirical knowledge, which often leads to the instability and inconsistency of quality between batches of Huangjiu products. Thus, deeply understanding the microflora structures of the yeast starter and wheat Qu and application of defined starter cultures will be helpful to standardise the fermentation process and stabilise and improve the organoleptic properties and quality of Huangjiu.
Different approaches have been used for exploring the microbial diversity of the conventional fermentation starters for Huangjiu, including culture-dependent and independent methods (Cai et al., 2018; Ji et al., 2018; Chen et al., 2021; Yang et al., 2022). Culture-dependent methods can successfully analyse the taxonomy and metabolism of any material to a certain depth (Liu et al., 2016). Due to the difficulties in isolating strains of uncultivable bacteria, these techniques have not been able to accurately portray the whole microflora information. High-throughput sequencing (HTS) technologies, like Illumina sequencing and 454 pyrosequencing, have many benefits over culture-dependent methods and can reveal a wealth of information about microbial diversity in wine starters without the need for strain isolation and cultivation (Fang et al., 2015; Chen et al., 2021). As a culture independent approach, HTS technology has been developed and commonly used to make a thorough analysis of the microbial communities in various fermented food types (del Carmen Portillo & Mas, 2016; Liu et al., 2016, 2017).
In the current research, the fungal structures of the starter cultures containing wheat Qus and Lin-fan yeast starters collected from different manufacturers of Shaoxing Huangjiu were investigated and compared using HTS technology on Illumina HiSeq platform. Furthermore, the LEfSe algorithm was used to determine which fungal species were most characteristic of each kind of starter. Finally, the potential correlations among the dominant fungi in the starters were uncovered through Spearman's correlation analysis.
Materials and methods
Data collection
Eight traditional fermentation starter samples, including four wheat Qus (WQ) and four Lin-fan yeast starters (YS), were obtained from four typical Huangjiu manufacturers (companies K, Z, J and G) in Shaoxing, Zhejiang Province, China in November 2020. Three pieces of each wheat Qu sample were chosen at random from the top, middle and bottom of the storage area, pulverised into powder and mixed as a sample. The Lin-fan yeast starters were sampled after homogeneous mixing, then centrifuged at 4 °C and 8000 r min−1 for 10 min, after which we collected the precipitates. All the samples were refrigerated before additional analysis was done.
DNA extraction
Based on the manufacturer's instructions, total DNAs were extracted from 0.2 g of each powdered wheat Qu sample and each precipitate of the Lin-fan yeast starter sample using a FastDNA® SPIN Kit (MP Biomedicals, California, USA). An agarose gel electrophoresis analysis (1% agarose in 0.5× TBE buffer) and a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, USA) were used to assess the content and quality of the extracted DNA.
Illumina Miseq sequencing of the fungal communities
The primers ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS2 (5′-GCTGCGTTCTTCATCGATGC-3′) were applied for fungal ITS1 regions amplification (Adams et al., 2013). PCR was conducted in 20 μL volume by initial denaturation step at 95 °C for 3 min, 35 cycles of 95 °C for 30 s, 55 °C for 30 s and 72 °C for 45 s, and an extension stage at 72 °C for 10 min. AxyPrep DNA Gel Extraction Kit (Axygen Bioscience Inc, Tewksbury, MA, USA) was used to purify and quantify the amplicons. (Promega Corp., Fitchburg, WI, USA). After that, Shanghai Majorbio Bio-pharm Technology Co., Ltd. sequenced using an Illumina MiSeq PE300 platform. (Shanghai, China).
