





Abstract
The presence of an abnormal amount of single-stranded DNA in the bacterial cell constitutes a genotoxic alarm signal that induces the SOS response, a broad regulatory network found in most bacterial species to address DNA damage. The aim of this review was to point out that beyond being a repair process, SOS induction leads to a very strong but transient response to genotoxic stress, during which bacteria can rearrange and mutate their genome, induce several phenotypic changes through differential regulation of genes, and sometimes acquire characteristics that potentiate bacterial survival and adaptation to changing environments. We review here the causes and consequences of SOS induction, but also how this response can be modulated under various circumstances and how it is connected to the network of other important stress responses. In the first section, we review articles describing the induction of the SOS response at the molecular level. The second section discusses consequences of this induction in terms of DNA repair, changes in the genome and gene expression, and sharing of genomic information, with their effects on the bacteria's life and evolution. The third section is about the fine tuning of this response to fit with the bacteria's ‘needs’. Finally, we discuss recent findings linking the SOS response to other stress responses. Under these perspectives, SOS can be perceived as a powerful bacterial strategy against aggressions.
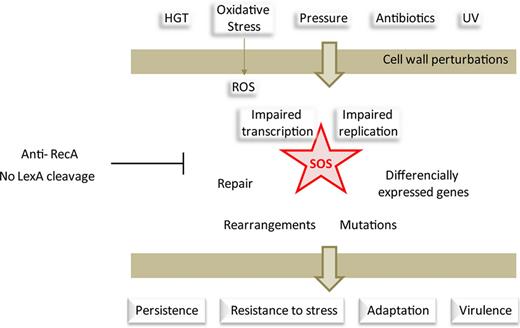
Beyond being a repair process, SOS induction leads to a very strong but transient response to genotoxic stress, which potentiates bacterial survival and adaptation to changing environments.
Introduction
SOS induction was first observed in cells where the replication fork encounters DNA lesions which it tries to replicate (Radman, 1975). Since these early investigations, a multitude of different studies have been carried out to decipher the triggers and components of the SOS response in a number of bacteria. We know now that the ultimate trigger is the formation of single-strand DNA (ssDNA), and many mechanisms other than those linked to the replication of DNA lesions have been characterized, through which ssDNA is formed in the cell and induce SOS.
The mechanism of induction of SOS by single-stranded DNA (ssDNA) is well understood (Walker, 1996; Michel, 2005; Kelley, 2006). Briefly, RecA is recruited on ssDNA by presynaptic complexes RecBCD or RecFOR. RecBCD recognizes double-strand DNA breaks (DSB) or double-strand ends (DSE). Its helicase and nuclease activities result in the formation of an ssDNA substrate for RecA. RecFOR recognizes DNA nicks and gaps, and recruits RecA to this ssDNA patch. RecA binds ssDNA in the form of a nucleofilament that catalyzes the auto-proteolysis of the repressor LexA. LexA represses the genes belonging to the SOS regulon by binding its cognate LexA box sequence on their promoters. LexA proteolysis thus leads to derepression of this regulon, comprised of about 40 genes in E. coli (Courcelle et al., 2001). The number and the type of genes found in the regulon vary extensively among bacteria. For instance, in the Gram-positive model bacterium Bacillus subtilis, the LexA regulon comprises 33 genes among which only eight are homologous counterparts of E. coli SOS genes (Au et al., 2005). In Pseudomonas aeruginosa, a Gammaproteobacterium distantly related to E. coli, the SOS regulon is limited to 15 genes and key repair genes such as ruvAB, uvrA, and uvrB escape LexA regulation (Cirz et al., 2006). The affinity of LexA for these genes is variable and depends on the LexA box sequence itself and on the number of LexA boxes present at the promoters of these genes. The LexA box consensus sequence was proposed to be 5′-CTGTN8ACAG-3′ (Butala et al., 2009) and can be extended to 5′-TACTGT(AT)4ACAGTA-3′ in E. coli (Fernandez De Henestrosa et al., 2000; Wade et al., 2005). LexA binding seems to be tolerant for variations in the sequence of eight base spacer region, but its length is invariant, as suggested by structural studies where the LexA–DNA complex was crystallized (Zhang et al., 2010). This leads to differential derepression (Courcelle et al., 2001)/re-repression of genes belonging to the SOS regulon as shown in vivo and in silico using an algorithm-based approach by Ronen et al. (2002). On the other hand, the LexA box consensus sequence varies among bacterial species (cyanobacteria, Gram-positives, Alphaproteobacteria, Gammaproteobacteria). The phylogeny of LexA boxes and the ability of LexA proteins from various bacterial species to bind various LexA boxes was studied in vitro and in silico (Mazon et al., 2004a,b) and shows that these LexA box sequences are related and in most cases, follow the phylogenic relationships of the cognate bacterial species. Examples of different LexA boxes are given in Table 1 – the list is nonexhaustive, but gives insight in the similarity of these sequences in different bacterial species.
LexA box sequences identified across different bacterial species
| Organism | LexA box consensus 5′-3′ | Method of determination | References |
| Gram-negatives | |||
| Beta- and Gammaproteobacteria | CTGTN8ACAG | In silico | Erill et al. (2003) |
| E. coli | TACTGTATATATATACAGTA | In vitro, in vivo | Fernandez De Henestrosa et al. (2000) and Wade et al. (2005) |
| Alphaproteobacteria | |||
| Rhodobacter sphaeroides | GTTCN7GTTC | In vitro | Fernandez de Henestrosa et al. (1998) |
| Rhizobium and Agrobacterium | GAACN7GTAC | In vitro, in vivo | Tapias & Barbe (1998) |
| Cyanobacteria | RGTACN3DGTWCB | In vitro | Mazon et al. (2004a,b) |
| Spirocheate | |||
| Leptospira interrogans | TTTGN5CAAA | In vitro | Fonseca et al. (2013) |
| Acidobacteria | |||
| Acidobacterium capsulatum | GTTCN7GTTC | In vitro, in vivo | Mazon et al. (2006) |
| Gram-positives | GAACN4GTTC | ||
| Firmicutes | |||
| Bacillus subtilis | CGAACRNRYGTTYC | In vitro, in silico | Winterling et al. (1998) |
| Chloroflexi | |||
| Dehalococcoides ethenogenes | GAACN4GTTC | In vitro | Fernandez de Henestrosa et al. (2002) |
| Actinobacteria | |||
| Mycobacterium | TCGAACN4GTTCGA | In vivo | Davis et al. (2002) |
| Deinococcus | |||
| Deinococcus radiodurans | CGAACRNRYGTTCG | Algorithm | Khan et al. (2008) |
| Integrons | ACTGTW8ACAGT | In silico | Cambray et al. (2011) |
| Organism | LexA box consensus 5′-3′ | Method of determination | References |
| Gram-negatives | |||
| Beta- and Gammaproteobacteria | CTGTN8ACAG | In silico | Erill et al. (2003) |
| E. coli | TACTGTATATATATACAGTA | In vitro, in vivo | Fernandez De Henestrosa et al. (2000) and Wade et al. (2005) |
| Alphaproteobacteria | |||
| Rhodobacter sphaeroides | GTTCN7GTTC | In vitro | Fernandez de Henestrosa et al. (1998) |
| Rhizobium and Agrobacterium | GAACN7GTAC | In vitro, in vivo | Tapias & Barbe (1998) |
| Cyanobacteria | RGTACN3DGTWCB | In vitro | Mazon et al. (2004a,b) |
| Spirocheate | |||
| Leptospira interrogans | TTTGN5CAAA | In vitro | Fonseca et al. (2013) |
| Acidobacteria | |||
| Acidobacterium capsulatum | GTTCN7GTTC | In vitro, in vivo | Mazon et al. (2006) |
| Gram-positives | GAACN4GTTC | ||
| Firmicutes | |||
| Bacillus subtilis | CGAACRNRYGTTYC | In vitro, in silico | Winterling et al. (1998) |
| Chloroflexi | |||
| Dehalococcoides ethenogenes | GAACN4GTTC | In vitro | Fernandez de Henestrosa et al. (2002) |
| Actinobacteria | |||
| Mycobacterium | TCGAACN4GTTCGA | In vivo | Davis et al. (2002) |
| Deinococcus | |||
| Deinococcus radiodurans | CGAACRNRYGTTCG | Algorithm | Khan et al. (2008) |
| Integrons | ACTGTW8ACAGT | In silico | Cambray et al. (2011) |
Even though the two LexA paralogues do not appear to play a role in the radioresistance of D. radiodurans (Narumi et al., 2001; Jolivet et al., 2006), several studies point to a role of RecA recruitment at DNA lesions and intracellular LexA levels (Satoh et al., 2006, 2012). Algorithm-based studies identified a conserved sequence related to the Bacillus subtilis LexA box upstream of the known SOS-regulated genes (Khan et al., 2008). Y: C or T; R: A or G; W: A or T; D: not C; B: not A; N: any base.
