





Abstract
As part of their life cycle, neutralophilic bacteria are often exposed to varying environmental stresses, among which fluctuations in pH are the most frequent. In particular, acid environments can be encountered in many situations from fermented food to the gastric compartment of the animal host. Herein, we review the current knowledge of the molecular mechanisms adopted by a range of Gram-positive and Gram-negative bacteria, mostly those affecting human health, for coping with acid stress. Because organic and inorganic acids have deleterious effects on the activity of the biological macromolecules to the point of significantly reducing growth and even threatening their viability, it is not unexpected that neutralophilic bacteria have evolved a number of different protective mechanisms, which provide them with an advantage in otherwise life-threatening conditions. The overall logic of these is to protect the cell from the deleterious effects of a harmful level of protons. Among the most favoured mechanisms are the pumping out of protons, production of ammonia and proton-consuming decarboxylation reactions, as well as modifications of the lipid content in the membrane. Several examples are provided to describe mechanisms adopted to sense the external acidic pH. Particular attention is paid to Escherichia coli extreme acid resistance mechanisms, the activity of which ensure survival and may be directly linked to virulence.
Neutralophilic bacteria, especially those affecting human health, possess mechanisms that enhance growth in moderate acid (pH 4–6) and enable survival in extreme acid (pH 1–3).
Biological context
Why should bacteria be armed with protection mechanisms against acid stress? Clearly, because these provide a way for them to withstand the deleterious effects of fluctuations in proton concentration to which they are exposed. Many bacteria that transit our gastrointestinal tract (GIT) are neutralophiles, often encountering strong and mild acidic environments: outside the host, in the preparation and preservation of foods and in the soil, but also inside the host, in the dental plaque, in the GIT (i.e. in the stomach or intestine) or, in case of intracellular pathogens, in the macrophage phagosome. For example, in the GIT, the differences in proton concentration in the different compartments are very large: ranging from the mildly acidic saliva (typical pH range 6.3–7.3) to the extremely acidic stomach (typical pH range 1.5–3.5) which means anything between a thousand to a millionfold increase in external proton concentration. Thus, such mechanisms are expected to be widespread.
The use of fermented foods, that is food products that are modified by the microbial growth, is, for humans, a common practice which dates back centuries (Hutkins, 2008). Acidic fermentation of dairy products, meat, fish and vegetables with the production of lactic acid, propionic acid, carbon dioxide and ethanol is in fact appreciated because it increases the flavour, digestibility and nutrition content of a specific food, but also because it enhances its preservation. Indeed, exposure to acid is an important part of the lactic acid bacteria (LAB) life cycle and their behaviour in acidic conditions can considerably affect the organoleptic and taste characteristics of fermented foods. However, the food industry has also to cope with an important issue: the preservation from spoilage and contamination caused by the overgrowth of food-borne pathogenic/toxigenic microorganism. The addition of organic acids (i.e. benzoate, sorbate, propionate), in addition to refrigeration, represents the best practice to protect food from pathogens overgrowth (Gould, 1996; Theron & Lues, 2010). The reasons why these organic acids are particularly effective at preventing bacterial growth is because at an external pH of 5, they conduct protons into the cytoplasm in the hydrophobic unionised form and then dissociate, thereby lowering the intracellular pH (see ‘’ and Hirshfield et al., 2003).
In soil, microbial communities live mainly in the rhizosphere, the zone surrounding the roots of plants, and they are affected from both the nutrients available and the root exudates. In particular, when plants use ammonium as nitrogen source, they release hydrogen ions which reduces the pH of the rhizosphere (Nye, 1981; Neumann & Martinoia, 2002; Hinsinger et al., 2003).
Acid mine drainage (AMD), originating from mining industry by exposure of the sulphide ore to atmospheric oxygen, causes environmental pollution in term of acidity (the pH of AMD can drop below 2) and metal content (aluminium, copper, zinc and manganese) (Johnson & Hallberg, 2005). In the last decade, bioremediation methods have been proposed that take advantage of microorganisms to neutralise the pH and decrease the heavy metal concentration. Thus, the bioreactors employed for the AMD bioremediation should contain bacteria capable of sulphate reduction and of acid tolerance (Lu et al., 2011; Ramond et al., 2013).
Hundreds of bacterial species reside in the oral cavity (Dewhirst et al., 2010), mostly in the dental plaque, which results from the adherence to a thin salivary pellicle on the enamel surface of several bacterial species that later develop into a biofilm community (Jenkinson, 2011). These bacteria when exposed to dietary carbohydrates produce acids, mainly lactic acid, causing a rapid fall of the micro-environmental pH. When food is masticated, the bicarbonate and other factors present in the saliva raise the pH. Thus, the dental plaque is subjected to frequent cycles of pH changes daily, with the absolute pH values reached being dependent on carbohydrates content of the food, metabolic activities of the bacterial plaque and teeth physiology (Loesche, 1986).
The human intestine, by far the largest body's surface (> 300 m2), is the site most heavily colonised by microbial communities, which find in it a stable and nutrient-rich environment (Flint et al., 2007, 2008). Sterile at birth, within a year the intestine becomes rapidly and abundantly colonised (up to 1012 cells per gram in the human colon) by hundreds of microbial species, most of which are obligate anaerobes belonging to the Bacteroidetes and Firmicutes phyla (Eckburg et al., 2005). The beneficial effects provided by the gut microbiota include key processes in human biology such as the fermentation of glycans into short-chain fatty acids (SCFAs: acetate, propionate, butyrate), the metabolism of amino acids and xenobiotics, and the biosynthesis of vitamins and isoprenoids (Neish, 2009). The mildly acidic pH of the distal gut (pH 5–6) is due to the accumulation of SCFAs which provide a chemical defence mechanism towards pathogen colonisation (see section ‘’).
Finally, orally acquired bacteria, including probiotics and food-borne pathogens, need to cope with the extreme acidic pH of the stomach (1.5–3.5), which acts as a bactericidal barrier (Giannella et al., 1972; Tennant et al., 2008). After residing in the stomach, bacteria pass into the small intestine where bicarbonate production neutralises the acid of the stomach, but then, they encounter the mildly acidic environment of the distal gut containing SCFAs.
Herein, we review the current knowledge of the molecular mechanisms adopted by Gram-positive and Gram-negative bacteria, mostly those affecting human health, for coping with acid stress. The four sections in which this review is organised are aimed at providing an overview of the different issues related to acid survival, which include (1) the effects of strong and weak acids on biological macromolecules; (2) the classification of the different protective mechanisms in neutralophilic bacteria; (3) the way acid is sensed and its negative effects counteracted and (4) the mechanism of action of the amino acid-dependent survival strategies activated under extreme acid stress. Other recent reviews have looked at different aspects of these processes (Foster, 2004; Slonczewski et al., 2009; Zhao & Houry, 2010; Hong et al., 2012; Kanjee & Houry, 2013).
Cellular pH and the potential effects of acidification
Bacteria, in general, are able to maintain a fairly constant internal pH (pHi) when grown in a wide range of media at different external pH (pHo) (reviewed in Slonczewski et al., 2009; Krulwich et al., 2011). Even acidophiles, which can only grow at low pHo, maintain a constant pHi as the pHo changes over several orders of magnitude. For example, the pHi of the acidophile Acidiphilium acidophilum (formerly known as Thiobacillus acidophilus) increases from 5.5 to 5.8 as the pHo changes from 1 to 4.5 (Matin et al., 1982). The same is true among those neutralophiles where this has been studied. The pHi of Escherichia coli, for example, changes only from 7.2 to 7.8 over a pHo range of 5.5 to 9 (Slonczewski et al., 1981), and the pHi of Bacillus subtilis is constant at 7.4 between pHo of 6 to 8 and only drops slightly below 7 when the pHo is 5.5 (Shioi et al., 1980). The outcome is that the pHi is kept higher than pHo < 7.5 or lower than pHo > 7.5.
This implies that larger fluctuations in pHi are undesirable and two observations indeed show that low pH is bad for bacteria that are not specifically adapted for it. One is the simple fact that bacteria are limited in the range of acidic pH values which they can survive without growth and even more limited in the range of pH values at which they are able to grow. The second is that bacteria generally show a transcriptional and translational response to a drop in pH (see sections Acid tolerance response (ATR) and acid resistance as protective mechanisms and ), and if this response is prevented or reduced, then growth and survival of these bacteria are impaired at lower pH (see section ‘Amino acid-dependent extreme acid resistance (XAR) in E. coli: chemical and physiological issues’). Presumably, essential components of the cell that in the absence of protective mechanisms are damaged by low pH lose their ability to function in cases where the response does not occur. But what are these targets and where in the cell does the damage take place?
A first step in addressing the question of where acid causes cellular damage is to determine the pH of different cellular compartments when cells are acidified. It is often stated that in Gram-negative bacteria, the outer membrane is not a physical barrier to the movement of protons, as the diameters of the porins are large enough to allow these ions to pass through. The situation is not necessarily this simple, however, as the physical state of the protons in acidified medium is not as simple as free protons or free hydronium ions (H3O+), and the mechanism(s) by which protons move across membranes is as yet not fully understood (Deamer, 1987; Swanson & Simons, 2009). Nonetheless, it has been shown by direct measurement in E. coli that when the external medium is acidified, the pH in the periplasm falls very rapidly to a level roughly the same as that outside the cell, consistent with the hypothesis that the outer membrane is not a significant barrier to proton movement (Wilks & Slonczewski, 2007). Once the pH in the periplasm has dropped, it remains low, as expected if it is effectively continuous with the external medium, and showing that the periplasm has limited buffering capacity.
The situation is different in the cytoplasm. The inner membrane is a major barrier to strong acids which are highly ionised, although even for strong acids, some molecules may cross the inner membrane in the nonionised form and subsequently dissociate in the periplasm (Gutknecht & Walter, 1981), and protons may also enter the cytoplasm through protein channels, transient water chains or damaged membranes (Deamer, 1987; Foster, 2004). Following external acidification by a strong acid such as hydrochloric acid, the cytoplasm of planktonically grown E. coli shows a transient drop in pH but rapidly (< 4 min) returns to neutral, when the fall in pHo is not too drastic (Wilks & Slonczewski, 2007). The same effect is seen for B. subtilis, although the recovery of pHi is not as complete as in E. coli (Kitko et al., 2009). These data were derived from studies on large numbers of cells and hence represent population averages. Broadly, similar results were obtained when studies were carried out at the single cell level by combining ratiometric GFP measurements with fluorescence microscopy, although tethered cells and cells in biofilms showed slower recovery and more examples of cells that fail to recover (Martinez et al., 2012). The transient drop and rapid correction of pHi seen when bacterial cells are exposed to a strong acid at moderate pH is likely to result from intrinsic buffering by cellular components or alterations in the flux of other ions, as the transcriptional response to acidification is not rapid enough to account for it.