Bio-informatics analysis
Raw sequencing reads were merged by the FLASH software (version 1.2.11), and quality-filtered using FASTP software (version 0.19.6). The chimeras were distinguished and deleted using uchime denovo methods. Afterwards, the UPARSE (version 7.0.1090) was used to cluster sequences with a 97% sequence similarity into operational taxonomic units (OTUs) (Edgar, 2013). At a confidence level of 70%, all OTUs were taxonomically categorised using a QIIME-based wrapper of RDP-classifier (version 2.11) and annotated using the UNITE (version 8.0) database in conjunction with fungal ITS sequence (Kõljalg et al., 2013). MOTHUR (version 1.30.2) was used to conduct a diversity analysis with the Shannon and Simpson indices, as well as a richness analysis with the ACE and Chao 1 indices, on the alpha diversity indices. The microbial composition was compared across samples using the PCoA and the beta diversity was determined using R software. Heatmap and hierarchical clustering (with full linkage) were used in R software (version 3.3.1) with the ‘vegan’ package to further display the relative abundances of the representative species. In addition, the linear discriminant analysis (LDA) effect size (LEfSe) method was used to determine which fungal species were most indicative of each base ingredient.
Results and discussion
The assessment of fungal diversity in traditional Huangjiu starters
The fungal composition of the traditional Huangjiu starters in the Shaoxing region was investigated by HTS technology on Illumina HiSeq platform. A total of 1 511 560 quality filtered sequencing reads corresponding to the fungal ITS rRNA genes were generated from the four wheat Qus (KWQ, ZWQ, JWQ, GWQ) and four Lin-fan yeast starters (KYS, ZYS, JYS, GYS) of four different Huangjiu manufacturers in Shaoxing (Table 1). Using a similarity threshold of 97%, all sequences were placed into one of 904 OTUs with a size between 65 (GYS) and 182 (JWQ). Good's coverage estimation values were within the range of 99.93%–99.99%, indicating sufficient sequence coverage to accurately characterise the whole fungal communities present in all of the samples (Liu et al., 2018).
Summary of the sequencing results and the alpha diversity indices in the eight starter samples
| Sample | Sequence number | Average length (bp) | OUT number | Shannon | Simpson | Chao1 | ACE | Good's coverage (%) |
|---|---|---|---|---|---|---|---|---|
| KWQ | 214 833 | 678 | 167 | 2.07 | 0.19 | 129.11 | 145.94 | 99.93 |
| KYS | 137 667 | 1283 | 73 | 0.72 | 0.69 | 57.18 | 58.78 | 99.97 |
| ZWQ | 218 350 | 654 | 86 | 2.13 | 0.22 | 50.08 | 55.32 | 99.99 |
| ZYS | 203 400 | 1132 | 100 | 1.44 | 0.38 | 76.66 | 80.97 | 99.96 |
| JWQ | 213 333 | 659 | 182 | 2.23 | 0.17 | 127.57 | 130.7 | 99.95 |
| JYS | 160 354 | 1256 | 87 | 0.69 | 0.73 | 77.75 | 92.1 | 99.95 |
| GWQ | 218 874 | 670 | 144 | 1.57 | 0.35 | 111.63 | 117.39 | 99.95 |
| GYS | 144 749 | 1271 | 65 | 0.88 | 0.56 | 54.07 | 78.06 | 99.96 |
| Sample | Sequence number | Average length (bp) | OUT number | Shannon | Simpson | Chao1 | ACE | Good's coverage (%) |
|---|---|---|---|---|---|---|---|---|
| KWQ | 214 833 | 678 | 167 | 2.07 | 0.19 | 129.11 | 145.94 | 99.93 |
| KYS | 137 667 | 1283 | 73 | 0.72 | 0.69 | 57.18 | 58.78 | 99.97 |
| ZWQ | 218 350 | 654 | 86 | 2.13 | 0.22 | 50.08 | 55.32 | 99.99 |