LexA box sequences identified across different bacterial species
| Organism | LexA box consensus 5′-3′ | Method of determination | References |
| Gram-negatives | |||
| Beta- and Gammaproteobacteria | CTGTN8ACAG | In silico | Erill et al. (2003) |
| E. coli | TACTGTATATATATACAGTA | In vitro, in vivo | Fernandez De Henestrosa et al. (2000) and Wade et al. (2005) |
| Alphaproteobacteria | |||
| Rhodobacter sphaeroides | GTTCN7GTTC | In vitro | Fernandez de Henestrosa et al. (1998) |
| Rhizobium and Agrobacterium | GAACN7GTAC | In vitro, in vivo | Tapias & Barbe (1998) |
| Cyanobacteria | RGTACN3DGTWCB | In vitro | Mazon et al. (2004a,b) |
| Spirocheate | |||
| Leptospira interrogans | TTTGN5CAAA | In vitro | Fonseca et al. (2013) |
| Acidobacteria | |||
| Acidobacterium capsulatum | GTTCN7GTTC | In vitro, in vivo | Mazon et al. (2006) |
| Gram-positives | GAACN4GTTC | ||
| Firmicutes | |||
| Bacillus subtilis | CGAACRNRYGTTYC | In vitro, in silico | Winterling et al. (1998) |
| Chloroflexi | |||
| Dehalococcoides ethenogenes | GAACN4GTTC | In vitro | Fernandez de Henestrosa et al. (2002) |
| Actinobacteria | |||
| Mycobacterium | TCGAACN4GTTCGA | In vivo | Davis et al. (2002) |
| Deinococcus | |||
| Deinococcus radiodurans | CGAACRNRYGTTCG | Algorithm | Khan et al. (2008) |
| Integrons | ACTGTW8ACAGT | In silico | Cambray et al. (2011) |
| Organism | LexA box consensus 5′-3′ | Method of determination | References |
| Gram-negatives | |||
| Beta- and Gammaproteobacteria | CTGTN8ACAG | In silico | Erill et al. (2003) |
| E. coli | TACTGTATATATATACAGTA | In vitro, in vivo | Fernandez De Henestrosa et al. (2000) and Wade et al. (2005) |
| Alphaproteobacteria | |||
| Rhodobacter sphaeroides | GTTCN7GTTC | In vitro | Fernandez de Henestrosa et al. (1998) |
| Rhizobium and Agrobacterium | GAACN7GTAC | In vitro, in vivo | Tapias & Barbe (1998) |
| Cyanobacteria | RGTACN3DGTWCB | In vitro | Mazon et al. (2004a,b) |
| Spirocheate | |||
| Leptospira interrogans | TTTGN5CAAA | In vitro | Fonseca et al. (2013) |
| Acidobacteria | |||
| Acidobacterium capsulatum | GTTCN7GTTC | In vitro, in vivo | Mazon et al. (2006) |
| Gram-positives | GAACN4GTTC | ||
| Firmicutes | |||
| Bacillus subtilis | CGAACRNRYGTTYC | In vitro, in silico | Winterling et al. (1998) |
| Chloroflexi | |||
| Dehalococcoides ethenogenes | GAACN4GTTC | In vitro | Fernandez de Henestrosa et al. (2002) |
| Actinobacteria | |||
| Mycobacterium | TCGAACN4GTTCGA | In vivo | Davis et al. (2002) |
| Deinococcus | |||
| Deinococcus radiodurans | CGAACRNRYGTTCG | Algorithm | Khan et al. (2008) |
| Integrons | ACTGTW8ACAGT | In silico | Cambray et al. (2011) |
Even though the two LexA paralogues do not appear to play a role in the radioresistance of D. radiodurans (Narumi et al., 2001; Jolivet et al., 2006), several studies point to a role of RecA recruitment at DNA lesions and intracellular LexA levels (Satoh et al., 2006, 2012). Algorithm-based studies identified a conserved sequence related to the Bacillus subtilis LexA box upstream of the known SOS-regulated genes (Khan et al., 2008). Y: C or T; R: A or G; W: A or T; D: not C; B: not A; N: any base.
The SOS-induced genes favor the repair of DNA lesions. Figure 1 shows simplified representations of the three main DNA repair pathways induced by SOS in E. coli and many other bacteria: homologous recombination, nucleotide excision repair, and translesion synthesis. In these three pathways, RecA nucleofilaments are central for the induction of the SOS response. However, in the homologous recombination (HR) pathway, RecA is also involved in the recruitment of the other homologous recombination proteins such as RecBCD and RecFOR, which allow the repair of single-stranded lesions. In addition to local repair, induction of HR can lead to rearrangements in the chromosome through recombination between similar sequences (see section ).
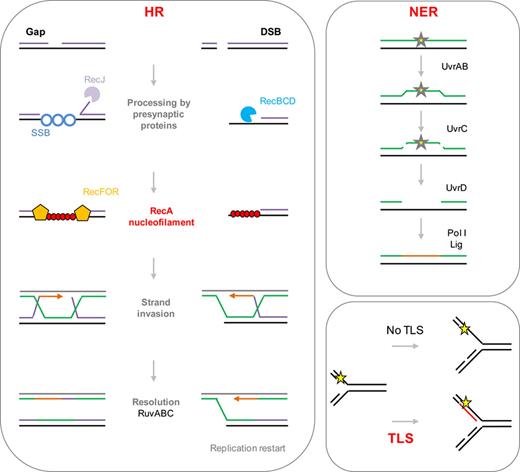
Simplified representations of DNA repair pathways induced by SOS (described in the introduction section). HR, NER, and TLS are induced during the SOS response. HR: homologous recombination. Single-strand nicks (gaps) are transformed into larger gaps by the RecJ exonuclease. Gaps are recognized by RecFOR presynaptic proteins. Double-strand breaks (DSB) are recognized by the presynaptic RecBCD exonuclease/helicase complex. RecFOR/RecBCD recruit RecA to initiate homologous recombination through strand invasion with the sister chromosome, usually resulting in mutation-free repair. DSB repair results in the formation of a replication fork. NER: nucleotide excision repair. The UvrAB complex recognizes the DNA lesion; UvrC proceeds with a double incision on both sides of the lesion and the ssDNA carrying the lesion is removed by the UvrD helicase. DNA polymerase I (Pol I) resynthesizes the missing DNA and the ligase ligates the newly synthesized DNA to the template, usually resulting in mutation-free repair. If the SOS-inducing signal persists, translesion synthesis (TLS) polymerases are induced. In the absence of TLS, the replicative DNA polymerase (Pol III) cannot replicate damaged DNA, leading to fork stalling. TLS polymerases (Pol IV, Pol V) can replicate damaged DNA in a mutagenic manner. Yellow stars represent DNA lesions.
The second pathway, nucleotide excision repair (NER), is driven by UvrABC (Sancar and Rupp, 1983) and allows the repair of lesions where the DNA is double stranded (like a mismatch). Briefly, UvrABC endonuclease recognizes the lesion and nicks the DNA, and UvrD helicase removes the DNA patch carrying the lesion (Kumura et al., 1985). DNA polymerase Pol I then fills the gap (Husain et al., 1985).
Translesion synthesis (TLS) can be performed by different specific DNA polymerases, PolV (encoded by umuCD), PolII (polB), and PolIV (dinB). Similarly to LexA, UmuD undergoes proteolytic cleavage catalyzed by the RecA nucleofilament (Nohmi et al., 1988). The active UmuD' forms the translesion synthesis DNA polymerase Pol V in complex with UmuC (UmuD'2C). PolV proceeds with DNA replication on damaged DNA by incorporating any base across from the DNA lesion that the proofreading proficient Pol III cannot replicate. As PolV, the other TLS polymerases allow the replication of damaged DNA in a mutagenic manner (Napolitano et al., 2000; Pages & Fuchs, 2003). TLS can be highly mutagenic as these polymerases can incorporate a correct or incorrect base across from the lesion on the template strand (Friedberg et al., 2002). Unlike Pol III, TLS polymerases do not have a proofreading activity, except for Pol II that has a high level of fidelity on undamaged DNA. Thus, induction of TLS leads to an increased frequency of spontaneous mutations (see section ).