When acid stress is more severe, the pHi falls to levels which are too low to correct by buffering or ionic flux, and inducible responses (further described ‘Amino acid-dependent XAR in E. coli: chemical and physiological issues’) become important in determining the cytoplasmic pH. In E. coli exposed to a pHo of 2.5, the pHi of the cell may fall as low as 3.5 if these inducible mechanisms are not operating, and cell survival under these conditions is very low. However, even when these systems are operative, the pHi was reported to fall to < 5 (Richard & Foster, 2004).
Weak organic acids also cause acid stress in bacteria, but here, the nature of the stress is more complicated. Because they are less dissociated at any given pH than strong acids such as HCl, organic acids can cross the inner membrane more freely in the uncharged form. Not only can they then dissociate in the cytoplasm, but will cause partially collapse of the pH gradient across cells, as they can combine with external protons and carry these back into cells, without them having to pass through the normal FoF1-ATPase channels. It has been shown that for both E. coli and B. subtilis, the presence of membrane-permeant organic acids such as acetic or benzoic acid prevents or significantly delays the normal recovery of cytoplasmic pH that occurs following acidification (Wilks & Slonczewski, 2007; Kitko et al., 2009). Moreover, the undissociated acids themselves may be inhibitory for growth (Salmond et al., 1984). Thus, the effect of weak acids – many of which will be encountered by gut microorganisms as the products of bacterial metabolism – depends in complicated ways on the nature of the particular acid and the pH of the surrounding medium.
To summarise, the periplasm is unable to resist changes in pHo and is hence a potential major site where damage by low pH may be serious. The cytoplasm is more protected, by its intrinsic buffering capacity, by the relative impermeability of the cytoplasmic membrane to protons and by inducible acid stress responses, but significant reduction in pH can occur here too. Likewise, proteins embedded in the membranes will be exposed to the detrimental effect of acidic pH due to their cellular location. For example, the domains of inner membrane proteins which are in the periplasm will be exposed to the low pH of the periplasm if the external medium is acidified.
What are the likely consequences of these reduced pH values for different components of the bacterial cell? Working out the cellular consequences of low pH is complex, as they depend again on the nature of the acid studied (including the nature of the cation, which may itself have significant cellular effects unrelated to changes in pH), and there are many potential problems that a decrease in pH could cause in the cell. Four possible targets will be considered briefly below – lowered enzyme activity, acid-induced protein unfolding, membrane damage and DNA damage – but this is by no means an exhaustive list.
In the cytoplasm, decreased enzyme activity is one serious potential effect of lowered pH, simply because the pH is below the optimum range of many important metabolic enzymes. It has been shown for example in anaerobically grown E. coli that glycolysis is strongly inhibited when pHi drops much below 7, with the rate at pH 6 being about 20% of that at pH 7.5 (Ugurbil et al., 1978; Hayes et al., 2006). A prolonged drop in pHi will thus compromise essential cellular processes including central metabolic pathways and ATP production. In contrast with this, the activity of enzymes which are needed to function at low pH during acid stress, such as the amino acid decarboxylases discussed in the following sections, often displays unusually low pH optima regardless of whether they are assayed in intact cells or following purification (Gale, 1946).
Another potential consequence of acidification could in principle be protein unfolding: low pH has often been used to induce protein denaturation in experiments in vitro, as it causes increased charge repulsion as more residues become fully protonated (Goto et al., 1990). Although the pH values used in vitro are generally lower than those which will occur in the cytoplasm, array data from E. coli exposed to acid do not show any consistent upregulation of the standard cytoplasmic chaperones such as GroE, DnaK or IbpB chaperone machines, all of which are strongly induced by the presence of unfolded cytoplasmic proteins. It therefore seems unlikely that acidification leads to significant protein unfolding in the cytoplasm in this organism (Arnold et al., 2001; Tucker et al., 2002; Maurer et al., 2005). However, induction of some or all of the genes encoding these chaperones has been reported for some other bacteria, as shown in Table 1, supporting the hypothesis that at least in some bacterial species, the unfolding of proteins in the cytoplasm can indeed occur after acidification.
Occurrence of mechanisms protecting Gram-negative (grey) and Gram-positive (white) neutralophilic bacteria from acid stress
| Protective mechanisms | F1-F0 ATPase | Amino acid-dependent decarboxylase/antiporter systems | Deiminase and deaminase systems | Urease | Protein repair and proteases | Modifications of cell membrane | |
| Escherichia coli | XAR and ATR | Foster (2004) and Sun et al. (2012a) | Glutamate (Castanie-Cornet et al., 1999; De Biase et al., 1999); arginine (Iyer et al., 2003); lysine (Meng & Bennett, 1992b) | Adenosine deaminase (Sun et al., 2012b); Glutaminase (Lu et al., 2013) | HdeA, HdeB (Kern et al., 2007); Hsp31 (Mujacic & Baneyx, 2007) | Chang & Cronan (1999) and Brown et al. (1997) | |
| Salmonella enterica var. Typhimurium | XAR and ATR | Foster & Hall, (1991, 1990) | Arginine (Kieboom & Abee, 2006); lysine (Park et al., 1996); ornithine (Viala et al., 2011) | DnaK (Bearson et al., 2006) | Alvarez-Ordonez et al. (2009) and Kim et al. (2005) | ||
| Vibrio cholerae | ATR | Lysine (Merrell & Camilli, 1999) | |||||
| Helicobacter pylori | Acid acclimation | Mobley et al. (1995) and Weeks et al. (2000) | GroEL, GroES (Zanotti & Cendron, 2010) | Haque et al. (1996) | |||
| Brucella spp. | XAR and ATR | Glutamate: B. microti (Occhialini et al., 2012) | All known species but B. ovis (Bandara et al., 2007; Sangari et al., 2007) | DnaK: B: melitensis (Teixeira-Gomes et al., 2000) HdeA: B. abortus (Valderas et al., 2005) | |||
| Proteus mirabilis | ATR | Glutamate (De Biase & Pennacchietti, 2012) | Mobley et al. (1995) | ||||
| Yersinia enterocolitica | ATR | Glutamate (De Biase & Pennacchietti, 2012) | Young et al. (1996) | ||||
| Listeria monocytogenes | XAR and ATR | Cotter et al. (2000) | Glutamate (Cotter et al., 2001) | ADI (Ryan et al., 2009) | Clp protease (Wemekamp-Kamphuis et al., 2004); GrpE (Ivy et al., 2012) | ||
| Lactococcus lactis | XAR and ATR | O'Sullivan & Condon (1999) and Amachi et al. (1998) | Glutamate (Sanders et al., 1998) | ADI (Budin-Verneuil et al., 2006) | ClpE, ClpP, GroEL, GroES, DnaK and GrpE (Frees et al., 2003) | Budin-Verneuil et al. (2005) | |
| Lactobacillus spp. | XAR and ATR | L. acidophilus and L. rhamnosous (Kullen & Klaenhammer, 1999; Koponen et al., 2012) | Glutamate: L. reuteri (Su et al., 2011);histidine: L. 30a and L. buchneri (Molenaar et al., 1993);ornithine: L. acidophilus (Azcarate- Peril et al., 2004) | ADI: L. sakei and L. reuteri; (Champomier Verges et al., 1999; Rollan et al., 2003) AgDI: L. brevis; (Lucas et al., 2007) | ClpE : L. rhamnosus; (Koponen et al., 2012) ClpL: L. reuteri (Wall et al., 2007); GrpE, GroES, DnaJ: L. acidophilus and L. reuteri (Lorca et al., 2002; Lee et al., 2008a) | L. casei (Fozo et al., 2004; Broadbent et al., 2010) | |
| Bacillus cereus | ATR | Arginine and lysine (Senouci-Rezkallah et al., 2011) | DnaK, GroES and Clp protease (Mols et al., 2010a) | ||||
| Streptococcus spp. | ATR | S. mutans, S. salivaris, S. sanguis and S. faecalis (Bender et al., 1986; Kobayashi et al., 1986; Kuhnert et al., 2003) | ADI:S. rattus, S. sanguis, S. pyrogene and S. suis (Casiano-Colon & Marquis, 1988; Degnan et al., 2000; Gruening et al., 2006) AgDI: S. mutans (Griswold et al., 2004) | S. salivaris (Chen et al., 1996, 2000) | RecA, AP endonuclease, Ssb, UvrA, DnaK, RopA, GroEL and ClpL: S. mutans; (Jayaraman et al., 1997; Hahn et al., 1999; Hanna et al., 2001; Kajfasz et al., 2009) | S. gordonii, S. salivary and S. mutans (Quivey et al., 2000; Fozo et al., 2004) | |
| Bifidobacterium spp. | ATR | B. lactis, B. animalis (Matsumoto et al., 2004; Ventura et al., 2004) | Glutamate: B. dentium (Ventura et al., 2009) | ||||
| Clostridium spp. | ATR | Glutamate: C. welchii, C. aerofoetidum and C. perfringens (Gale, 1940) | dnaK, groES, groEL, hsp90, hsp18, clpC, and htrA in C. acetobutylicum (Alsaker et al., 2010) |
| Protective mechanisms | F1-F0 ATPase | Amino acid-dependent decarboxylase/antiporter systems | Deiminase and deaminase systems | Urease | Protein repair and proteases | Modifications of cell membrane | |
| Escherichia coli | XAR and ATR | Foster (2004) and Sun et al. (2012a) | Glutamate (Castanie-Cornet et al., 1999; De Biase et al., 1999); arginine (Iyer et al., 2003); lysine (Meng & Bennett, 1992b) | Adenosine deaminase (Sun et al., 2012b); Glutaminase (Lu et al., 2013) | HdeA, HdeB (Kern et al., 2007); Hsp31 (Mujacic & Baneyx, 2007) | Chang & Cronan (1999) and Brown et al. (1997) | |
| Salmonella enterica var. Typhimurium | XAR and ATR | Foster & Hall, (1991, 1990) | Arginine (Kieboom & Abee, 2006); lysine (Park et al., 1996); ornithine (Viala et al., 2011) | DnaK (Bearson et al., 2006) | Alvarez-Ordonez et al. (2009) and Kim et al. (2005) | ||
| Vibrio cholerae | ATR | Lysine (Merrell & Camilli, 1999) | |||||
| Helicobacter pylori | Acid acclimation | Mobley et al. (1995) and Weeks et al. (2000) | GroEL, GroES (Zanotti & Cendron, 2010) | Haque et al. (1996) | |||
| Brucella spp. | XAR and ATR | Glutamate: B. microti (Occhialini et al., 2012) | All known species but B. ovis (Bandara et al., 2007; Sangari et al., 2007) | DnaK: B: melitensis (Teixeira-Gomes et al., 2000) HdeA: B. abortus (Valderas et al., 2005) | |||
| Proteus mirabilis | ATR | Glutamate (De Biase & Pennacchietti, 2012) | Mobley et al. (1995) | ||||
| Yersinia enterocolitica | ATR | Glutamate (De Biase & Pennacchietti, 2012) | Young et al. (1996) | ||||
| Listeria monocytogenes | XAR and ATR | Cotter et al. (2000) | Glutamate (Cotter et al., 2001) | ADI (Ryan et al., 2009) | Clp protease (Wemekamp-Kamphuis et al., 2004); GrpE (Ivy et al., 2012) | ||
| Lactococcus lactis | XAR and ATR | O'Sullivan & Condon (1999) and Amachi et al. (1998) | Glutamate (Sanders et al., 1998) | ADI (Budin-Verneuil et al., 2006) | ClpE, ClpP, GroEL, GroES, DnaK and GrpE (Frees et al., 2003) | Budin-Verneuil et al. (2005) | |
| Lactobacillus spp. | XAR and ATR | L. acidophilus and L. rhamnosous (Kullen & Klaenhammer, 1999; Koponen et al., 2012) | Glutamate: L. reuteri (Su et al., 2011);histidine: L. 30a and L. buchneri (Molenaar et al., 1993);ornithine: L. acidophilus (Azcarate- Peril et al., 2004) | ADI: L. sakei and L. reuteri; (Champomier Verges et al., 1999; Rollan et al., 2003) AgDI: L. brevis; (Lucas et al., 2007) | ClpE : L. rhamnosus; (Koponen et al., 2012) ClpL: L. reuteri (Wall et al., 2007); GrpE, GroES, DnaJ: L. acidophilus and L. reuteri (Lorca et al., 2002; Lee et al., 2008a) | L. casei (Fozo et al., 2004; Broadbent et al., 2010) | |
| Bacillus cereus | ATR | Arginine and lysine (Senouci-Rezkallah et al., 2011) | DnaK, GroES and Clp protease (Mols et al., 2010a) | ||||
| Streptococcus spp. | ATR | S. mutans, S. salivaris, S. sanguis and S. faecalis (Bender et al., 1986; Kobayashi et al., 1986; Kuhnert et al., 2003) | ADI:S. rattus, S. sanguis, S. pyrogene and S. suis (Casiano-Colon & Marquis, 1988; Degnan et al., 2000; Gruening et al., 2006) AgDI: S. mutans (Griswold et al., 2004) | S. salivaris (Chen et al., 1996, 2000) | RecA, AP endonuclease, Ssb, UvrA, DnaK, RopA, GroEL and ClpL: S. mutans; (Jayaraman et al., 1997; Hahn et al., 1999; Hanna et al., 2001; Kajfasz et al., 2009) | S. gordonii, S. salivary and S. mutans (Quivey et al., 2000; Fozo et al., 2004) | |
| Bifidobacterium spp. | ATR | B. lactis, B. animalis (Matsumoto et al., 2004; Ventura et al., 2004) | Glutamate: B. dentium (Ventura et al., 2009) | ||||
| Clostridium spp. | ATR | Glutamate: C. welchii, C. aerofoetidum and C. perfringens (Gale, 1940) | dnaK, groES, groEL, hsp90, hsp18, clpC, and htrA in C. acetobutylicum (Alsaker et al., 2010) |
Direct involvement in survival to acid stress not experimentally demonstrated.