| ZYS | 203 400 | 1132 | 100 | 1.44 | 0.38 | 76.66 | 80.97 | 99.96 |
| JWQ | 213 333 | 659 | 182 | 2.23 | 0.17 | 127.57 | 130.7 | 99.95 |
| JYS | 160 354 | 1256 | 87 | 0.69 | 0.73 | 77.75 | 92.1 | 99.95 |
| GWQ | 218 874 | 670 | 144 | 1.57 | 0.35 | 111.63 | 117.39 | 99.95 |
| GYS | 144 749 | 1271 | 65 | 0.88 | 0.56 | 54.07 | 78.06 | 99.96 |
Summary of the sequencing results and the alpha diversity indices in the eight starter samples
| Sample | Sequence number | Average length (bp) | OUT number | Shannon | Simpson | Chao1 | ACE | Good's coverage (%) |
|---|---|---|---|---|---|---|---|---|
| KWQ | 214 833 | 678 | 167 | 2.07 | 0.19 | 129.11 | 145.94 | 99.93 |
| KYS | 137 667 | 1283 | 73 | 0.72 | 0.69 | 57.18 | 58.78 | 99.97 |
| ZWQ | 218 350 | 654 | 86 | 2.13 | 0.22 | 50.08 | 55.32 | 99.99 |
| ZYS | 203 400 | 1132 | 100 | 1.44 | 0.38 | 76.66 | 80.97 | 99.96 |
| JWQ | 213 333 | 659 | 182 | 2.23 | 0.17 | 127.57 | 130.7 | 99.95 |
| JYS | 160 354 | 1256 | 87 | 0.69 | 0.73 | 77.75 | 92.1 | 99.95 |
| GWQ | 218 874 | 670 | 144 | 1.57 | 0.35 | 111.63 | 117.39 | 99.95 |
| GYS | 144 749 | 1271 | 65 | 0.88 | 0.56 | 54.07 | 78.06 | 99.96 |
| Sample | Sequence number | Average length (bp) | OUT number | Shannon | Simpson | Chao1 | ACE | Good's coverage (%) |
|---|---|---|---|---|---|---|---|---|
| KWQ | 214 833 | 678 | 167 | 2.07 | 0.19 | 129.11 | 145.94 | 99.93 |
| KYS | 137 667 | 1283 | 73 | 0.72 | 0.69 | 57.18 | 58.78 | 99.97 |
| ZWQ | 218 350 | 654 | 86 | 2.13 | 0.22 | 50.08 | 55.32 | 99.99 |
| ZYS | 203 400 | 1132 | 100 | 1.44 | 0.38 | 76.66 | 80.97 | 99.96 |
| JWQ | 213 333 | 659 | 182 | 2.23 | 0.17 | 127.57 | 130.7 | 99.95 |
| JYS | 160 354 | 1256 | 87 | 0.69 | 0.73 | 77.75 | 92.1 | 99.95 |
| GWQ | 218 874 | 670 | 144 | 1.57 | 0.35 | 111.63 | 117.39 | 99.95 |
| GYS | 144 749 | 1271 | 65 | 0.88 | 0.56 | 54.07 | 78.06 | 99.96 |
The indices of Chao 1, ACE, Simpson and Shannon were diverse among the eight samples, indicating the remarkable variability in the richness and diversity of these starters (Table 1). The sample JWQ had the highest Shannon index and lowest Simpson index, which showed a greater diversity of fungal communities than other samples. Besides, the sample KWQ had the richest fungal genera abundance based on the Chao 1 and ACE indices. Regarding these observations, discrimination involving the geographical origins and manufacturing environment as well as technological factors of different manufacturers may explain the diversity and richness variation in these starters (Zhou et al., 2022), but it should be confirmed through a much wider sampling in further studies. In addition, the diversity of fungal community in wheat Qus (JWQ, ZWQ, KWQ and GWQ) appeared to be higher than those in Lin-fan yeast starters (ZYS, GYS, KYS and JYS). Lin-fan yeast starter is manufactured by fermentation on the steamed rice using the yeast starters which being rich in mainly S. cerevisiae and bacteria (Tian et al., 2022). On the other hand, traditional wheat Qu is naturally produced using the raw crushed wheat under non-sterile fermentation conditions (Zhang et al., 2019). In consequence, a wide variety of microbes originating from both indigenously present in the wheat and the local processing environments can be presented in wheat Qu (Zhang et al., 2012; Yang et al., 2022), which may result in its greater fungal diversity than Lin-fan yeast starter.