LexA is a late-induced gene, and can stop the SOS induction when the genotoxic signal disappears and LexA cleavage is not favored anymore. Ongoing biochemical and sequence analysis studies aim at shedding light into the dynamics of SOS induction through the study of RecA–LexA interactions (Kovacic et al., 2013) or biochemical and structural bases of auto-proteolysis and damage sensing (Butala et al., 2009; Aravind et al., 2013). In an in vitro report about the timing of ssDNA-RecA-mediated self-cleavage of LexA, Butala et al. (2011) showed that a LexA repressor which is bound to its target lexA box is insensitive to auto-proteolysis. This observation explains how the binding affinity of LexA for different promoters correlates with the timing of the gene's expression.
SOS induction is a well-described cellular response. A comprehensive review about the mechanisms of SOS induction and the phylogenetic conservation of this cellular stress response was published in 2007 by Erill and collaborators (Erill et al., 2007). Comparison of the SOS regulon among the various bacterial species and genera allowed the description of a LexA-regulated core regulon comprising few genes (recA, uvrA, ruvAB and recN), which could correspond to the ancestral core set (Erill et al., 2007). However, the description of a mutagenic cassette originally characterized in Mycobacterium tuberculosis (Davis et al., 2002), but later found to be present in a wide range of bacterial species, has complexified the evolutionary scheme of the regulon. Indeed, this mutagenic cassette, which consists in three genes encoding a DNA polII α error-prone subunit and its two auxiliary proteins (Warner et al., 2010), is in most cases associated with a second copy of the lexA gene. The distribution of this gene set has been found to be broader than that of the core regulon gene set described above, and it has been proposed to be the original core set of the SOS regulon (Erill et al., 2007).
In light of subsequent studies, more knowledge has been accumulated on the conditions leading to the induction of the SOS response and its outcomes, such as virulence, persistence, emergence of multiple resistances, and, more generally, adaptation. We focus in this review on new data uncovering the SOS-related strategies developed by bacteria in response to various aggressions.
Origin of SOS-inducing single-stranded DNA (ssDNA)
Origin of ssDNA in the absence of external damaging agents
Most ssDNA originates from double-strand breaks (DSBs). As described in the introduction, ssDNA is the sole inducer of the SOS response. Experiments measuring the formation of DNA lesions capable of inducing SOS in E. coli using flow cytometry led to the observation that most of the chromosomal ssDNA (two-thirds according to the authors) comes from the action of RecBCD on DSBs. The remaining third are ssDNA stretches formed independently of RecBCD (Pennington & Rosenberg, 2007). Such DSBs mostly originate from spontaneous DNA breakage during replication, most probably after fork stalling (Fig. 2a). Several conditions leading to replication arrest are described below.
![Origins of ssDNA-inducing SOS. (a) Replication fork stalling often leads to spontaneous ssDNA nicks. These breaks are eventually transformed into double-strand breaks (DSBs) when replication restarts, and DSBs are potent inducers of SOS as described in section Origin of SOS-inducing single stranded DNA (ssDNA). (b) Replication–transcription collisions. Adapted from (Helmrich et al., 2013). Codirectional collisions may happen because of the difference of velocity of the replisome and the transcription complex, and lead to DSB formation (Dutta et al., 2011). Head-on collisions occur when transcription and replication progress in opposite directions. Both types of collisions cause DSB formation through either R-loop formation or topological stress, if rescue pathways are insufficient. (c) Transcription stalling. Adapted from (Wimberly et al., 2013). Elongating RNA polymerase (RNAP) can stall upon encounter with a DNA lesion or bulky protein complexes. In this case, transcribed RNA can anneal to the template DNA forming a structure called R-loop. R-loops are another type of SOS inducers as described in section Origin of SOS-inducing single stranded DNA (ssDNA). R-loops can lead to re-priming of a replication fork. If the re-primed replication fork encounters a nick, it can stall and lead to DSBs [as in (a)].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femsre/38/6/10.1111_1574-6976.12077/1/m_fmr12077-fig-0002-m.jpeg?Expires=1749871196&Signature=dBBA97f1vNphOsvKOiBXlMZI1MJsweL-PeZyfN3M-6KeYqX9Psp30ssXeDwunWq8yMGOZbv6Ol3gN4f6CAB4MtS-fO~9iDpc-NqOkSQzAjm6JVB7vPaRx9hHyaB1VOvlBEUBXGCNTJtJW1HWnCLoplr7xEyZyEGb2aPx9VFBsR55o3zDNuMFPw0872ZOuUkJx3JOQlUz9j8Uo9goH1YJREr2Z9aqRzGOHnt3FvGtdknLxSXICNzXUfN9YX7RjjufVXO2GexzmFyHZyWUVSEnxvu9RfsgdtEVkxUL-SdsN6vWdATVSZtnfzqX2toDhKLKzr97dsHNUnGBJqtctqkcvw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Origins of ssDNA-inducing SOS. (a) Replication fork stalling often leads to spontaneous ssDNA nicks. These breaks are eventually transformed into double-strand breaks (DSBs) when replication restarts, and DSBs are potent inducers of SOS as described in section Origin of SOS-inducing single stranded DNA (ssDNA). (b) Replication–transcription collisions. Adapted from (Helmrich et al., 2013). Codirectional collisions may happen because of the difference of velocity of the replisome and the transcription complex, and lead to DSB formation (Dutta et al., 2011). Head-on collisions occur when transcription and replication progress in opposite directions. Both types of collisions cause DSB formation through either R-loop formation or topological stress, if rescue pathways are insufficient. (c) Transcription stalling. Adapted from (Wimberly et al., 2013). Elongating RNA polymerase (RNAP) can stall upon encounter with a DNA lesion or bulky protein complexes. In this case, transcribed RNA can anneal to the template DNA forming a structure called R-loop. R-loops are another type of SOS inducers as described in section Origin of SOS-inducing single stranded DNA (ssDNA). R-loops can lead to re-priming of a replication fork. If the re-primed replication fork encounters a nick, it can stall and lead to DSBs [as in (a)].
Replication fork problems can lead to DSB formation. It is known that replication arrest or stalling leads to formation of ssDNA on the lagging strand template, which constitutes a substrate for the formation of the RecA nucleofilament and SOS induction in the absence of DNA lesions (Walker, 1996). Such an event can happen when replication is impaired, as is the case for E. coli conditional replication mutants harboring thermo-sensitive alleles of genes coding for different subunits of the replicative DNA polymerase Pol III (Flores et al., 2005; Lestini & Michel, 2007). Figure 2a shows a synthetic representation of how ssDNA that induces SOS can be formed during replication. Replication arrests may also occur upon transcription replication collisions (Fig. 2b), such as in conditions where bulky factors block the DNA polymerase complex. Such an example is described in an E. coli strain where the highly transcribed ribosomal rrn operons were inverted to be transcribed in the opposite direction of replication (Boubakri et al., 2010). In this case, head-on collisions of the replisome with the bulky transcription complexes hinder the progression of the replication fork and lead to SOS induction. In the absence of stalled RNA polymerase (RNAP) removal systems, the DNA replication is impaired and SOS is induced (Boubakri et al., 2010; McGlynn et al., 2012).
R-loop formation in nonreplicating bacteria also leads to DSB formation (Fig. 2c). The conditions described above involve replication fork stalling, which implies active replication of the bacterial chromosome. However, DSBs are also formed in nonreplicating conditions. The origin of these DSBs was found to lie in R-loop formation during transcription in E. coli (Wimberly et al., 2013). R-loops are RNA–DNA hybrids that are formed when the RNAP is stalled on DNA and the RNA in synthesis anneals to the homologous transcribed ssDNA template in E. coli (reviewed in McGlynn et al., 2012) and eukaryotes (Helmrich et al., 2011). This can happen, for example, when the RNA polymerase encounters a roadblock and backtracks (Dutta et al., 2011). In growing cells, this can lead to replication–transcription conflicts as described above (for a review, see Merrikh et al., 2012). R-loops were shown to lead to genomic instability in E. coli, as in eukaryotes (Gan et al., 2011), by impairing DNA replication.