Occurrence of mechanisms protecting Gram-negative (grey) and Gram-positive (white) neutralophilic bacteria from acid stress
| Protective mechanisms | F1-F0 ATPase | Amino acid-dependent decarboxylase/antiporter systems | Deiminase and deaminase systems | Urease | Protein repair and proteases | Modifications of cell membrane | |
| Escherichia coli | XAR and ATR | Foster (2004) and Sun et al. (2012a) | Glutamate (Castanie-Cornet et al., 1999; De Biase et al., 1999); arginine (Iyer et al., 2003); lysine (Meng & Bennett, 1992b) | Adenosine deaminase (Sun et al., 2012b); Glutaminase (Lu et al., 2013) | HdeA, HdeB (Kern et al., 2007); Hsp31 (Mujacic & Baneyx, 2007) | Chang & Cronan (1999) and Brown et al. (1997) | |
| Salmonella enterica var. Typhimurium | XAR and ATR | Foster & Hall, (1991, 1990) | Arginine (Kieboom & Abee, 2006); lysine (Park et al., 1996); ornithine (Viala et al., 2011) | DnaK (Bearson et al., 2006) | Alvarez-Ordonez et al. (2009) and Kim et al. (2005) | ||
| Vibrio cholerae | ATR | Lysine (Merrell & Camilli, 1999) | |||||
| Helicobacter pylori | Acid acclimation | Mobley et al. (1995) and Weeks et al. (2000) | GroEL, GroES (Zanotti & Cendron, 2010) | Haque et al. (1996) | |||
| Brucella spp. | XAR and ATR | Glutamate: B. microti (Occhialini et al., 2012) | All known species but B. ovis (Bandara et al., 2007; Sangari et al., 2007) | DnaK: B: melitensis (Teixeira-Gomes et al., 2000) HdeA: B. abortus (Valderas et al., 2005) | |||
| Proteus mirabilis | ATR | Glutamate (De Biase & Pennacchietti, 2012) | Mobley et al. (1995) | ||||
| Yersinia enterocolitica | ATR | Glutamate (De Biase & Pennacchietti, 2012) | Young et al. (1996) | ||||
| Listeria monocytogenes | XAR and ATR | Cotter et al. (2000) | Glutamate (Cotter et al., 2001) | ADI (Ryan et al., 2009) | Clp protease (Wemekamp-Kamphuis et al., 2004); GrpE (Ivy et al., 2012) | ||
| Lactococcus lactis | XAR and ATR | O'Sullivan & Condon (1999) and Amachi et al. (1998) | Glutamate (Sanders et al., 1998) | ADI (Budin-Verneuil et al., 2006) | ClpE, ClpP, GroEL, GroES, DnaK and GrpE (Frees et al., 2003) | Budin-Verneuil et al. (2005) | |
| Lactobacillus spp. | XAR and ATR | L. acidophilus and L. rhamnosous (Kullen & Klaenhammer, 1999; Koponen et al., 2012) | Glutamate: L. reuteri (Su et al., 2011);histidine: L. 30a and L. buchneri (Molenaar et al., 1993);ornithine: L. acidophilus (Azcarate- Peril et al., 2004) | ADI: L. sakei and L. reuteri; (Champomier Verges et al., 1999; Rollan et al., 2003) AgDI: L. brevis; (Lucas et al., 2007) | ClpE : L. rhamnosus; (Koponen et al., 2012) ClpL: L. reuteri (Wall et al., 2007); GrpE, GroES, DnaJ: L. acidophilus and L. reuteri (Lorca et al., 2002; Lee et al., 2008a) | L. casei (Fozo et al., 2004; Broadbent et al., 2010) | |
| Bacillus cereus | ATR | Arginine and lysine (Senouci-Rezkallah et al., 2011) | DnaK, GroES and Clp protease (Mols et al., 2010a) | ||||
| Streptococcus spp. | ATR | S. mutans, S. salivaris, S. sanguis and S. faecalis (Bender et al., 1986; Kobayashi et al., 1986; Kuhnert et al., 2003) | ADI:S. rattus, S. sanguis, S. pyrogene and S. suis (Casiano-Colon & Marquis, 1988; Degnan et al., 2000; Gruening et al., 2006) AgDI: S. mutans (Griswold et al., 2004) | S. salivaris (Chen et al., 1996, 2000) | RecA, AP endonuclease, Ssb, UvrA, DnaK, RopA, GroEL and ClpL: S. mutans; (Jayaraman et al., 1997; Hahn et al., 1999; Hanna et al., 2001; Kajfasz et al., 2009) | S. gordonii, S. salivary and S. mutans (Quivey et al., 2000; Fozo et al., 2004) | |
| Bifidobacterium spp. | ATR | B. lactis, B. animalis (Matsumoto et al., 2004; Ventura et al., 2004) | Glutamate: B. dentium (Ventura et al., 2009) | ||||
| Clostridium spp. | ATR | Glutamate: C. welchii, C. aerofoetidum and C. perfringens (Gale, 1940) | dnaK, groES, groEL, hsp90, hsp18, clpC, and htrA in C. acetobutylicum (Alsaker et al., 2010) |
| Protective mechanisms | F1-F0 ATPase | Amino acid-dependent decarboxylase/antiporter systems | Deiminase and deaminase systems | Urease | Protein repair and proteases | Modifications of cell membrane | |
| Escherichia coli | XAR and ATR | Foster (2004) and Sun et al. (2012a) | Glutamate (Castanie-Cornet et al., 1999; De Biase et al., 1999); arginine (Iyer et al., 2003); lysine (Meng & Bennett, 1992b) | Adenosine deaminase (Sun et al., 2012b); Glutaminase (Lu et al., 2013) | HdeA, HdeB (Kern et al., 2007); Hsp31 (Mujacic & Baneyx, 2007) | Chang & Cronan (1999) and Brown et al. (1997) | |
| Salmonella enterica var. Typhimurium | XAR and ATR | Foster & Hall, (1991, 1990) | Arginine (Kieboom & Abee, 2006); lysine (Park et al., 1996); ornithine (Viala et al., 2011) | DnaK (Bearson et al., 2006) | Alvarez-Ordonez et al. (2009) and Kim et al. (2005) | ||
| Vibrio cholerae | ATR | Lysine (Merrell & Camilli, 1999) | |||||
| Helicobacter pylori | Acid acclimation | Mobley et al. (1995) and Weeks et al. (2000) | GroEL, GroES (Zanotti & Cendron, 2010) | Haque et al. (1996) | |||
| Brucella spp. | XAR and ATR | Glutamate: B. microti (Occhialini et al., 2012) | All known species but B. ovis (Bandara et al., 2007; Sangari et al., 2007) | DnaK: B: melitensis (Teixeira-Gomes et al., 2000) HdeA: B. abortus (Valderas et al., 2005) | |||
| Proteus mirabilis | ATR | Glutamate (De Biase & Pennacchietti, 2012) | Mobley et al. (1995) | ||||
| Yersinia enterocolitica | ATR | Glutamate (De Biase & Pennacchietti, 2012) | Young et al. (1996) | ||||
| Listeria monocytogenes | XAR and ATR | Cotter et al. (2000) | Glutamate (Cotter et al., 2001) | ADI (Ryan et al., 2009) | Clp protease (Wemekamp-Kamphuis et al., 2004); GrpE (Ivy et al., 2012) | ||
| Lactococcus lactis | XAR and ATR | O'Sullivan & Condon (1999) and Amachi et al. (1998) | Glutamate (Sanders et al., 1998) | ADI (Budin-Verneuil et al., 2006) | ClpE, ClpP, GroEL, GroES, DnaK and GrpE (Frees et al., 2003) | Budin-Verneuil et al. (2005) | |
| Lactobacillus spp. | XAR and ATR | L. acidophilus and L. rhamnosous (Kullen & Klaenhammer, 1999; Koponen et al., 2012) | Glutamate: L. reuteri (Su et al., 2011);histidine: L. 30a and L. buchneri (Molenaar et al., 1993);ornithine: L. acidophilus (Azcarate- Peril et al., 2004) | ADI: L. sakei and L. reuteri; (Champomier Verges et al., 1999; Rollan et al., 2003) AgDI: L. brevis; (Lucas et al., 2007) | ClpE : L. rhamnosus; (Koponen et al., 2012) ClpL: L. reuteri (Wall et al., 2007); GrpE, GroES, DnaJ: L. acidophilus and L. reuteri (Lorca et al., 2002; Lee et al., 2008a) | L. casei (Fozo et al., 2004; Broadbent et al., 2010) | |
| Bacillus cereus | ATR | Arginine and lysine (Senouci-Rezkallah et al., 2011) | DnaK, GroES and Clp protease (Mols et al., 2010a) | ||||
| Streptococcus spp. | ATR | S. mutans, S. salivaris, S. sanguis and S. faecalis (Bender et al., 1986; Kobayashi et al., 1986; Kuhnert et al., 2003) | ADI:S. rattus, S. sanguis, S. pyrogene and S. suis (Casiano-Colon & Marquis, 1988; Degnan et al., 2000; Gruening et al., 2006) AgDI: S. mutans (Griswold et al., 2004) | S. salivaris (Chen et al., 1996, 2000) | RecA, AP endonuclease, Ssb, UvrA, DnaK, RopA, GroEL and ClpL: S. mutans; (Jayaraman et al., 1997; Hahn et al., 1999; Hanna et al., 2001; Kajfasz et al., 2009) | S. gordonii, S. salivary and S. mutans (Quivey et al., 2000; Fozo et al., 2004) | |
| Bifidobacterium spp. | ATR | B. lactis, B. animalis (Matsumoto et al., 2004; Ventura et al., 2004) | Glutamate: B. dentium (Ventura et al., 2009) | ||||
| Clostridium spp. | ATR | Glutamate: C. welchii, C. aerofoetidum and C. perfringens (Gale, 1940) | dnaK, groES, groEL, hsp90, hsp18, clpC, and htrA in C. acetobutylicum (Alsaker et al., 2010) |
Direct involvement in survival to acid stress not experimentally demonstrated.