The fungal communities of traditional Huangjiu starters
The fungal OTU sequences were annotated into variable taxa from the Unite Database. The taxonomic information of fungi at the phylum and genus level is presented graphically in Fig. 1. As shown in Fig. 1a, three fungal phyla were found in all eight starter samples and had a relative abundance over 1%, including Ascomycota, Basidiomycota and Mucoromycota. Among these, Ascomycota represented the largest fraction in the starter samples, which accounted for 79.68% (JWQ) to 98.66% (KYS) of the total fungal sequences, illustrating its predominance in both wheat Qus and Lin-fan yeast starters. The findings were in line with the previous studies of Yang et al. (2022) and Zhang et al. (2022). However, genus-level findings demonstrated a significant variation of the dominant fungal genus among the traditional starters (Fig. 1b). In wheat Qu samples, there were 17 genera with more than 1% of relative abundance, including Fusarium, Epicoccum, Aspergillus, Alternaria, Rhizomucor, Trichomonascus, Candida, Rhizopus, Cutaneotrichosporon, Cladosporium, Cryptococcus, Filobasidium, Wickerhamomyces, Phaeosphaeriopsis, Hyphopichia, Wallemia and Botrytis. These microorganisms comprised from 91.11% (ZWQ) to 98% (GWQ) of total fungi. The most predominant fungal genera that detected in all four samples of wheat Qus were Fusarium, Epicoccum and Alternaria. This result was comparable with previous studies on traditional wheat Qus performed by Yang et al. (2022) and Ji et al. (2018). Aspergillus, which had been demonstrated as a predominant fungal genus in wheat Qus according to recent reports (Zhang et al., 2019; Yang et al., 2022), had high relative abundances across the wheat Qu samples GWQ (11.95%), KWQ (23.78%) and JWQ (23.94%). As the filamentous fungi in wheat Qus, the genus Aspergillus can synthesise and secrete a broad range of different enzymes, which could make a great contribution to the starch saccharification in wine mash (Zheng et al., 2011; Liu et al., 2018). Candida, a non-Saccharomyces yeast, had a high relative abundance in ZWQ (12.77%). This genus, mainly originates from the workers' hands (Zhang et al., 2021), can remarkably cause alcohol content regulation and rice wines aromatic quality improvement by co-fermenting with Saccharomyces (Englezos et al., 2016; Yang et al., 2022). The results of our studies indicated that moulds were predominant of the fungal microorganisms in wheat Qus. Traditional wheat Qu has a loose structure and relatively high internal oxygen content. Therefore, this might be conducive to forming and proliferating filamentous fungal spores (Zhang et al., 2019).
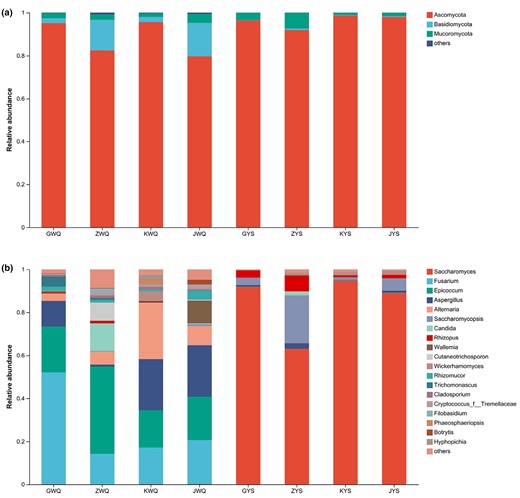
The fungal community abundance of the traditional wheat Qus (GWQ, ZWQ, KWQ and JWQ) and Lin-fan yeast starters (GYS, ZYS, KYS and JYS) at the phylum (a) and genus (b) level.