Formation of such R-loop structures was very recently shown to also lead to stress-induced mutations in nonreplicating E. coli (Wimberly et al., 2013), suggesting that problems at transcribed regions leading to RNAP stalling can constitute a genotoxic signal.
ssDNA formation due to external damaging agents
DNA damage can be formed upon physical insults, and ssDNA can also originate from DNA damage provoked by external agents. For instance, UV irradiation causes DNA lesions that need to be repaired in order for the cell to survive. The repair of damaged DNA by HR or TLS requires the initial recruitment of RecA to the site of lesion (Fujii et al., 2006). Hence, inactivating the recruitment of RecA, for instance in a mutant deleted for recA, leads to cell death because of lack of repair and possibly because of inefficient TLS in the absence of SOS induction. As mentioned in the introduction, SOS induction leads to the expression of TLS polymerases; hence, the repair of UV lesions is highly mutagenic.
Another damaging factor described by Aertsen and colleagues is pressure. High pressure indeed induces SOS through DSB formation through the Mrr nuclease (Aertsen & Michiels, 2005), in E. coli strains carrying the mrr gene (Aertsen et al., 2008). In turn, SOS can induce high-pressure resistance through spontaneous mutations inactivating the mrr gene (Aertsen & Michiels, 2005). What is remarkable in this example is how the induction of the SOS response is a subtle way for the cell to turn an external aggression into an advantage by becoming better adapted to its environment.
Another study on the effect of gamma radiation on E. coli reports a link between the SOS response and radiation-induced cell death, a bacterial mechanism similar to the eukaryotic programmed cell death (Wadhawan et al., 2013). Moreover, SOS-dependent DNA repair by HR and TLS was found to be necessary for cell recovery after gamma irradiation.
Reactive oxygen species (ROS) can also lead to DSB formation. Recently, reactive oxygen species and their various effects on cellular mechanisms have drawn much attention. ROS-like superoxide or hydroxyl radicals (OH−) are potent bacterial stressors, which can directly damage DNA. For instance, superoxide leads to the release of free iron ions from proteins containing iron (Fe-S cluster proteins; Keyer & Imlay, 1996; Imlay, 2003). Iron can also localize along the phosphodiester backbone of nucleic acids. Moreover, high concentrations of free ferrous ion lead to the production of hydroxyl radicals (OH−) through the Fenton reaction (Henle et al., 1999; Rai et al., 2001; Daly, 2009). OH− attacks the DNA backbone and ultimately causes DSBs (Jena, 2012). Moreover, superoxide and OH− are also formed after poisoning of the DNA gyrase (by fluoroquinolones or protein inhibitors like the CcdB toxin), an essential protein for chromosome replication (Dwyer et al., 2007).
Another type of DNA damage caused by oxidative attack is the incorporation of oxidized guanine residues during replication (7,8-Dihydro-8-oxo-guanine or 8-oxo-G; Sussenbach & Berends, 1964). Bacteria possess a defense system against such damage, first by hydrolyzing the oxidized guanine present in the nucleotide pool and second, by affecting the incorporated G through the base excision repair pathway (BER; Lu et al., 2001), by limiting its incorporation and the formation of mismatches (Sobol, 2012). Incomplete action of the BER system possibly leads to DSBs that are cytotoxic if unrepaired (Foti et al., 2012).
ROS can also indirectly cause DNA damage (Farr & Kogoma, 1991). OH− radicals can damage proteins (Ide et al., 1985) and lipids (Schaich & Borg, 1988) as shown in E. coli and Salmonella enterica (Paez et al., 2011; also reviewed in Dwyer et al., 2009). Additionally, it is known that mistranslated and misfolded proteins are more susceptible to oxidation (Dukan et al., 2000). Mistranslational corruption of proteins may lead to replication fork collapse and induction of the SOS response in E. coli (Balashov & Humayun, 2002; Al Mamun et al., 2006). Interestingly, in Deinococcus radiodurans, cell death by radiation is not caused by direct DNA damage but primarily by oxidative damage of proteins, which eventually results in the abolition of DNA repair (Krisko & Radman, 2010). The legendary resistance of D. radiodurans to ionizing radiations is actually a consequence of a more efficient proteome protection against ROS rather than a more efficient genome protection, and cell death correlates with protein carbonylation due to oxidative damage. Preserving cell integrity in times of oxidation seems to depend as much on proteome protection against ROS then on DNA protection. Furthermore, affecting the integrity of the proteome can result in the induction of the SOS response through impaired DNA replication and repair.
Antibiotics can trigger bacterial stress at both lethal and sublethal concentrations. Antibiotics may induce SOS through DNA damage or through replication arrest. The fluoroquinolone (FQ) family of antibiotics which causes replication arrest by blocking DNA gyrase (Pohlhaus & Kreuzer, 2005), and trimethoprim (Lewin & Amyes, 1991) which affects purine and pyrimidine synthesis do not directly generate lesions but rather perturb DNA replication. Mitomycin C (MMC) leads to T-T cross-links that lead to DNA break formation. β-lactams were also reported to induce SOS in some conditions by an unusual mechanism. In E. coli, β-lactams cause cell wall damage, which induces the two-component system proteins DpiBA. DpiA can bind the origin of replication of the chromosome and, together with DpiB, impair replication, which results in the induction of the SOS response (Miller et al., 2003, 2004).
Interestingly, SOS is also induced by low doses of several antibiotics, termed sub-MIC for subminimal inhibitory concentration. The evidence that sub-MICs of antibiotics play an important role in the appearance and dissemination of antibiotic resistance has emerged in recent years (Kohanski et al., 2010a; Gullberg et al., 2011). This is especially relevant considering that a large proportion of ingested antibiotics are released intact in the environment (Liu et al., 1999a,b) and that low levels of antibiotics are found in wastewater plants, hospitals, and soil (Fick et al., 2009; Haggard & Bartsch, 2009; Kummerer, 2009a,b).
First, sub-MICs FQs have been shown to induce SOS in Salmonella enterica, even if bacterial growth is not affected by these low concentrations (Yim et al., 2011). Ciprofloxacin at sub-MIC induces SOS and increases the frequency of point mutations in S. aureus (Mesak et al., 2008; Mesak & Davies, 2009). Moreover, ampicillin at sub-MIC was found to downregulate mismatch repair in E. coli, P. aeruginosa, and Vibrio cholerae, hence increasing mutation frequencies due to SOS (Gutierrez et al., 2013). This brings along a different problem than SOS induction by the same antibiotics at high concentrations: in fact, at high concentration, bacteria either acquire mutations that help them to survive or die, the latter possibility being the most likely. However, at low antibiotic concentrations, bacteria do not die and SOS induction leads to an increase in bacterial genome variability. Mutations acquired in such a way can then be fixed in populations if they provide a fitness advantage. More and more reports now suggest that these low concentrations of antibiotics and the consequent induction of SOS are one of the factors contributing to the acquisition of multiple resistances and adaptation factors by bacteria.
Moreover, the effects of sub-MICs of antibiotics from different families – such as antibiotics that target translation and not DNA (aminoglycosides, chloramphenicol, and tetracycline) – on the SOS response were further addressed. These antibiotics are not expected to induce SOS. Interestingly, V. cholerae, but not E. coli, was found to induce SOS in response to these antibiotics, which favors the appearance of resistant derivatives (Baharoglu et al., 2010a). Moreover, aminoglycoside (AG)-mediated SOS induction is conserved among distantly related Gram-negative pathogens (Klebsiella pneumoniae and Photorhabdus luminescens), suggesting that E. coli is more of an exception than a paradigm for the physiological response to sub-MICs of antibiotics (Baharoglu et al., 2013). It is also interesting to mention here that AGs block translation. In fact, translating ribosomes control the transcription elongation rate: active ribosomes can prevent RNAP from backtracking (Proshkin et al., 2010; Potenski & Klein, 2011). Consequently, inhibiting protein synthesis increases levels of transcription-generated R-loops. Formation of transcriptional R-loops in bacteria is indeed shown to be inhibited by ribosomes on the nascent transcript (Masse & Drolet, 1999). As described in section , R-loops have the potential to induce SOS. Aminoglycosides target protein translation, but they clearly also have a negative effect on transcription and replication at sub-MIC, which leads to SOS induction in several bacterial species.
It can sound puzzling that sub-MICs of AGs induce SOS in several bacteria for they do not directly target DNA synthesis or the DNA molecule. These observations point to a role for intermediate factors that cause DNA damage. One of the reasons for this SOS induction was actually found to be that sub-MIC AG treatment leads to the formation of ROS (Baharoglu et al., 2013, 2014). Further studies suggest a role for RNA polymerase stalling at AG-mediated DNA lesions and formation of R-loops with the consequences on SOS as described in the previous paragraph (Baharoglu et al., 2014).