As noted above, the periplasm is more susceptible to acid-induced damage because this compartment is subject to greater extremes of pH change. It is therefore not surprising to find in several acid-resistant species of Gram-negative enteric bacteria, which have to face the extremely acidic environment of the mammalian stomach, that specific periplasmic chaperones play a major role in protecting periplasmic and membrane proteins against damage by low pH (recently reviewed by Hong et al., 2012). These are the HdeA and HdeB proteins. These proteins contribute to acid resistance in a range of different species, including E. coli, Shigella flexneri and Brucella abortus. In these species (all of which can infect with a low infectious dose), loss of one or both of the HdeA and HdeB proteins impairs growth at low pH (Waterman & Small, 1996; Gajiwala & Burley, 2000; Valderas et al., 2005; Kern et al., 2007). The molecular details of their mode of action are beginning to be elucidated. Both proteins are dimeric and have to dissociate into monomers to exert their activity, which consists of blocking the aggregation of acid-unfolded proteins and in assisting their renaturation by promoting a folding-competent state, which would allow other periplasmic chaperones (i.e. DegP and SurA) to assist the refolding of periplasmic proteins during the acid stress-recovery phase (Tapley et al., 2010; Zhang et al., 2011).
The cytoplasmic membrane is a major barrier to proton influx in acid-treated cells, but damage to the membrane caused by the acid treatment itself may cause this barrier to break down. A key component of membranes protecting against damage by acid appears to be cyclopropane fatty acids (CFAs), the levels of which have been shown to correlate well with acid resistance in E. coli (Brown et al., 1997). In support of this, E. coli mutated in the cfa (cyclopropane fatty acid synthase) gene becomes very sensitive to a shift to low pH, and this sensitivity can be overcome in part by supplying CFAs exogenously (Chang & Cronan, 1999). The transcription of the cfa gene is also upregulated under acidic conditions (Chang & Cronan, 1999), showing that changing the membrane's content of fatty acids is an adaptive response to acid stress. E. coli membranes lacking CFAs have been shown to be more permeable to protons, again supporting the hypothesis that the presence of CFAs is important at maintaining membrane integrity under acidic conditions (Shabala & Ross, 2008). Mutations in genes that change membrane composition and architecture have also been implicated in increased acid sensitivity of a number of Gram-positive bacteria (reviewed by Cotter & Hill, 2003). Thus, it can be hypothesised that bacterial membranes have evolved protective mechanisms that are necessary to maintain their integrity and thus reduce proton influx, under acidic conditions.
Finally, DNA is another cellular macromolecule which may be damaged by acid. DNA extracted from E. coli cells treated at extremely low pH values showed evidence of increased damage over time (4 h) with an increasing number of strand breaks (Jeong et al., 2008). A key cellular component that protects against this damage is the Dps protein, the levels of which increase enormously as cells move into stationary phase (at which point, they also often become much more resistant to acid). In dps mutants, which show heightened sensitivity to acid, DNA damage induced by acid is significantly increased (Choi et al., 2000; Jeong et al., 2008). The acidic pH can favour a process which occurs at very slow rate in physiological conditions: the depurination of the DNA, consisting in the loss of purines which results in the formation of apurinic sites (Lindahl & Nyberg, 1972). This event occurs more rapidly at acidic pH when the nitrogenous bases become protonated. Because the depurination can cause loss of genetic information, repair systems are induced in bacteria. Indeed, several bacterial mutants in these repair systems also show heightened acid sensitivity, consistent with DNA damage being a consequence of acidification (Quivey et al., 1995; Hanna et al., 2001).
ATR and acid resistance as protective mechanisms
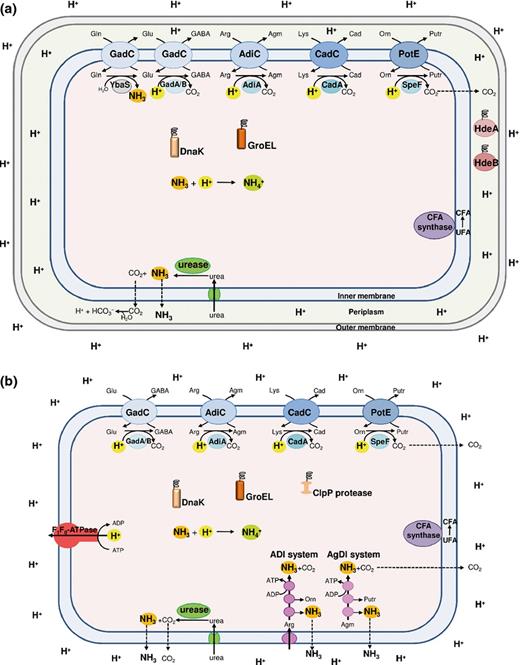
When at the beginning of the 1990s, scientists begun to study how bacteria cope with extreme and mild acid stress, they distinguished between the ATR and XAR (Lin et al., 1995; Bearson et al., 1997). ATR indicates an adaptive response at a nonlethal, mildly acidic pH that produces an enhanced tolerance to severe acidic challenge, that is, up to pH 3.0. Most of the microorganisms coping with acidic stress possess at least one mechanism classified as ATR. However, some bacteria possess also XAR mechanisms which allow unadapted cells to survive at levels too acidic to permit growth, that is, at pH ≤ 2.5 (Foster, 2001). Thus, the ATR towards mild acid stress involves mechanisms that maintain intracellular pH homoeostasis, whereas the XAR response to extreme acid stress involves mechanisms that prevent the intracellular pH from falling to life-threatening levels. Different organisms employ a variety of strategies, which may include (1) actively expelling protons out of the cell (via the F1F0-ATPase); (2) sequestering the intracellular protons via biochemical reactions that either consume protons (i.e. amino acid decarboxylation) or generate ammonia (i.e. amino acid deiminases, deaminases and urease); (3) repairing or preventing acid damage in macromolecules; and (4) modifying the proton permeability of the cellular membrane. Figure 1 provides an overview of these different mechanisms, operating in Gram-negative (a) and Gram-positive (b) bacteria, that are briefly described below. Table 1 gives detailed information about which system operates in which bacterial species.
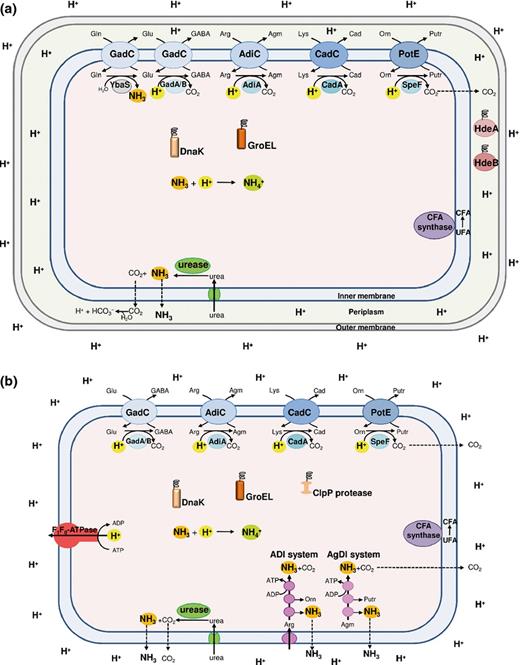
Schematic representation of all the mechanisms of protection against acid stress that can occur in Gram-negative (a) and Gram-positive (b) neutralophilic bacteria. Note that not all the mechanisms are present in a same microorganism, but that each microorganism possesses specific ones, as listed in detail in Table 1. The amino acid decarboxylase/antiporter systems (blue), dependent on glutamate (Glu; GadAB/GadC), arginine (Arg; AdiA/AdiC), lysine (Lys; CadA/CadC) and ornithine (Orn; SpeF/PotE), consume intracellular protons. The glutamine (Gln)-dependent system, consisting of glutaminase YbaS (grey) and glutamine/glutamate antiporter GadC, produces NH3. The F1F0-ATPase (red) pumps out protons via ATP hydrolysis. The amino acid deiminase pathways (magenta), arginine deiminase (ADI) and agmatine deiminase (AgDI), yield NH3 and ATP. The urease/urea transporter system (green) imports and hydrolyses urea yielding NH3. The cytoplasmic DnaK and GroEL (orange) and periplasmic HdeA and HdeB (dark red) chaperones protect denatured proteins, whereas Clp protease (brown) remove damaged proteins. The CFA synthase enzyme (purple) converts unsaturated fatty acids (UFA) into cyclopropane fatty acids (CFA).
F1F0-ATPase
This is a sophisticated molecular machine localised in the plasma membrane, which can either synthesise ATP (ATP synthase), using the energy released by the exergonic entry of protons from the extracellular space into the cell, or conversely pump out protons using the energy provided by ATP hydrolysis (ATPase). The involvement of F1F0-ATPase in pH homoeostasis was first observed in Streptococcus spp., which do not have the respiratory chain and use this enzymatic complex to expel protons when the cytoplasmic pH decreases (Kobayashi et al., 1986). Thereafter, several lines of evidence using mutant strains or enzymatic inhibitors have suggested a role of F1F0-ATPase in ATR in several other bacterial species (Table 1). Whether it acts by directly expelling protons or by producing energy to support the activity of protection or repair systems has still to be conclusively demonstrated.