In contrast, Lin-fan yeast starters possessed relatively simple fungal communities. As presented in Fig. 1b, the relative abundances of 6 fungal genera were higher than 1% in the four samples of Lin-fan yeast starters, including Saccharomyces, Saccharomycopsis, Rhizopus, Aspergillus, Candida and Wickerhamomyces. These microorganisms comprised from 97.75% (ZYS) to 99.34% (GYS) of total fungi. The genera that dominated in all the four Lin-fan yeast starters were Saccharomyces and Saccharomycopsis. Clearly, the dominant fungi of wheat Qu and Lin-fan yeast starter were significantly different, and in particular, Saccharomyces strongly dominated the microflora of Lin-fan yeast starters (from 63.02% (ZYS) to 94.77% (KYS)). Various genus Saccharomyces members have been found important for food fermentation, particularly S. cerevisiae, the major active yeast species and ethanol producer during the fermentation of Huangjiu (Tian et al., 2022), and other traditional fermented foods (Liu et al., 2018; Zhou et al., 2022). In addition, Zhang et al. (2012) reported that S. cerevisiae was the commonest yeast in the representative Shaoxing Huangjiu fermentation process. It is likely that this species' competitive growth in the presence of fermentable carbohydrates and tolerance to ethanol makes it the species of choice in alcoholic fermentation procedures (Zhou et al., 2022). The yeast Saccharomyces populations skyrocket throughout the brewing process, as does the alcohol content (Huang et al., 2019). In this investigation, the genus Saccharomycopsis was also observed as predominant in the samples of Lin-fan yeast starters. Saccharomycopsis fibuligera, the species within the genus of Saccharomycopsis, was identified in all Lin-fan yeast starter samples with more than 1% of relative abundance. As a non-Saccharomyces yeast, this species can degrade native starch into dextrin, maltose and glucose through the production of various enzymes, including acid protease, β-glucosidase, glucoamylase and α-amylase (Chi et al., 2009; Li et al., 2018). Therefore, S. fibuligera has been reported as the main amylolytic yeast in fermenting food (Nie et al., 2013; Jiang et al., 2019). According to recent reports, an increasing number of studies indicated the contributing impact of the non-Saccharomyces yeasts on the flavour and taste of wine (Nuñez-Guerrero et al., 2016). Nonetheless, the work and function mechanisms in non-Saccharomyces yeasts and interaction between non-Saccharomyces yeasts and S. cerevisiae during the fermentation processes of Huangjiu are still barely understood (Huang et al., 2019). Rhizopus, which was detected across all the Lin-fan yeast starter samples and had a relative abundance of more than 1% in GYS, ZYS and JYS, has a strong ability to produce amylase and is commonly presented in various types of amylolytic fermentation starters for rice wine (Dung et al., 2007; Liu et al., 2018). Moreover, this genus can also greatly contribute to improving the flavour of rice wine through the generation of volatile compounds such as ethanol, 3-methylbutanol and 2-methylpropanol (Christen et al., 2000; Abedinifar et al., 2009; Cai et al., 2018).
Fungal community comparisons of different starters
The OTUs shared among the different starter samples were estimated. As shown in Fig. 2, 22, 6, 28 and 29 unique OTUs were obtained, respectively, from the wheat Qu samples of GWQ, ZWQ, KWQ and JWQ, while 3, 7, 3 and 7 OTUs were, respectively, acquired from the samples GYS, ZYS, KYS and JYS of Lin-fan yeast starters. Twenty-five OTUs were common to all the samples, which accounted for 8.22% of total OTUs. The common OTUs mainly belonged to Saccharomycopsis (39.38%), Fusarium (13.79%) and Epicoccum (12.96%) at the genus level.