Several points are striking in these latter findings. First, low concentrations of antibiotics that do not affect cell growth – even those that do not target DNA or its replication – have the power of inducing the bacterial SOS response and as such increasing mutation frequencies of bacteria (Baharoglu & Mazel, 2011). Second, formation of ROS is the factor leading to the actual DNA damage, as discussed above. Finally, and once again, a tight connection between transcription blocks and triggering of the SOS response is seen. Induction of SOS by low levels of AGs is important for growth under these conditions. This aspect of SOS induction needs further work to be fully understood.
ssDNA formation during ssDNA uptake
Transformation is a mechanism of HGT that relies on ssDNA uptake and processing (Dubnau, 1999). Transformation occurs when a bacterial cell reaches a competent state rendering it capable of taking up DNA present in its environment and, in some cases, of integrating the acquired DNA in its genome by recombination (Claverys et al., 2009). It was observed long ago in Bacillus subtilis that when lysogenic strains were rendered competent, the induction of a prophage led to the reduction in the frequency of transformation (Yasbin et al., 1975; McVeigh & Yasbin, 1996). This was not due to reduced transformation efficiency but rather to the induction of a prophage in the recipient cells leading to the lysis of the cell and thus a drop in the number of viable transformants. Prophage induction is in many cases SOS dependent. It is likely that the reduced transformation efficiency of lysogenic cells compared to nonlysogenic cells is the result of induction of the SOS response by the ssDNA acquired during transformation. Furthermore, it was shown in naturally competent V. cholerae that ssDNA uptake during competence induces the SOS response (Baharoglu et al., 2012).
In addition to transformation, conjugation is another HGT mechanism where plasmid or chromosomal DNA from a donor strain enters the recipient cell in a single-stranded fashion. Like transformation, conjugation also induces SOS in E. coli and V. cholerae (Baharoglu et al., 2010a).
Consequences of SOS induction
Repair
As mentioned earlier, the SOS response triggers homologous recombination, required especially for the repair of double-strand ends/breaks (DSB/DSE). A recent study in E. coli proposed that instead of a genome-wide search for homology, genotoxic stress-induced, and RecA-dependent condensation of sister chromosomes, which are otherwise segregated at cell poles, allow repair of DSBs (Shechter et al., 2013). Such genome packaging is also observed in E. coli in response to oxidative stress (Ko et al., 2012). Moreover, the importance of RecN was recently discovered for the repair of DSBs after treatment with MMC, a DNA-damaging agent that leads to DSB formation (Keyamura et al., 2013) and induction of the SOS response. RecN is an SMC (structural maintenance of the chromosome) family protein induced by SOS, underlining the significance of the SOS response in the repair of lethal chromosomal DSBs.
Moreover, the importance of SOS induction in the case of replication–transcription conflicts described above is notable. Indeed, in E. coli strains in which the transcription of rrn operons leads to replication arrest after stalling of the RNAP, preventing induction of the SOS response results in a decrease in fitness (or even absence of growth; Boubakri et al., 2010). RNAP stalling can be an obstacle to the progression of the replication fork and needs to be dislodged (McGlynn et al., 2012). Intermediate DNA structures, R-loops, capable of inducing SOS are formed at the stalled RNA polymerase. By inducing expression of genes coding for helicases such as DinG, SOS also allows the processing and removal of bulky protein complexes that prevent DNA polymerase action (Baharoglu et al., 2010b). Additionally, DinG was proposed to inhibit the formation and accumulation of R-loops in vivo in E. coli cells (Boubakri et al., 2010; De Septenville et al., 2012).
Recent data also show that transcription stalling is involved in SOS induction in response to DNA damage by sub-MIC AGs in V. cholerae (Baharoglu et al., 2014), with a role for the RNAP-binding protein Mfd. In fact, the helicase Mfd can recognizes R-loops at sites where the RNAP is stalled on damaged DNA and releases the transcription complex (Roberts & Park, 2004; Deaconescu et al., 2006; Savery, 2007). Interestingly, Mfd is also required for the repair of DSBs caused by MMC in Helicobacter pylori, where an mfd mutant is susceptible to antibiotics and has a DNA repair defect (Lee et al., 2009). Mfd is involved in increased mutation frequencies after FQ treatment in Campylobacter jejuni as well (Han et al., 2008). All of these observations point to Mfd as a possible partner of the SOS response. There is also evidence that the NusA protein, a component of the elongating RNA polymerase, is involved in stress-induced mutagenesis by interacting with the TLS polymerase Pol IV in E. coli (Cohen & Walker, 2010). This is once again evidence highlighting an undeniable connection between transcription and induction of the SOS response.
Change: outcome of SOS induction in terms of genome plasticity and gene expression
As described in the introduction, SOS induction increases the levels of proteins involved in HR. Hence, one of the consequences of this SOS induction is to increase intrachromosomal recombination of homologous DNA sequences after DSB formation. This was observed for instance in E. coli through facilitated reconstitution of a functional lacZ gene from the fragments lacZ' and ‘lacZ flanked by homologous regions in conditions inducing SOS (after treatment with sub-MIC of FQ; Lopez & Blazquez, 2009). Interestingly, it was found in V. cholerae that homologous recombination plays an unsuspected role in chromosome fusions. Vibrio cholerae has two circular chromosomes with different origins of replication. Under genetic conditions leading simultaneously to impaired replication from the origin of the second chromosome and to induction of the SOS response, it was found that homologous recombination proceeds to the fusion of the two chromosomes through homologous IS sequences present in both chromosomes, giving rise to changes in gene dosage (Val et al., 2014). In fact, the closer a gene is to the origin of replication, the more its copy number in a given cell increases due to multiple firings of replication. When two chromosomes are fused, only one origin is active; thus, the gene dosage is subject to change. This suggests that in the wild-type strain, such fusions can happen even if they are not easily detected and that SOS induction can favor their occurrence, which can influence gene expression and cell physiology in general.
Interestingly, DSB formation and subsequent SOS induction can also constitute an advantage in harsh conditions: it was shown in pathogenic Pseudomonas aeruginosa biofilm communities that ROS-mediated formation of DSBs was beneficial to bacteria in the presence of antibiotics as they allowed genome rearrangements and resistance (Boles & Singh, 2008).
In addition to HR, SOS leads to increased mutation frequencies through TLS. Induction of SOS leads to a high state of mutability in the bacterial cells, for example after antibiotic treatment in E. coli (Ysern et al., 1990). This allows them to increase their chances of generating mutations that will permit them to survive under the stress conditions they are submitted to. Indeed, point mutations can result in antibiotic resistance acquisition. For instance, resistance to ciprofloxacin (FQ) and to rifampicin is due to mutations caused during the induction of the SOS response through the action of the error-prone polymerases IV/V (Cirz & Romesberg, 2006). Sub-MICs of FQs were clearly shown to cause the development of resistance in S. aureus (Didier et al., 2011) and S. enterica (Hughes & Andersson, 2012). The increased mutation frequency in the presence of FQs was proposed to lead to the overload of MutS-dependent mismatch repair, causing the accumulation of unrepaired DNA and the appearance of point mutations that lead to antibiotic resistance.
A recent study achieved mathematical modeling of the link between DNA damage after UV irradiation, SOS induction and mutation frequency by measuring the quantity of the TLS polymerase UmuD'2C (Pol V) and corresponding mutation frequencies in E. coli (Ni et al., 2008). The authors conclude that there is a close correlation between UmuD'2C levels and mutation frequency (for a review on UV-mediated DNA damage and repair, see Rastogi et al., 2010). DSB-dependent stress-induced mutagenesis described in starving E. coli also requires the SOS response and DinB (Pol IV; Shee et al., 2011). Interestingly, even in mismatch repair-deficient hyper-mutator strains, inactivation of the SOS response leads to reduced development of antibiotic resistant bacteria (Cirz & Romesberg, 2006), showing the importance of SOS induction in the appearance of point mutations.
It was found that SOS induction mediated by sub-MIC antibiotics is also coupled with an increased frequencies of spontaneous mutation in E. coli and V. cholerae (the appearance of rifampicin resistance increased from 10−9 to around 10−8; Baharoglu & Mazel, 2011). Such a modest increase in mutation frequency is nonetheless of high importance as it was shown to influence the evolution of multidrug resistance in E. coli (Denamur et al., 2005). Strains characterized by low or high mutation rates actually have a lower resistance to antibiotics than strains that have an intermediate rate of mutation (around 10−8), regardless of the antibiotic tested (Denamur et al., 2005). In fact, high mutation frequencies probably more often lead to deleterious mutations, demonstrating that SOS induction levels can be subtly fine-tuned to achieve adapted levels of mutability.