Amino acid-dependent decarboxylase/antiporter systems
The amino acid decarboxylases are cytosolic enzymes, mostly using pyridoxal 5′-phosphate (PLP), a derivative of vitamin B6. They perform a proton-consuming decarboxylation reaction on specific amino acids, such as glutamate, lysine, arginine and ornithine (Gale, 1946). These enzymes have an acidic pH optimum (< 6) that varies from enzyme to enzyme, thereby covering the whole range of acidity, from 4 to 6. Each decarboxylase works in strict association with a cognate antiporter, which is localised in the cell membrane and becomes active only when the extracellular pH drops below threshold levels, thereby providing a selective gate for entry of the amino acid substrates and exit of the decarboxylation products. A description of how these systems work is provided in the section ‘Amino acid-dependent XAR in E. coli: chemical and physiological issues’ and has also been reviewed recently (Kanjee & Houry, 2013). Table 1 lists the microorganisms possessing these systems.
Deiminase and deaminase systems
These systems share the ability to produce ammonia (NH3) which combines with intracellular protons to yield the ammonium ion ( ; pKa = 8.95 at 35 °C) (Martinelle & Haggstrom, 1997), thereby raising pHi. Two deiminases (Fig. 1) involved in protection from acid stress have been described in many bacteria, especially in those inhabiting the oral cavity in dental plaque.
; pKa = 8.95 at 35 °C) (Martinelle & Haggstrom, 1997), thereby raising pHi. Two deiminases (Fig. 1) involved in protection from acid stress have been described in many bacteria, especially in those inhabiting the oral cavity in dental plaque.
The arginine deiminase (ADI; not to be confused with the E. coli Adi system) system is composed of three enzymes, which are active at pH 3.1 or lower (Casiano-Colon & Marquis, 1988): the arginine deiminase, the ornithine transcarbamoylase and the carbamate kinase catalyse the overall conversion of arginine into ornithine, NH3 and carbon dioxide (CO2) with the formation of 1 ATP/arginine (Cunin et al., 1986). Beside its energy-generating function, the ADI system can further contribute to pH homoeostasis because ATP can be used to extrude cytoplasmic protons by the F1F0-ATPase (see above).
Some bacteria possess the agmatine deimination (AgDI) pathway, which yields putrescine from agmatine, the decarboxylation product of arginine (Jones et al., 2010).
Recently, glutaminase (see section ‘Amino acid-dependent XAR in E. coli: chemical and physiological issues’) and adenosine deaminases were shown to contribute to XAR in E. coli (Sun et al., 2012b; Lu et al., 2013). These enzymes use the relevant substrates to release NH3 in the cytoplasm.
Urea breakdown
The enzyme urease catalyses the hydrolysis of urea, yielding NH3 and carbamate, which spontaneously decomposes into a further molecule of NH3 and CO2 (Mobley et al., 1995). Also in this case, NH3 production results in relief from acid stress because this molecule combines with intracellular protons to yield  . Urease requires nickel for catalytic activity and consists of three subunits, a, b and c, encoded by the ureCBA operon. The gene cluster contains other genes (ureDEFG), encoding proteins required for the incorporation of nickel, urease biogenesis and urea metabolism.
. Urease requires nickel for catalytic activity and consists of three subunits, a, b and c, encoded by the ureCBA operon. The gene cluster contains other genes (ureDEFG), encoding proteins required for the incorporation of nickel, urease biogenesis and urea metabolism.
Mechanisms of repair or damage prevention of proteins
As stated in the section ‘’, exposure to acidic pH can also lead to the accumulation of damaged proteins in the cytoplasm. Depending on the bacterial species (Table 1), the induction of cytoplasmic (DnaK and GroEL) and periplasmic (HdeA and HdeB) chaperones occurs upon acid stress. In addition, in several microorganisms, the induction of components of the Clp protease complex, which by removing damaged proteins participate in protein homoeostasis, was observed. It should be noted, however, that the involvement of cytoplasmic chaperones and proteases in counteracting the acid stress has not been proved by genetic evidence for all the organisms listed in Table 1.
Modification of the cell membrane
The modification of the phospholipids in the internal membrane is also a mean of decreasing proton permeability. Indeed, in several bacteria, the resistance to acidic pH is associated with the conversion of unsaturated fatty acids (UFAs) into CFAs through a postsynthetic addition of a methyl group from S-adenosyl-methionine to the double bond of UFA (Chang & Cronan, 1999; Kim et al., 2005). In other microorganisms, such as oral bacteria, the acidic pH induces a shift in the membrane composition from short-chained saturated fatty acids to long-chained mono UFAs (Fozo et al., 2004).
Bacteria have evolved to adapt to environments with varying levels of acidity. However, while some, like E. coli, possess most (if not all) of the above-described mechanisms, others are armed only with some of them as the outcome of the best adaptation to the type of acid challenge they encounters. Below, we provide an overview of the occurrence of such mechanisms in a number of neutralophilic bacteria which infect domesticated animals and humans and are employed in food preparations. Key information is summarised in Table 1.
Escherichia coli
In pathogenic, commensal and laboratory strains of E. coli, ATR and XAR mechanisms have been intensively investigated (Table 1). The level of survival depends on the strain under analysis and the growth conditions preceding the acidic challenge (Lin et al., 1995, 1996). When E. coli is exposed to mild acidic conditions, the expression of several proteins is induced (Heyde & Portalier, 1990; Blankenhorn et al., 1999; Tucker et al., 2002). However, the peculiarity of this bacterium stands in its ability to mount an efficient XAR response, resulting from several different mechanisms. The XAR mechanism referred to as oxidative (glucose-repressed) acid resistance (AR1) is induced under oxidative growth conditions, that is, when the cells are grown to the stationary phase in complex media buffered at a pH of 5.5, with no glucose added (Lin et al., 1995). AR1 is dependent on RpoS, repressed by glucose and operative during pH 2.5 challenge in minimal medium as such, i.e. no addition of exogenous molecules (Lin et al., 1995; Castanie-Cornet et al., 1999). The activity of the F1F0-ATPase is important for protection by AR1 (Richard & Foster, 2004) as confirmed by the finding that this protein complex is still working during acid challenge at pH 2.5 (Sun et al., 2012a). To date, the mechanism behind AR1 has not been fully elucidated.
Under fermentative growth conditions, at least three XAR mechanisms are operative, which rely on the activity of amino acid-dependent decarboxylase/antiporter systems, namely the glutamate-, arginine- and lysine-dependent systems (Foster, 2004). The glutamate-dependent system (AR2) is by far the most effective (Lin et al., 1996; Diez-Gonzalez & Karaibrahimoglu, 2004) and relies on the action of the two isoforms of glutamate decarboxylase, that is, GadA and GadB, and the glutamate/γ-amino butyric acid (GABA) antiporter GadC (see section ‘Amino acid-dependent XAR in E. coli: chemical and physiological issues’). The arginine decarboxylase AdiA and the arginine/agmatine antiporter AdiC are the components of the arginine-dependent system (AR3), whereas the lysine decarboxylase CadA and the lysine/cadaverine antiporter CadB are the components of the lysine-dependent system (AR4). The latter two systems are typically induced by low pH, under anaerobic conditions and in the presence of the corresponding amino acid (Meng & Bennett, 1992a; Gong et al., 2003). Notably, full induction of the AR3 and AR4 systems under anaerobiosis requires the outer membrane, acid-inducible porins OmpC and OmpF, probably mediating the transport of the relevant amino acid under acidic conditions (Bekhit et al., 2011).
Besides the already mentioned periplasmic chaperones HdeA and HdeB (see section ‘’), Hsp31 might act as their cytoplasmic counterpart, in particular in AR2 and AR3 (Mujacic & Baneyx, 2007).
Salmonella enterica serovar Typhimurium
In the 1990s, several experiments provided evidence of increased synthesis in this organism of several acid shock proteins (ASP) during adaptation at mildly acidic pH (Foster & Hall, 1991, 1990; Foster, 1993). Both log-phase and stationary-phase grown cells can exhibit this adaptative process, but stationary-phase cells are more tolerant to acid pH than exponential-phase cells. Several regulatory proteins are involved in the induction of different subsets of ASPs, depending on the stage of growth. The alternative sigma factor RpoS and the regulator OmpR are responsible for induction of ASP in stationary phase ATR. On the contrary, Fur and PhoPQ (see section ‘’) trigger the expression of ASPs involved in the exponential-phase ATR. In particular, Fur controls a subset of ASPs in an iron-independent manner, contributing to ATR under organic acid stress (Foster, 1993; Hall & Foster, 1996), whereas PhoPQ is involved in the response to inorganic acid stress (Bearson et al., 1998).
A positive correlation between the ATR and virulence was observed in Salmonella, in that virulent strains are more acid tolerant than nonvirulent strains (Garcia-del Portillo et al., 1993; Wilmes-Riesenberg et al., 1997; Berk et al., 2005).
In S. enterica serovar Typhimurium, protection from acid stress by the inducible arginine, lysine and ornithine decarboxylases was demonstrated (Kieboom & Abee, 2006; Lee et al., 2007; Viala et al., 2011). It is notable that S. enterica serovar Typhimurium does not possess a glutamate-dependent system like the E. coli AR2 system.
The arginine-dependent system was first characterised in cells growing anaerobically, indicating that the presence of oxygen is detrimental for arginine-dependent survival at extreme acid pH (Kieboom & Abee, 2006). A greater level of arginine-dependent resistance to acid stress was observed also under aerobic conditions, but only when cells were acid-adapted prior to exposure to minimal medium at a pH of 2.5 (Alvarez-Ordonez et al., 2010).
A comparative study on the amino acid decarboxylase-dependent systems demonstrated a different contribution in ATR (Viala et al., 2011). In fact, the arginine decarboxylase system plays a predominant role at extreme acidic pH, but is ineffective during growth at moderate acidic pH; the ornithine decarboxylase only improves growth at moderate acidic pH in the absence of oxygen, but plays a minor role during survival; the lysine decarboxylase has a broader range of actions and confers both significant survival at pH 2.3 and growth improvement at pH 4.5 in an O2-independent manner.
The lysine and ornithine decarboxylase-dependent systems are also involved in the response to intracellular acidic pH in the Salmonella containing vacuole in macrophages, as demonstrated by the finding that when these two amino acids are added in the culture medium, the acidification of the vacuole was significantly delayed (Viala et al., 2011).
The proton translocation activity of the F1F0-ATPase also has an important role in the Salmonella ATR: mutants lacking this activity are acid sensitive and less virulent (Foster & Hall, 1991, 1990; Garcia-del Portillo et al., 1993).