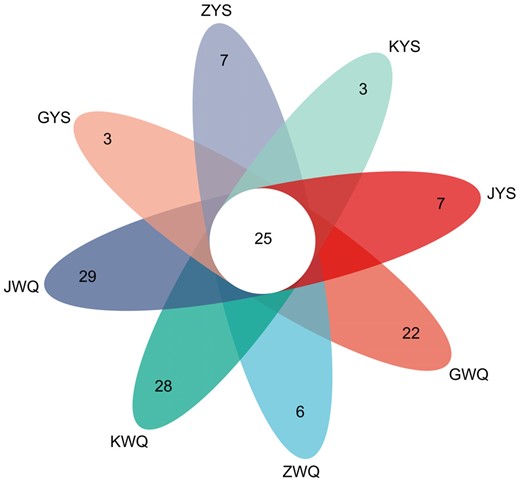
Venn diagrams of different traditional starter samples according to fungal communities, with the evaluation of the shared OTUs.
According to the relative abundance of the top 50 abundant fungal genera across all samples, heatmap plots and hierarchical clustering were further performed to outline the differences and similarities in the composition of the fungi of the traditional starters (Fig. 3). The classification of the starter samples mainly included two clear separated groups. The wheat Qus clustered together, meanwhile, the samples of Lin-fan yeast starters clustered independently. In terms of wheat Qus, there were differences in fungal genera and quantities among the samples of ZWQ, GWQ, JWQ and KWQ. Moreover, the sample ZWQ had a more distinctive fungal flora and was less similar to the samples of GWQ, JWQ and KWQ. On the other hand, the Lin-fan yeast starter samples KYS, JYS and GYS were gathered and distinguished from ZYS, indicating that these three samples had similar fungal structures. In order to provide a visual comparison of the fungal communities among the eight starter samples, principal coordinates analysis (PCoA) was then employed, based on the information of total OUTs' relative abundance (Fig. 4). PC1 and PC2, the principal coordinate components, explained 72.86% and 8.74% of the variation in the fungal community respectively. As shown in Fig. 4, the samples of Lin-fan yeast starters (GYS, ZYS, KYS and JYS) exhibited a homologous cluster and were obviously separated from the wheat Qus, which revealed relatively low similarities in fungal community between wheat Qus and Lin-fan yeast starters. In addition, the close position among JWQ, KWQ and GWQ showed their analogous principal fungal composition, whereas the group of ZWQ samples was generally separated in the PCoA plot. The PCoA score plot results were consistent with those of hierarchical clustering analysis. It has been reported that the geographical origin has a considerable influence on the fungal flora of wheat Qus (Ji et al., 2018; Chen et al., 2021). In the present study, however, significant differences were also found among the wheat Qu samples that were all collected from Shaoxing, Zhejiang. As a starter culture in the process of Huangjiu brewing, the traditional wheat Qu was produced from raw wheat in an open work environment (Xie et al., 2007; Zhang et al., 2012). Therefore, the manufacturing environments such as the indoor and outdoor ground surfaces, the surfaces of tools and the air, and the workers' hands (Zhou et al., 2022), as well as the technological factors and ingredients, might be the reason that resulted in the variations of fungal communities among the wheat Qus in our study.
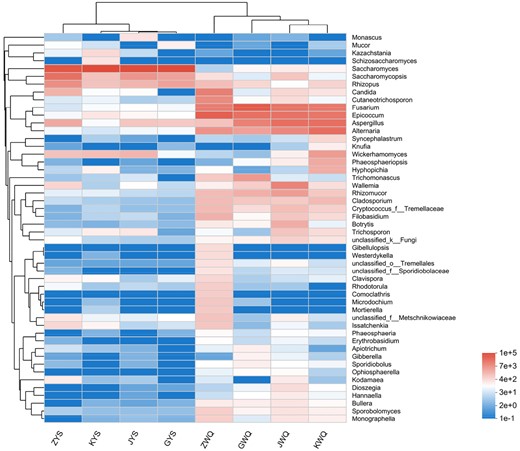
Heatmap comparison and hierarchical clustering dendrogram of the top 50 abundant fungal genera found within some traditional starters. Intensity of colour reflects the abundance of different fungus species.