SOS induction in response to HGT also leads to genome rearrangements. As mentioned in section , during conjugation and transformation, plasmid or chromosomal DNA enters the recipient cell in a single-stranded fashion. The fact that HGT (i.e. incoming DNA) induces SOS in the recipient is not trivial: by doing so, incoming DNA may induce its own integration in the recipient bacterium's genome, highlighting once more the role of the SOS response as an initiator of genome plasticity and in the acquisition of adaptation factors. Interestingly, even narrow host-range conjugative plasmids which cannot replicate or be maintained in the recipient bacteria induce SOS upon entry into the cell (Baharoglu et al., 2010a). This means that even though the incoming plasmid is not maintained and the incoming DNA is eventually degraded, conjugation still leaves an imprint in the recipient by inducing SOS-dependent genome plasticity such as point mutations or integron rearrangements, as discussed in the following paragraph.
As depicted above, SOS is a stress response inducing the expression of recombination and repair genes. But other genes are also part of the SOS regulon and induced following genotoxic stress. For example, apart from inducing mutation frequency, another way for the SOS response to confer antibiotic resistance and adaptive responses is through increasing the expression of resistance and adaptation genes. It was described for instance that SOS induction after quinolone treatment could in turn induce the expression of plasmid-borne quinolone resistance determinants in Enterobacteria (Da Re et al., 2009). Resistance to quinolones is here triggered by the presence of quinolones themselves, at sub-inhibitory concentrations, or by other antibiotics that induce the SOS response. This latter point is important. It means that bacteria not only become resistant to the factor triggering the genotoxic stress, but also modulate their gene expression and modify their genome (spontaneous mutations, genome rearrangements) so that they become able to resist to other stresses when they come along. In this way, SOS is a powerful strategy of bacteria to turn external aggressions in their favor. SOS is also involved in other bacterial processes such as persistence, virulence, and biofilm formation.
Persistence is a nonhereditary and reversible state. Persisters are antibiotic-tolerant cells that are not killed during treatment and resume growth when antibiotics are removed (for a review Lewis, 2010). Dorr et al. showed that E. coli persisters are not preexisting dormant cells but that their formation is rather induced by the SOS response after FQ treatment (Dorr et al., 2009). Interestingly, the appearance of persister cells was shown to be much higher during treatment with a sub-MIC of FQ than when higher antibiotic concentrations were used (stronger SOS induction). SOS induction was thus necessary, but a low level of induction was found to be more favorable to the development of persistence. Interestingly, in a report about the dynamics of SOS induction, Butala et al. (2011) observed that the dissociation rate of LexA from the promoters it regulates – which correlates with the levels of SOS induction – directly influences the formation of E. coli persisters.
SOS-induced dormancy was shown to be due to TisB toxin induction by SOS (Dorr et al., 2010). Indeed, persister cell formation can occur through the induction of toxins from the toxin–antitoxin family, such as TisB from the SOS regulon, which decreases the growth rate (drop of ATP levels, no active peptidoglycan synthesis, no ribosome formation, no replication), causing tolerance to multiple antibiotics. Interestingly, toxin–antitoxin modules are found in many bacterial genomes (Pandey & Gerdes, 2005; Leplae et al., 2011), and TisB may not be the only toxin that leads to persistence. Therefore, the use of SOS-inducing antibiotics at sub-MICs may lead to persistence and eventually contribute to the development of multidrug resistance. In fact, other examples of SOS-related TA modules exist, like the E. coli YafNO system which is induced by SOS (Singletary et al., 2009), or CcdAB from the F plasmid which induces SOS through poisoning of the gyrase (Karoui et al., 1983; Bernard & Couturier, 1992).
In addition to resistance to stress and adaptation, SOS induction also triggers virulence in some bacteria. Indeed, SOS can directly regulate virulence factors: for example, LexA regulates the expression of the V. cholerae prophage CTX genes leading to production of the cholerae toxin (Quinones et al., 2005; Kimsey & Waldor, 2009). SOS induced in response to sub-MIC of antibiotics also triggers the production of the shiga-toxin (Stx) in pathogenic E. coli (Nassar et al., 2013). Furthermore, SOS induces horizontal dissemination of virulence factors present in pathogenicity islands in S. aureus (Ubeda et al., 2005), which can happen for instance in response to antibiotic treatment (Maiques et al., 2006).
Biofilm formation and quorum sensing can also be linked to the SOS response and DNA damage in some cases. For instance, in V. harveyi, induction of bioluminescence was shown to be important for repair of DNA damage caused by UV irradiation (Czyz et al., 2000). Bioluminescence is regulated by quorum sensing, and Czyz et al. found that UV irradiation induces bioluminescence. In subsequent studies, the authors showed that while bioluminescence has a fitness cost on growing V. harveyi, it confers a selective advantage under conditions of DNA damage (caused by low doses of UV irradiation; Czyz et al., 2003). Although these studies do not directly demonstrate a link between the SOS response and quorum sensing in the repair of damaged DNA, together with data suggesting that quorum sensing is involved in SOS induction by sub-MIC of antibiotics (Baharoglu & Mazel, 2011), such a link seems plausible. Quorum sensing also regulates biofilm formation in V. cholerae (Hammer & Bassler, 2003; Zhu & Mekalanos, 2003). On the basis that replication-inhibiting antibiotics induce biofilm formation in various bacterial species (Hoffman et al., 2005; Kaplan, 2011), Gotoh et al. (2010) tested and found that the SOS response is involved in biofilm formation by P. aeruginosa. Moreover, taking into account reports showing that natural conjugation in E. coli (Ghigo, 2001) and extracellular DNA uptake in Staphylococcus epidermidis, which induce SOS, also induce biofilm formation (Kaplan et al., 2011) and that the pathways regulating biofilm formation and quorum sensing are intertwined, it is appealing to address in the future the possible links between quorum sensing, biofilm formation, and the SOS response.
It was mentioned earlier how SOS can be used by bacteria to turn external aggressions in their favor. The examples given were that of resistance to high pressure or to antibiotics conferred by mutations acquired after SOS induction. In the same line of thought, increased resistance to one antibiotic can lead to the development of resistance to other classes of antibiotics through SOS-mediated integron rearrangements. Hocquet et al. (2012) show that metronidazole treatment of a patient infected with P. aeroginosa induces the SOS response and integron recombination leading to β-lactam and ceftazidime resistance. Moreover, in their recent study, they report the action of metronidazole on antibiotic resistance acquisition in P. aeruginosa. They show that metronidazole, which is used against anaerobic bacteria and not against P. aeruginosa, triggers nevertheless the SOS stress response in this bacterium. In turn, SOS can trigger the appearance of resistances to multiple antibiotics in P. aeruginosa. The authors clearly show the induction of SOS by metronidazole and correlate this with increased resistance to aminoglycosides and fluoroquinolones in vitro (Hocquet & Bertrand, 2014).
An integron is a genetic platform of promoterless open reading frames called gene cassettes, separated by recombination sites. The integron integrase, which encodes a site-specific recombinase, catalyzes excision and integration of these cassettes in a single-stranded circular form (for a review, see (Mazel, 2006). Other than integron rearrangements, the integrase also allows the capture of exogenous circular promoterless gene cassettes. Gene cassettes placed at the first positions of the integron can be expressed from a constitutive promoter (Levesque et al., 1994; Jove et al., 2010). Importantly, all known integron integrases are regulated by the SOS response (Guerin et al., 2009; Cambray et al., 2011), which highlights the integron as an evolutionary tool containing a pool of unexpressed genes able to be recombined and expressed when needed, that is in response to stress. The SOS control of integrase expression not only means that numerous rearrangements may take place within the chromosomal integrons when they are present, but also that plasmid-borne integrases are likely to be induced when their host's SOS response is triggered. To date, hundreds of cassettes encoding resistance to antibiotics have been characterized in integrons (Partridge et al., 2009). The induction of the integrase by SOS also explains how resistance and adaptation genes can be recruited in such structures during stressful growth, and shows the power of this structure coupled to SOS in the defense of bacteria harboring integrons.