Vibrio cholerae
In this Gram-negative bacterium, cells adapted to mildly acidic conditions (pH 5.7) can survive exposure to both inorganic and organic acid shocks (pH 4.5) and show higher colonisation of suckling and adult mice than do unadapted cells (Merrell & Camilli, 1999). When V. cholerae is exposed to organic acid challenge, c. 60 proteins are upregulated and 50 downregulated (Merrell et al., 2001). The lysine decarboxylase system plays an important role in the ATR (Merrell & Camilli, 1999; Kovacikova et al., 2010). Expression of the cadBA operon, encoding the antiporter and the decarboxylase, respectively, is induced at acidic pH through the ToxR-like regulator CadC; however, pH-independent and CadC-independent basal expression has also been observed (Merrell & Camilli, 2000). Low pH and low oxygen tension (anaerobiosis) also trigger the expression of AphB, a LysR-type activator, which in turn is responsible for the activation of the expression of cadC (Kovacikova et al., 2010). However, overlapping ATR effectors may be present because a cadA mutant, although impaired in an ATR assay, showed no decrease in colonisation of the suckling or adult mouse intestines (Merrell & Camilli, 1999). ToxR, a transmembrane DNA-binding protein that regulates expression of many virulence factors of V. cholerae, is also involved in the ATR response to organic acid shock, suggesting a possible link between ATR response and virulence in V. cholerae (Merrell & Camilli, 2002).
Helicobacter pylori
This microorganism has the remarkable ability to colonise the stomach, through which other bacteria only transit. Helicobacter pylori is primarily responsible for peptic ulcer disease (Marshall & Warren, 1984) and often associated with gastric carcinomas (Parsonnet et al., 1991). To move from the acidic stomach lumen to the colonisation site, the mucus layer, H. pylori, utilises the flagellar motility apparatus (Ottemann & Lowenthal, 2002), which is upregulated upon shift to acidic pH (Merrell et al., 2003; Wen et al., 2003).
To colonise the human stomach, H. pylori develops an adaptative response called acid acclimation, which differs from XAR and ATR and enables this bacterium not only to survive but also to grow in an acidic environment (Sachs et al., 2005). This mechanism is based on its ability to maintain its periplasmic pH close to neutrality even in the presence of an extreme external acidity, thereby allowing the maintenance of cytoplasmic pH also at near neutrality (Sachs et al., 2003; Marcus et al., 2005). The cytoplasmic urease (Mobley et al., 1995) and the proton-gated urea channel UreI, which increases urea entry into the cytoplasm at acidic pH (Weeks et al., 2000), are essential players for acid acclimation (Fig. 2). In fact, at neutral pH, the activity of urease (with an apparent Km of urease > 200 mM) is limited by urea entry which occurs by passive diffusion across the inner membrane. At acidic pH, the activation of a UreI-dependent transport of urea allows maximal urease activity (with an apparent Km ∼1 mM).
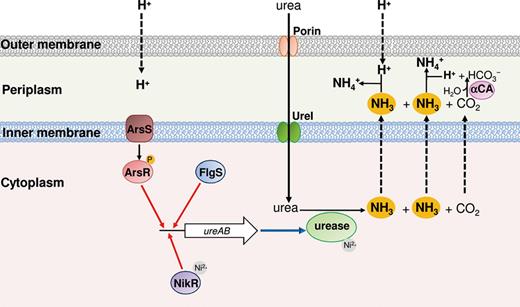
Scheme representing detection of low pH and acid acclimation in Helicobacter pylori during acid stress. The ArsR (red) and FlgS (blue) response regulators as well as the NikR transcriptional regulator (violet) bind the ureAB promoter, activating the expression of urease (red arrows). The urea dispersed in the periplasm moves into the cytoplasm through the activated UreI channel (green). The NH3 and CO2 produced by the urease (light green) diffuse in the periplasm, where the latter gas is hydrated by the α-type carbonic anhydrase (α-CA; pink).
Another enzymatic activity important for the survival of H. pylori in the acid environment of the stomach is the periplasmic α-type carbonic anhydrase (Fig. 2), an enzyme that converts the CO2 produced by urease and freely diffusing in the periplasm, into bicarbonate (Marcus et al., 2005; Scott et al., 2010). Thus, when H. pylori is in the stomach, (1) urea, which is 1–3 mM in the gastric juice, is taken up into the cytoplasm through the activated UreI channel; (2) CO2 and NH3 produced by urease diffuse rapidly across the inner membrane into the periplasm; and (3) CO2 is rapidly converted into bicarbonate by the periplasmic α-type carbonic anhydrase. In addition, NH3 is used to neutralise both the protons produced by this reaction and those entering into the periplasm from the environment. The outcome is that the pH of the periplasmic space of the bacterium is kept around 6.1, even if the pHo is at much lower values. The ureA and ureB genes encoding the urease structural components, the ureI gene and other genes coding for urease accessory components are all upregulated under acidic challenge (Sachs et al., 2005).
The pH sensing and the acid-induced transcription of the urease-encoding operons is mediated by the ArsRS two-component histidine kinase system and the cytoplasmic histidine kinase FlgS (Fig. 2; see section ‘’). Besides the urease gene cluster, the H. pylori ArsRS regulon includes more than 100 genes, among which are the amiE and amiF genes (Pflock et al., 2006), encoding the corresponding NH3-producing enzymes, AmiE and AmiF, which are aliphatic amidases probably with an active role in H. pylori acid acclimation (Zanotti & Cendron, 2010).
The activity of urease is dependent on nickel ion (Ni2+), which is inserted in the active site of the enzyme in the stoichiometry ratio of 24 Ni2+ per urease, and the expression of ureAB genes is induced by NikR (van Vliet et al., 2002). This latter transcriptional regulator binds Ni2+ that is free in the cytoplasm and maintains Ni2+ homoeostasis, by activating genes encoding the nickel-storage proteins and by repressing its own transcription and that of the genes encoding nickel uptake components (van Vliet et al., 2002; Muller et al., 2011). Because the solubility of nickel increases at low pH, NikR is indirectly involved in the response to acidic pH (van Vliet et al., 2004). However, the binding of this transcriptional regulator to the ureA promoter was observed at a low pH even in the absence of nickel, suggesting an active role of NikR in acid-induced expression of urease (Li & Zamble, 2009).
Brucella spp.
Brucellae are Gram-negative facultative intracellular bacteria highly pathogenic for animals and humans. They are transmitted by consumption of nonpasteurised milk and dairy products or by direct contact with infected animals or carcasses.
Brucella species encounter acidic environments in food, during the transit in the GIT and vagina of hosts, and inside the macrophages phagosome. It was suggested that the low pH acts as an intracellular signal for the regulation of genes involved in survival and multiplication within the phagocytic cell (Porte et al., 1999). In fact, in the phagosomes of murine macrophages containing live B. suis, the pH was found to be 4.0–4.5 1 h after uptake and to stay at this value until 5 h postinfection. Notably, the addition of vacuolar pH-neutralising reagents causes a strong reduction of intracellular bacterial viability.
Brucella exhibits potent urease activity that has been suggested to protect the bacteria during their passage through the stomach when they are acquired by the oral route (Sangari et al., 2007). Indeed, B. ovis, the only Brucella species lacking urease activity, is preferentially transmitted by sexual route. Moreover, analysis of B. abortus and B. suis mutant strains showed that urease is involved in XAR using survival assays performed at pH 2.0 and not at pH 4, and when the bacteria were administered to mice by the oral route and not by intraperitoneal injection (Bandara et al., 2007; Sangari et al., 2007).
Brucellae contain two urease operons, both located in chromosome I. The ure1 operon contains the genes ureDABCEFG, and it is the main genetic locus for the expression of the urease activity (Bandara et al., 2007; Sangari et al., 2007). The ure2 locus is composed of 13 genes, forming a single transcriptional unit, involved in urease production (ureABCEFGD), urea transport (ureT) and nickel transport (nikKMLQO). The analysis of the ureT mutant strain, which showed impaired urease activity and survival under acid exposure at low urea concentrations, provided evidence that UreT is an acid-activated urea transporter, like H. pylori UreI (Sangari et al., 2010).
Recently, it has been demonstrated that an active glutamate decarboxylase-dependent system is present in B. microti, a new Brucella species isolated from the common vole, red fox, and from the soil (Occhialini et al., 2012). Brucella microti contains a gadB gene located upstream of gadC, coding for the glutamate decarboxylase and glutamate/GABA antiporter, respectively. Interestingly, in the classical pathogenic Brucella species, either one or both genes are inactivated by stop codons and/or frameshift mutations. XAR assays showed that the resistance of B. microti to a pH of 2.5 is dependent on the presence of glutamate and on functional gadB and gadC genes (Occhialini et al., 2012). Moreover, B. suis, which is killed at a pH of 2.5 even in the presence of glutamate, displays an in vitro acid-resistant phenotype by heterologous complementation with the gadBC locus of B. microti. The reduced viability of a B. microti gadBC mutant relative to wild type in spleens and livers of Balb/c mice suggests that the glutamate decarboxylase-dependent system contributes to the protection of this Brucella species during passage through the host stomach (Occhialini et al., 2012).
Similarly, to E. coli, B. abortus possesses the periplasmic chaperone HdeA, which contributes to protection of B. abortus from the acidic conditions encountered in the phagosomal compartment of host macrophages (Valderas et al., 2005).
Listeria monocytogenes
In this food-borne pathogen, the alternative sigma factor σB is necessary for the full level of acid resistance observed in several strains of L. monocytogenes (Davis et al., 1996; Ferreira et al., 2003; Wemekamp-Kamphuis et al., 2004). Listeria stationary-phase cells are typically more resistant to acid stress than log-phase cells (Ferreira et al., 2003; Ivy et al., 2012). Indeed, stationary-phase cells have higher transcript levels for σB-dependent acid response genes than log-phase cells, although the latter cells show a more rapid induction of the stress response following acid shock (Ivy et al., 2012).
The glutamate decarboxylase-dependent system is required for survival in synthetic and ex vivo porcine gastric fluid (Cotter et al., 2001). In most L. monocytogenes strains, there is redundancy of decarboxylase and antiporter genes: gadT1 (lmo448), gadT2 (lmo2362) encode the antiporters, while gadD1 (lmo447), gadD2 (lmo2363) and gadD3 (lmo2434) encode glutamate decarboxylase isoforms. The five genes are localised in three separate genetic loci on the L. monocytogenes chromosome: gadD1T1, gadT2D2 and gadD3 (Conte et al., 2002; Cotter et al., 2005). Glutamate decarboxylase activity is subjected to strain variation and is correlated with the observed levels of ATR. The gadT2D2 locus plays an important role in survival under extreme acidic conditions, whereas the gadD1T1 locus facilitates growth under mild acidic conditions (Cotter et al., 2001, 2005). Recently, it was shown that the synthesis of GABA can be uncoupled from its efflux (Karatzas et al., 2012, 2010). According to a current model, under mild acidic conditions, the intracellular pool of glutamate is instantly used by the GadD2 enzyme, leading to a steady-state intracellular GABA levels. Indeed, the intracellular accumulation of GABA in different L. monocytogenes strains nicely correlates with their viability at pH 3.2. The activity of GadD1 appears only when the pH reaches 4.0, at which point, GABA is exported in the extracellular medium in an amount depending on the medium and the strain used (Karatzas et al., 2012).