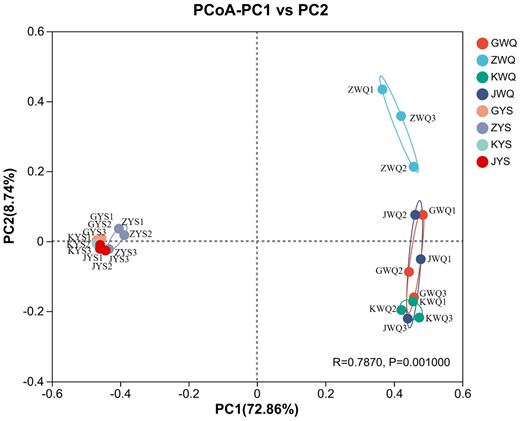
Principal coordinate analysis (PCoA) of fungal communities among some starter samples based on fungal ITS gene OTUs relative abundance.
LEfSe analysis of the fungal taxa in different types of starters
To investigate the influence of each component's abundance on the differential impact, LEfSe was performed through linear discriminant analysis (LDA) with a threshold of 3 (Fig. 5). In comparison of the fungal communities in wheat Qus and Lin-fan yeast starters, nine genus-level fungal taxa were verified as being differentially abundant in these two particular types of starters, indicating the most representative microflora of wheat Qus or Lin-fan yeast starters in this study. Of these, eight taxa were enriched in wheat Qus, including Epicoccum, Fusarium, Alternaria, unclassified_o_Tremellales, Filobasidium, Cryptococcus, Botrytis and Cladosporium (Fig. 5b). Genus Epicoccum had the highest LDA score of 5.11, followed by Fusarium with a LDA of 5.09 and Alternaria with a LDA of 4.76. On the other hand, only genus Saccharomyces was enriched in Lin-fan yeast starters, with a LDA score of 5.63.
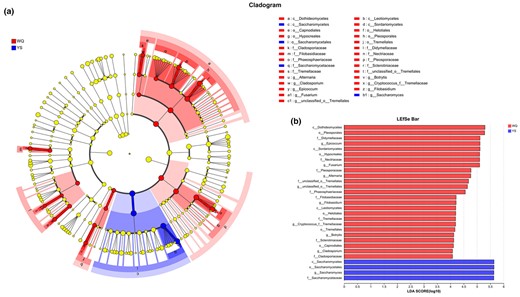
LEfSe taxonomic cladogram (a) and bar (b) of the fungal taxa of wheat Qus and Lin-fan yeast starters.
Correlation analysis among the different microbes of traditional starters
Correlation-based network analysis was explored to analyse the interaction among the dominant fungi at the genus level of the traditional starters using the Spearman correlation test (¦r¦ > 7 with P < 0.05). As shown in Fig. 6, the network led to the identification of 70 edges, including 42 positive and 28 negative correlations, which revealed that the relationships among the dominant fungi were complicated. Alternaria, Rhizomucor, Cladosporium and Fusarium were regarded as positive hubs with the largest number of 11 positive links to other genera. Rhizomucor, which reported to generate organic acids such as succinic acid and oxalic acid thus improving the flavour of Huangjiu (Millati et al., 2005; Ji et al., 2018), positively correlated with Aspergillus, Epicoccum, Cryptococcus, Fusarium, Cladosporium, Alternaria, Botrytis, Cutaneotrichosporon, Wallemia, Phaeosphaeriopsis and Filobasidium. Fusarium exhibited positive correlations with Rhizomucor, Aspergillus, Epicoccum, Cryptococcus, Cladosporium, Botrytis, Alternaria, Filobasidium, Phaeosphaeriopsis, Trichomonascus and Cutaneotrichosporon. This genus was also considered the predominant fungi in Hong Qu (Liu et al., 2018; Huang et al., 2019). On the contrary, Saccharomyces showed significant negative correlations with 12 fungal genera and was considered as the negative hubs, indicating that this genus possessed the ability to inhibit the growth of most fungi in the traditional starters. Saccharomyces plays a great role in efficiently metabolising sugars to alcohol (Thanh & Tuan, 2008), which may lead to the inhibition of other microorganisms with alcohol intolerance. The result was consistent with those of Huang et al. (2019) and Liu et al. (2018). These fungal hubs are still the dynamic stability of the microecosystem of traditional starters by interspecific interactions. Therefore, the microbial interaction was also a considerable factor that influencing the microbial structure of traditional starters, and should be deeply understood in further studies.