Share
We saw earlier that HGT induces SOS. In turn, SOS induces HGT and consequently induces the transfer of genetic information, as it was mentioned for the dissemination of virulence determinants (sections , ). It was found for instance that SOS induction triggers the transfer of integrating conjugative elements (ICEs) from a donor to a recipient cell. An example of this is the transfer of the V. cholerae ICE SXT, in which the two main ICE-encoded transcriptional activators SetC and SetD are derepressed when the host's SOS response is induced (Beaber et al., 2004). It is thought that the ICE-encoded repressor, SetR, undergoes autoproteolysis in the presence of the ssDNA-RecA nucleofilament similarly to LexA. ICEs such as SXT are self-transmissible bacterial mobile elements that play a major role in the dissemination of antibiotic resistance genes in bacterial populations. They transfer by conjugation in a process similar to that of many conjugative plasmids, and their transfer was shown to induce the SOS response in recipients to the same extent as was observed for conjugative plasmids (Baharoglu et al., 2010a). In those terms, the SOS response is a mean for bacteria not only to survive or change, but also to share information with neighboring cells.
Fine tuning of SOS induction: attaining balance
Bacteria can temper their SOS induction
Not all ssDNA induces SOS. In an original recent study, it was observed in growing E. coli using immunofluorescence microscopy that not all ssDNA formed after antibiotic treatment induces SOS (Kohiyama et al., 2013). This means that bacteria have a way of insuring genome stability by neutralizing chromosomal ssDNA formed after fluoroquinolone treatment, preventing SOS induction as long as its induction is not required.
Mechanisms actively preventing RecA nucleofilament formation on ssDNA exist. One of them is through the action of RecX. The recX gene is located downstream of recA in the E. coli chromosome and was proposed by Fuchs and colleagues to regulate RecA (Pages et al., 2003). RecX actually possesses an affinity for RecA and inhibits both its recombinase and coprotease functions without interacting with ssDNA or double-stranded DNA (Stohl et al., 2003). RecX was found to destabilize ssDNA–RecA nucleofilaments in log-phase cells in a genetic context where RecA was over expressed (Massoni et al., 2012). Another protein that turns off the SOS responses through inhibition of the RecA coprotease is DinI (Yasuda et al., 1998). A higher mutation frequency was observed in an E. coli strain deleted for the dinI gene after treatment with the SOS-inducing agent MMC. In vitro studies suggest that, conversely to RecX, DinI does not prevent RecA nucleofilament assembly but stabilizes it, thus ‘hiding’ ssDNA (Lusetti et al., 2004). DinI and RecX are themselves under the control of the SOS response, affect the stability of the RecA filament, and may thus participate in regulating the SOS response. Another protein able to antagonize RecA is UvrD, which is also induced by SOS and involved in the last steps of NER (Lestini & Michel, 2007; Centore et al., 2008). Furthermore, when DinB (Pol IV) is over produced, the active TLS polymerase Pol IV can impede replication fork progression in such a way that fork arrest does not lead to additional SOS induction (Mori et al., 2012).
Finally, ‘Trashing’, which consists in a sequestration of genotoxic ssDNA observed in vivo in E. coli cells, may represent a new facet of the regulation of the SOS response (Kohiyama et al., 2013). In this case, ssDNA is somewhat ‘hidden’ from RecA and no induction of the SOS response takes place. Bacteria have thus developed strategies to attenuate SOS to decrease fitness cost and more importantly to mutate less to survive.
Decreasing the fitness cost of SOS induction
Apart from the evolution of bacterial resistance described in section , moderate SOS induction is also involved in the conservation of already existent multiple resistances by the bacteria through the reduction of the fitness cost. For instance, bacteria that show slightly increased frequencies of mutation and harbor antibiotic resistances are found in greater proportions in the commensal flora of cystic fibrosis patients subjected to prolonged antibiotic treatment (Gustafsson et al., 2003). A recent report even proposes that upon acquisition of resistance (to a β-lactam in this case), E. coli can reorganize its entire metabolic network to reduce the fitness costs associated with the acquisition of this resistance (Handel et al., 2013). The authors observed there that the SOS response itself was downregulated in response to the acquisition of the resistance. SOS induction thus has a fitness cost better to avoid if the induction is unnecessary.
It also was mentioned in section that the entry of conjugative plasmids into a recipient cell induces the SOS response. The exceptions to this are the plasmids carrying the psiB (protein for SOS inhibition) gene. In fact, some narrow host-range conjugative plasmids harbor this gene, which is transferred early during conjugation and which inhibits induction of the SOS response in the recipient cell (Bagdasarian et al., 1992), probably by binding to RecA (Petrova et al., 2009). The advantage of this could be to avoid inducing a stress response in the host: as narrow host-range plasmid can replicate only in a small number of hosts, they may have evolved to prevent unnecessary mutations and fitness costs.
Attenuate SOS induction to mutate less and survive
In several reports, intermediate levels of SOS induction (for instance in response to sub-MICs of antibiotics) were found to be a potent strategy of bacteria to become persistent (see section ) or acquire spontaneous mutations that allow them to survive (see section ). It was also observed in E. coli that the mismatch repair system tends to attenuate the appearance of mutations during UV-induced SOS by removing those introduced by the UmuD'2C (PolV) translesion polymerase (Belov et al., 2013).
We can speculate that bacteria use the SOS response and its mutagenic influence as a ‘last resort’, avoiding its induction when other mechanisms are sufficient to repair the damaged DNA (Delmas & Matic, 2005).
SOS and other stress responses
We described how SOS is linked with transcription-related insults to DNA. SOS is actually connected with various stress responses, highlighting that responses to these various stresses are integrated into a larger network ensuring survival of the bacterial cells.
Cell wall stress
The most straightforward example of a connection between cell wall stress and SOS is the effect of β-lactams on bacteria: β-lactams induce the SOS response and prophage induction in S. aureus (Maiques et al., 2006) and E. coli (Wadhawan et al., 2013). β-lactams actually induce the SOS response by triggering a cell wall stress through the DpiAB two-component system (Miller et al., 2004).
In the absence of β-lactams, SOS induction by cell wall stress through the DpiAB system can also be observed by inactivating the genes coding for components of the cell membrane (such as penicillin binding proteins or PBP; Miller et al., 2004). Furthermore, the RpoE envelope stress response sigma factor of E. coli is also required for the increase in frequency of stress-induced mutations through formation of DSBs (Gibson et al., 2010). All of these observations point to a link between cell wall stress and SOS induction.
One consequence of this is that SOS, in conjunction with the DpiAB system, confers β-lactam resistance by transiently halting cell division (Miller et al., 2004). Once again, we observe that antibiotics trigger the bacterial stress response, which in turns allows the bacterial cell to resist to this external stress. According to recent reports, the resistance to β-lactams conferred by SOS induction is synergistic with alterations in the cell wall and in the TCA cycle in S. aureus (Keaton et al., 2013; Plata et al., 2013), which implies a link with oxidative stress.
Oxidative stress
In fact, reactive oxygen species (O2−, H2O2, OH−) formed during oxidative stress have the potential to damage DNA, and damaged DNA is a potent inducer of the SOS response. Our group very recently showed that sub-MICs of AGs lead to the incorporation of oxidized guanine in DNA, suggesting the occurrence of oxidative stress at concentrations 100-fold below the MIC, a concentration at which no killing is observed. Subsequently, sub-MICs AGs mediate induction of the SOS response mostly through oxidized guanine incorporation in DNA (Baharoglu et al., 2013). As ROS can damage DNA and proteins and induce mutagenesis (Mcbride et al., 1991; Nunoshiba et al., 1999), we proposed these as the missing links between sub-MIC antibiotic treatment and guanine oxidation.
The link between bactericidal antibiotics, ROS formation, and cell death has lately been subject to debate. To relieve possible confusion, it may be worth mentioning here that the above-described connection between ROS and SOS happens in conditions where ROS are created in the bacterial cell. For instance, we know that in the presence of sub-MICs of antibiotics, ROS are formed in V. cholerae and not in E. coli (Baharoglu et al., 2013). On the other hand, striking studies presented ROS formation as the key step leading to cell death by β-lactams, FQs, and AGs (Kohanski et al., 2007, 2010a,b). This study by Kohanski et al. suggests that all bactericidal antibiotics, regardless of their cellular target, have the potential to induce ROS formation, which kills bacteria. Subsequent studies challenged this hypothesis as no cell death due to ROS formation occurred using these antibiotics in different experimental conditions (Keren et al., 2013; Liu & Imlay, 2013). Finally, a recent report elegantly demonstrates that in E. coli, it is not ROS that kill bacteria upon AG treatment but rather increased AG uptake due to differential intracellular iron levels and synthesis of iron–sulfur clusters (Ezraty et al., 2013). In these latter studies (Kohanski et al., 2007; Ezraty et al., 2013; Keren et al., 2013; Liu & Imlay, 2013), antibiotics were used at lethal concentrations because the lethality of antibiotics was what the authors wanted to address. Conversely, in the study about ROS formation and SOS (Baharoglu et al., 2013, 2014), concentrations 100-fold below the MIC were used. Hence, these studies differ in the fact that they address either lethality or only SOS induction by antibiotics through ROS formation in nonlethal conditions.