Listeria monocytogenes possesses a functional ADI (i.e. arginine deiminase) system and putative AgDI (i.e. agmatine deiminase) enzymes (Ryan et al., 2009). Analysis of both the growth rate at sublethal pH and the rate of survival at lethal pH of mutants of the relevant genes indicated a prominent role for the ADI system in the ATR of L. monocytogenes (Ryan et al., 2009; Chen et al., 2011). The expression of the system, induced by low pH, anaerobiosis and addition of arginine, is mediated by a specific transcriptional activator ArgR, which is encoded by a distantly located gene. Under ADI-inducing conditions, the genes required for arginine biosynthesis are simultaneously upregulated, suggesting that for optimal operation of the listerial ADI system, de novo synthesis of arginine is also required (Ryan et al., 2009).
Interestingly, in L. monocytogenes acid shock, such as that encountered in stomach, upregulates regulons and specific genes involved in host invasion, intracellular survival and multiplication (Ivy et al., 2012). Thus, the transit through the stomach allows the bacterium to be better armed for the subsequent stages of infection, including intracellular growth and survival. Indeed, electron microscope analysis showed that, while unadapted L. monocytogenes cells are digested in the phagosome, those acid-adapted remain intact and in active multiplication within the phagosome or free in the cytoplasm (Conte et al., 2002).
It has recently been reported that during growth in brain heart infusion (BHI) medium, three genes coding for proteins participating in macromolecular repair, namely clpP, clpE and grpE, were induced after acid shock treatment (5–15 min) of log-phase cells grown at 37 °C (Ivy et al., 2012). This finding as well as previous ones (Karatzas et al., 2010) suggests that transcription can still occur at a pH of 3.5, which is nonlethal but does not support growth. This point may deserve further investigation in other bacterial species when subjected to acid stress.
Lactococcus lactis
This neutralophilic bacterium has GRAS status (‘generally regarded as safe’) and is mostly employed for the production and modification of food products, mainly in the dairy industry. L. lactis possesses ATR: just a 5-min exposure to mildly acidic pH (pH 5) enables cells to become more resistant to acid (pH 4), heat, NaCl, H2O2 and ethanol (O'Sullivan & Condon, 1997). During the adaptation period, a small subset of proteins, crucial for improved survival at low pH, are synthesised (Frees et al., 2003).
In L. lactis ssp. cremoris, there is a positive correlation between ATR, the cellular level of F1F0-ATPase and the internal pH acidification of cells in batch cultures induced with sublethal levels of acid; on the other hand, an inverse correlation is observed between the cytoplasmic levels of ATP and ATPase levels as a function of intracellular acidification (O'Sullivan & Condon, 1999). Indeed, a L. lactis mutant with reduced F1F0-ATPase activity is unable to maintain intracellular pH and is less viable at low pH (Amachi et al., 1998).
Another mechanism for maintaining pHi involves the glutamate decarboxylase-dependent system: mutants lacking either the gadB or the gadC gene products are acid sensitive (Sanders et al., 1998). Expression of L. lactis gadCB is high in cultures allowed to acidify during growth and depends on the presence of glutamate and chloride ions in the medium. Therefore, the gadCB-mediated XAR system may play a significant role for the survival of lactococcal cells in the stomach, where HCl is produced in large quantities by gastric cells, or during cheese production when high levels of both NaCl and glutamate are present (Sanders et al., 1998).
Lactobacillus spp.
Besides being the major components of the human microbiota, lactobacilli are also among the LAB commonly used in the food industry and as probiotics. To exert their health-promoting functions in the human gut, probiotic bacteria must recover viability after passage through the strongly acidic gastric compartment.
The involvement of an ADI system in the ATR was proved in the species L. sakei and L. reuteri (Champomier Verges et al., 1999; Rollan et al., 2003). The ADI pathway in L. sakei is induced by the presence of arginine, low oxygen and low glucose levels (Champomier Verges et al., 1999).
Several lines of evidence suggest that in LAB, the amino acid-dependent systems play a role as energy suppliers, rather than being used to neutralise the environmental pH. In L. curvatus, the action of the ornithine decarboxylase-dependent system results in a net efflux of positive charges with the consequent generation of an electrical gradient, that together with the transmembrane pH gradient (alkaline inside), gives rise to a proton motive force, used for generating metabolic energy (Cid et al., 2008). By measuring ΔpH and ΔΨ across the membrane in L. buchneri, it was demonstrated that histidine decarboxylase and histidine/histamine antiport also participate in energy production (Molenaar et al., 1993).
The glutamate decarboxylase-dependent system has also been extensively studied in LAB due to the link between decarboxylation of glutamate and ATP synthesis through the generation of a proton motive force (Higuchi et al., 1997) and to the beneficial role of GABA in food (Li & Cao, 2010). Recently, this system was analysed in L. reuteri 100-23 strain which is used in industrial sourdough fermentations (Su et al., 2011): this strain is characterised by a short period of growth, followed by an extended period of fermentation at pH 3.2–3.6. In the L. reuteri genome, the glutamate decarboxylase (gadB) gene is part of a cluster, containing genes encoding two glutamate/GABA antiporters (gadC1 and gadC2), and a glutaminase (gls3). Analysis of a gadB mutant demonstrated that this gene contributes to L. reuteri acid resistance at pH 2.5 and to the competitiveness of L. reuteri in sourdoughs. In the same work, it was proposed that the XAR of L. reuteri relies on glutamine, available in cereal fermentations, by means of the product of the gls3 gene, which converts glutamine into glutamate, thereby generating NH3 and supplying the glutamate decarboxylase-dependent system with its substrate (Su et al., 2011). This finding is probably not restricted to L. reuteri as a glutaminase-dependent system has recently been reported to provide XAR to E. coli (see section ‘Amino acid-dependent XAR in E. coli: chemical and physiological issues’).
The analysis of the fatty acid content of L. casei following growth at neutral and acidic pH showed an increase of long-chained, mono UFAs (Fozo et al., 2004), saturated fatty acids, and CFAs (Broadbent et al., 2010) in the latter condition.
Bacillus cereus
This is a food spoilage microorganism, which is also able to grow in soil (Vilain et al., 2006). When stationary-phase cells are pre-adapted to sublethal conditions (pH 5), B. cereus develops an ATR, which requires de novo protein synthesis (Thomassin et al., 2006), becoming more tolerant to an acid challenge at pH 4.0 (Browne & Dowds, 2002; Jobin et al., 2002).
Transcriptomic analysis and radical formation assays showed that in B. cereus, the exposure to acidic pH in aerobic conditions induces a secondary oxidative stress (Mols et al., 2010a, b): a perturbation of the electron transfer chain, with a premature leakage of electrons to oxygen, was suggested to be the primary cause of the formation of reactive oxygen species. Indeed, increased expression of genes encoding enzymes that can counteract the reactive oxygen species (i.e. superoxide dismutase, catalases and nitric oxide dioxygenase) and act via alternative electron donor and acceptor mechanisms (such as nitrate and nitrite reductase) occurs (Mols et al., 2010a).
Bacillus cereus does not use the F1F0-ATPase as a proton pump. Indeed, the genes encoding the subunits of the F1F0-ATPase are highly downregulated in cells exposed to sublethal pHs (Mols et al., 2010a). In these conditions, the chaperone-encoding gene dnaK and the protease-encoding gene clpC were also found to be upregulated.
In B. cereus ATCC14579, amino acids improve ATR: cells grown at pH 7.0 are more resistant to acid shock when glutamate, arginine or lysine are present in the medium. The amino acid-dependent pH homoeostasis in B. cereus relies on arginine and lysine decarboxylase (Senouci-Rezkallah et al., 2011). In fact, the transcription of the corresponding genes is activated during acid adaptation. Notably, glutamate decarboxylase (GadB) activity was assayed in this strain (Senouci-Rezkallah et al., 2011) although the corresponding gene is absent in this as well as in some other B. cereus strains and the gadC homologue is never present (Mols et al., 2010a). It was therefore proposed that glutamate might be decarboxylated by some other decarboxylases, that is, arginine decarboxylase (Senouci-Rezkallah et al., 2011).
The sporulating bacterium B. subtilis can live in different environments including the GIT. Indeed, ingested Bacillus spores upon exiting from the stomach can germinate in the gut at the level of the jejunum where cells are able to grow and resporulate (Tam et al., 2006). During vegetative life, B. subtilis maintains pH homoeostasis when the pHo decreases to 6 (Kitko et al., 2009) and growth in this moderate acid induces adaptation to a more acid medium (i.e. pH 4.5) (Wilks et al., 2009). Transcriptomic analysis showed that in these conditions, several genes encoding NAD(P)-dependent dehydrogenases were upregulated. This could imply that pH homoeostasis is provided by the proton-pumping activity of the electron transport chain. In analogy to B. cereus, low pH induces genes responding to oxidative stress (Wilks et al., 2009).
Streptococcus spp.
Members of the genus Streptococcus are the most abundant inhabitants of the oral plaque microbiota (Dewhirst et al., 2010).
Concerning the ATR, much interest has aroused by the S. mutans group which, due to its higher ability of producing acids via glycolysis (acidogenicity) and to tolerate exposure to acidic pH (aciduricity) with respect to other streptococci, represents the major causative agent for dental caries (Loesche, 1986). Cells adapted at pH values close to the minimum for growth (pH 5) better withstand potentially lethal acidification (pH 2.5) (Belli & Marquis, 1991).
The ATR in several Streptococcus species of dental plaque is mostly based on the F1F0-ATPase (Bender et al., 1986; Kobayashi et al., 1986; Kuhnert et al., 2003), and the pH profiles of this enzyme from different streptococci correlate well with the ATR of the different strains. Acidic pH also stimulates the expression of the F1F0-ATPase at the transcriptional level (Kuhnert et al., 2004; Len et al., 2004b).
The role of the ADI system in protecting cells from acid stress was demonstrated in S. rattus, S. sanguis (Casiano-Colon & Marquis, 1988), S. pyogenes (Degnan et al., 2000) and S. suis (Gruening et al., 2006). Streptococcus mutans, which does not possess the genes for ADI system in its genome, has been shown to express an AgDI system. The genes encoding the three enzymes of the latter system constitute an operon and also include a gene encoding an amino acid transporter and, nearby, a gene encoding AguR, a transcriptional regulator required for the induction of the system by low pH and agmatine (Griswold et al., 2004; Liu & Burne, 2009; Liu et al., 2009).