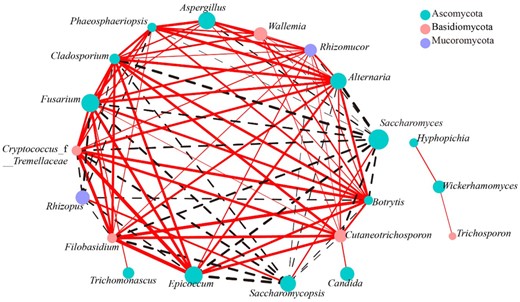
Correlation network analysis applied to the top 20 abundant fungal genera present in the traditional starters. The density of a certain genus determines the corresponding node's size. The phylum taxonomic categorisation is shown as the colour of the nodes. A solid red line indicates a positive association, whereas a dotted black line indicates a negative one. In addition, the edge thickness indicates the strength of the correlation.
Conclusions
This investigation described the fungal communities in four wheat Qus and four Lin-fan yeast starters used for manufacturing Huangjiu collected from different manufacturers in the Shaoxing region using HTS technology. The predominant phyla of the eight starters were Ascomycota, followed by Basidiomycota and Mucoromycota. Differentiation was found in the fungal community composition of wheat Qus and Lin-fan yeast starters, as well as the samples of the same type. Eight and one genus-level fungal taxa were found abundant within the microflora of wheat Qus and Lin-fan yeast starters, respectively, by LEfSe analysis. In addition, the interrelationship among the dominant fungi in traditional starters was also explored using Spearman's correlation analysis. Results contribute to a greater understanding of the microbial diversity of the traditional starters. It could also be helpful in screening fermentation strains and enhancing Shaoxing Huangjiu quality.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (No. 31870013) and Project funded by China Shaoxing Huangjiu Group Co., Ltd (08220102106).
Author contributions
Huanyi Yang: Formal analysis (equal); writing – original draft (equal); writing – review and editing (equal). Xiangwei Kong: Data curation (equal); methodology (equal). Guochang Sun: Resources (equal). Zhenna Du: Formal analysis (equal); investigation (equal). Zukang Fu: Resources (equal). Shengnan Shao: Data curation (equal); methodology (equal). Chi Shen: Data curation (equal); validation (equal). Wei Zang: Validation (equal). Jianqiu Sun: Conceptualization (equal); supervision (equal); validation (equal). Jiandi Zhou: Resources (equal); supervision (equal).
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
Ethical approval was not required for this research.
Data availability statement
The data that support the findings of this study are available from the corresponding author on reasonable request.
References
This study presented the dominant filamentous fungi in wheat Qu and the fermentation mashes of Chinese rice wine. The research provided background information for the discussion of the current study.
This study elucidated the microbial communities of ten traditional wheat Qus collected from different Chinese rice wine manufacturers in Shaoxing region, and presented evidence that supports the results of the current study.
This review details the manufacturing process, microbiotas, flavour, enzymes, and metabolites of wheat Qu. The reference provides a theoretical basis for our research.
This study analysed the fungal diversity of Chinese traditional Baijiu daqu made at three temperatures through high-throughput sequencing on Illumina MiSeq platform. The methodology used and the approach of bioinformatics analysis were similar to the current study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}