The fact that low doses of antibiotics can induce SOS through ROS formation in certain species, like V. cholerae, Klebsiella pneumoniae, and Photorhabdus luminescens, might indicate that some species overcome their poorly efficient protection system against oxidative stress by being more easily capable of modifying their gene expression patterns (Mcbride et al., 1991; Nunoshiba et al., 1999).
In the same line of thought, the DinF protein belonging to the SOS regulon was found to be involved in response to oxidative stress in E. coli (Rodriguez-Beltran et al., 2012): in fact, production of DinF protects against bile salts and H2O2 by decreasing the effects of ROS and protein carbonylation, showing that SOS and the oxidative stress response can be complementary mechanisms in bacterial survival.
General stress
SOS and RpoS were suggested to be complementary mechanisms in response to certain stresses. RpoS, the stationary phase sigma factor, is induced in response to various stresses during the exponential growth phase and increases resistance to stress in E. coli (Merrikh et al., 2009a,b). For instance, and similarly to the SOS response, the RpoS regulon can also be induced during oxidative stress as well as cell wall stress (Allen & Griffiths, 2012; Mika et al., 2012). Genes expressed following the induction of the RpoS regulon, namely catalases (KatE, KatG) and iron chelators, protect cells from ROS-related DNA damage such as DSBs, which are SOS inducers. The link between the SOS and RpoS responses lies in the fact that both can be considered as mechanisms providing bacteria with means to survive DNA damage (Shee et al., 2011). Other facts linking SOS and RpoS exist: for example, both regulate the dinB gene, which encodes the TLS polymerase Pol IV (Kim et al., 2001; Layton & Foster, 2003). Antibiotic-induced mutagenesis involves SOS-dependent dinB induction (Kohiyama et al., 2013) but also induction mediated by RpoS (Henle & Linn, 1997) or NusA (Cohen & Walker, 2010), a component of the RNA polymerase complex that interacts with DinB. In this sense, RpoS and SOS can also be thought of as complementary mechanisms that protect bacteria from DNA damage: when RpoS is not sufficient, SOS takes over.
Nutrient stress
A connection between nutrient stress and SOS is also conceivable. For instance, cAMP-dependent SOS induction and mutagenesis were first observed in resting E. coli populations (Taddei et al., 1995). cAMP in complex with the CRP protein regulates a set of genes involved in the response to carbon source stress, called carbon catabolite regulation. More recently, it was found that the cAMP–CRP complex represses SOS-mediated mutations in stationary cell cultures (Macphee & Ambrose, 2010). However, the mechanism of such a regulation remains to be elucidated. cAMP–CRP-dependent induction of SOS has also been observed in E. coli cells starved for arginine, but only when these cells resume growth (Janion et al., 2002). The authors propose here that these cells accumulate DNA damage that induces SOS upon replication in a yet undiscovered mechanism implicating carbon catabolite control.
SOS-like responses
As described above, transformation is a mechanism of HGT that relies on ssDNA uptake and processing (Dubnau, 1999). Transformation occurs when a bacterial cell reaches a competent state rendering it capable of taking up external DNA present in its environment. Several mechanisms, such as special growth conditions or stress, lead to the induction of competence for natural transformation in B. subtilis and Streptococcus pneumoniae (Claverys et al., 2009). Competence has been suggested to be a stress response that could substitute for the SOS response in some bacterial species that lack an SOS regulon but in which the DNA repair genes are part of the competence regulon (for a review see Charpentier et al., 2012). For instance in S. pneumoniae which lacks LexA, it was found that AGs, FQs, and MMC induce competence and RecA, as well as the rate of genetic exchange in response to antibiotics, which is reminiscent of the E. coli SOS response (Prudhomme et al., 2006). However, recent data tone down this view: in S. thermophilus which carries the LexA-like repressor HdiR, competence and SOS were shown to be antagonistic mechanisms (Boutry et al., 2013). In this bacterium, SOS-inducing agents induce the SOS regulon genes such as RecA but decrease cellular transformability. As a corollary, induction of competence negatively affects DNA repair under these circumstances. In V. cholerae, SOS-inducing agents do not induce competence either (Baharoglu et al., 2012). However, DNA uptake during competence does induce SOS. Nevertheless, transformation seems to be induced in response to stress in many bacteria (Charpentier et al., 2012), suggesting it as an alternative to SOS induction in several cases.
An SOS-like response was also reported in Acinetobacter baumanii, where DNA damage induces genes involved in response to DNA damage, namely recA, TLS polymerases, and NER genes (Norton et al., 2013). However, the regulation of this SOS response does not involve LexA, which is absent from the A. baumanii genome. Nevertheless, induction of this stress response is responsible for damage repair and increase of mutation frequencies leading to antibiotic resistance acquisition, as it happens for E. coli with SOS induction. The increased mutation frequency was recently found to be the result of the A. baumanii TLS polymerase UmuDAb which recognizes palindromic sequences at the promoters of eight different DNA repair genes and TLS polymerases and regulates their expression in a RecA-dependent manner (Aranda et al., 2013).
Strategies to inhibit SOS in the battle against bacterial resistance
In the search for compounds that can potentiate the effect of antibiotics on bacteria, an engineered bacteriophage that suppresses the SOS response by over-expressing a noncleavable LexA repressor has also been reported to enhance killing by quinolones, AGs, and β-lactams in E. coli, to reduce the number of resistant bacteria that arise from antibiotic treatment and to increase survival of infected mice (Lu & Collins, 2009). According to the authors, these observations would be the result of disabling DNA damage repair. Such phages could be used in combination with antibiotics as adjuvants that suppress SOS induction and sensitize the bacterial cell to DNA damage. Another type of antimicrobial molecule was recently identified as a repressor of RecA expression: the aminocoumarins. The use of these gyrase inhibitors prevented SOS induction and decreased mutation frequency and recombination even in the presence of FQs in S. aureus (Schroder et al., 2013). Another approach is based on inhibition of RecA expression by engineered artificial small RNAs that are complementary to the recA mRNA (Sharma et al., 2013). The use of such sRNAs resulted in an increased sensitivity of E. coli to FQs in laboratory conditions. Finally, RNase E was proposed as a possible target to prevent SOS induction because inactivation of RNase E expression in E. coli impedes SOS induction by MMC (Manasherob et al., 2012).
A recent review also discusses various steps in food processing which trigger SOS induction in bacteria and the impact on food spoilage (van der Veen & Abee, 2011).
As depicted in this review, SOS is a transient but very strong stress response used by bacteria to increase their chances to adapt to changing environments and to survive. This is synthesized in the graphical abstract. The stress signal can be a genotoxic one, such as during direct exposure to DNA-damaging agents, but it appears here that many stresses causing changes in a given bacterial lifestyle can be the origin of such a stress. SOS induction can thus be seen as a bacterial adaptation mechanism, which has to be tightly controlled by bacteria to prevent a decrease of fitness in conditions where it is not needed. It can be conceived to use SOS modulation as a weapon against bacteria, through the adoption of better policies of antibiotic usage and food processing to avoid SOS-dependent adaptation and resistance of bacteria.
Acknowledgements
We thank Geneviève Garriss and Jason Bland for the helpful reading of the manuscript. Our work was supported by the Institut Pasteur, the Centre National de la Recherche Scientifique (CNRS-UMR3525); the European Union Seventh Framework Programme (FP7-HEALTH-2011-single-stage) ‘Evolution and Transfer of Antibiotic Resistance’ (EvoTAR); and the French Government's Investissement d'Avenir program Laboratoire d'Excellence ‘Integrative Biology of Emerging Infectious Diseases’ (Grant ANR-10-LABX-62-IBEID). ZB was supported by a DIM Malinf postdoctoral fellowship (Conseil régional d’Île-de-France) and EvoTAR. The authors declare no conflict of interest.
References
Author notes
Editor: Dieter Haas

{kind=link}
{kind=link}
{kind=link}
{kind=link}