In S. salivaris, urease plays an important role in protecting from acidic stress. The functioning of the system is ensured by that the fact that the oral cavity contains 3–10 mM urea in the saliva (Chen et al., 2000). Indeed, the viability of wild-type S. salivaris cell at pH 3 increases by increasing the concentration of urea, whereas a ureC-deficient strain does not survive (Chen et al., 2000). Notably, urease levels in the biofilm of the dental plaque are higher than those detected in planktonic cells growing in continuous culture, pointing to the influence of this enzymatic system in pH homoeostasis of the oral microbiota (Li et al., 2000).
An alteration in the content of membrane fatty acids was observed in S. gordonii, S. salivarius and S. mutans (Quivey et al., 2000; Fozo et al., 2004). In particular, in S. mutans, both the growth in acidic media and the acidification due to glucose metabolism give rise to a gradual increase in the proportion of long-chained mono UFAs in the membrane (Fozo & Quivey, 2004a). The presence of a fatty acid biosynthesis inhibitor or a mutation in the fabM gene, encoding an enzyme involved in UFA production, prevented the changes in membrane composition and rendered this microorganism more acid-sensitive (Fozo & Quivey, 2004a, b).
Transcriptomic and proteomic analyses revealed that the acidic environment causes changes in the cellular metabolism of Streptococcus (Len et al., 2004a; Martinez et al., 2010). In particular, in S. mutans and S. sobrinus, many glycolytic enzymes and those involved in malolactic fermentation, that is, conversion of l-malate into lactic acid and CO2, are upregulated by low pH and this can be useful to increase ATP production and control pHi homoeostasis by virtue of the proton-consuming decarboxylation reaction involved.
Also, the synthesis of branched amino acids is upregulated at pH 5. The proposed mechanism for acid resistance is directly via a decrease in the cytoplasmic concentration of protons, by removing reducing equivalents in the form of pyruvate and 2-oxobutanoate, and indirectly by the consumption of NADPH and by the production of NH3 (Len et al., 2004a; Martinez et al., 2010). Indeed, the ilvE gene, encoding the branched-chain amino acid aminotransferase, is upregulated at acidic pH and an ilvE mutant strain exhibits defects in the growth at pH 5.4 as well as in its ability to survive a challenge at pH 2.5 (Santiago et al., 2012).
Bifidobacterium spp.
Bifidobacteria are Gram-positive anaerobes and typical inhabitants of the distal gut. Even though much work has been carried out to investigate the potential benefits exerted by Bifidobacterium species on human health as probiotics, little is known about their acid survival mechanisms.
Bifidobacteria possess ATR mechanisms (Maus & Ingham, 2003; Waddington et al., 2010; Jin et al., 2011). Indeed, B. lactis was shown to better survive in synthetic human gastric fluid at pH 3.5, when pre-exposed to the low pH in a yogurt sample (pH from 6.5 to 4.7) (Maus & Ingham, 2003). Some species, such as B. lactis and B. animalis, are more acid tolerant than others, and this feature correlates with the activity of the F1F0-ATPase at pH 4 and the induction of the atp operon (Ventura et al., 2004). In B. longum, B. adolescentis and B. pseudocatenulatum, which are not acid tolerant, the F1F0-ATPase activity is high at less acidic pH (Matsumoto et al., 2004).
A proteomic analysis performed on B. longum showed that exposure to pH 4.8 induces not only the F1F0-ATPase, but also enzymes involved in carbohydrate metabolism, energy production and conversion, and amino acid metabolism. In particular, the enzymes of the biosynthesis pathway of branched-chain amino acids and glutamine synthetase were found to be upregulated. This analysis together with the higher concentration of valine and  ion detected after growth at pH 4.8 suggested a role of the deamination of branched-chain amino acids in maintaining the internal pH of the cells (Sanchez et al., 2007).
ion detected after growth at pH 4.8 suggested a role of the deamination of branched-chain amino acids in maintaining the internal pH of the cells (Sanchez et al., 2007).
Among Bifidobacteria species, B. dentium and B. longum are members of the oral microbiota implicated in human dental caries. They are able to survive and maintain their internal pH in acidic conditions in the absence of an extracellular energy source, an environment similar to that encountered in caries lesions, where energy sources such as carbohydrates are not always available and thus where the F1F0-ATPase does not function (Nakajo et al., 2010). The genome of B. dentium contains two adjacently located genes encoding glutamate decarboxylase (GadB) and its cognate antiporter (GadC), respectively, which are not present in other bifidobacterial genomes so far sequenced (Ventura et al., 2009). Both genes are significantly upregulated in response to growth at pH 4. Notably, the genes coding for molecular chaperones and participating in the deamination of branched-chain amino acids, proteolytic degradation, amino acids uptake and catabolism were also found to be upregulated (Ventura et al., 2009).
Clostridium spp.
Clostridium, with its more than 100 species, is an ubiquitous genus, isolated from soil, water, sewers and intestine. Notably, Clostridium is identified in natural AMD or in the bioreactors for AMD bioremediation, which typically include acidophiles. This may be due to the acid resistance feature of these species, associated with their metabolic ability to retrieve energy from different carbon sources such as cellulosic material (Porsch et al., 2009; Lu et al., 2011; Sanchez-Andrea et al., 2011).
The survival observed in the GIT of mice orally inoculated with vegetative cells of C. perfringens has provided an indication that this bacterium is able to survive passage through the stomach (Tennant et al., 2008). Moreover, a sublethal acid shock at pH 4.5 for 20 min was shown to increase the ATR at pH 3.5 at least 15-fold, thus suggesting that this microorganism is also able to develop an ATR (Villarreal et al., 2000).
Clostridial species were reported to possess glutamate decarboxylase activity (Gale, 1940), and the biochemical characterisation of this enzyme from C. perfringens provided evidence that its expression is affected by pH changes of the culture medium and that the optimum pH of the purified enzyme is 4.7 (Cozzani et al., 1970, 1975). These data suggest a role for glutamate decarboxylase-dependent system in survival of this bacterium during acid stress. This is supported by the recent observation that in C. perfringens, the gadC gene, coding for the glutamate/GABA antiporter, is adjacent to the gadB gene coding for glutamate decarboxylase on the chromosome (De Biase & Pennacchietti, 2012). Moreover, a C. acetobutylicum strain carrying a plasmid that contains a genomic fragment encoding small noncoding RNAs shows robust tolerance to a variety of carboxylic acids and an upregulation, via an unidentified mechanism, of a homologue of gadC (Borden et al., 2010).
Mechanisms of detection of low pH
In order for cells to turn on a response that enables them to adapt to the presence of potentially damaging levels of acid, they must constantly monitor the surrounding environment and be able to detect that acid, directly or indirectly. Any molecule with residues that are titratable by pH has the potential to detect changes in pH (Slonczewski et al., 2009), but the molecular details of exactly how detection of the low pH signal occurs and how it is converted into a genetic response are only recently beginning to be understood. In bacteria, the major players in the detection mechanism are two-component systems (TCS) where the histidine kinase protein is an integral membrane protein with a periplasmic domain. It is unsurprising that bacterial cells exposed to acidic stress use extra-cytoplasmic ‘sensors’, given the higher vulnerability of periplasmic proteins and the relative difficulty with which protons cross the cytoplasmic membrane. Below, several different detection systems are discussed, to illustrate some of the different principles involved in acid detection; the extent to which these different systems have been studied varies and this is reflected in the level of detail presented for each system.
The PhoPQ system of Salmonella
Salmonella Typhimurium exhibits a classic ATR, being quite sensitive to a low pH of 3.3 if grown at pH 7.6, but showing 1000-fold increased survival if grown for one generation at pH 5.8 (Foster & Hall, 1991). A phoP mutant showed greatly increased sensitivity to acid and decreased inducibility of the ATR. Because PhoP is the response regulator of the TCS system PhoPQ, this suggests that this system may have a role in detecting acidity. A point in the Salmonella life cycle where this ability could be particularly important would be in the macrophage, where cells are exposed to a pH of around 5.5, and indeed, S. typhimurium strains with mutations in phoP or phoQ have reduced survival in mouse macrophages and show reduced virulence in infection of mice (Miller et al., 1989). PhoQ detects several signals including the presence of divalent cations and antimicrobial peptides, but its specific ability to detect low pH directly has been demonstrated in vitro by reconstitution in vesicles with an acidified interior (Prost et al., 2007). The periplasmic domain of PhoQ has been purified, enabling the detailed study of its structure using NMR, and this showed that PhoQ undergoes a clear conformational change as the pH drops, with a maximally flexible state at pH 5–5.5. A similar, though, not identical structural change was seen in the structure of a His157Asn mutant in this domain, and this mutation when present in the full-length protein leads to derepression of a PhoP-regulated promoter even at a neutral pH. Thus, it has been proposed that the normal nonphosphorylated and inactive state of PhoQ is maintained in that state by a hydrogen-bonded network of amino acid residues including His157 and that either protonation or mutation of that residue disrupts the network, leading to a greater flexibility of the periplasmic domain of the protein. This in turn promotes a structural change in the protein that propagates through the membrane, causing activation of the autokinase activity of PhoQ followed by phospho-transfer to PhoP and the activation of the genes of the ATR (Prost et al., 2007; Prost & Miller, 2008). The fact that this change can be mimicked by a variety of mutations including His157Ala shows that it is the imidazole ring of this residue that is important in maintaining this network, and loss of this ring (rather than simply the loss of a positive charge) destabilises the network. This destabilisation is also likely to result from the effects of acidification on other residues in the network, including aspartate and glutamate residues, so a simple model where His157 is the sole acid-sensing residue is not thought to be correct, but it is clear that this residue plays a critical role.
The ArsRS system of H. pylori
Another example where a histidine residue is important but not the sole determinant in sensing low pH is provided by the ArsRS TCS of H. pylori. This TCS is one of several systems in H. pylori which help the organism to sense and respond to low pH, and it regulates the urease genes which are crucial for the ability of this organism to survive in the human stomach (Fig. 2; Pflock et al., 2005). At acidic pH, ArsR, phosphorylated by ArsS, binds to the ureA and ureI promoters. At neutral pH, ArsR is not phosphorylated and binds to the promoter of the gene encoding an antisense small RNA, 5′ureB-sRNA, targeted at the 5′ end of ureB, which promotes premature termination of transcription of ureAB mRNA (Wen et al., 2013). There is good evidence that a histidine residue in the sensor kinase ArsS plays a role in this activation. In this case, the fact that the periplasmic domain contains only seven histidine residues made it experimentally sensible to mutate each one individually and record the effects on acid-inducible gene expression in H. pylori. Mutation of His94 (but none of the other histidine residues) to glutamine was found to very substantially reduce the expression of two ArsRS regulated genes at pH 5 (Muller et al., 2009). Double mutation of both His94 and His44 to alanine led to restoration of acid inducibility, suggesting that under certain circumstances, other residues can take on the role of detecting acid. The structural changes caused by these mutations have not been characterised.
The cytoplasmic histidine kinase FlgS is required for periplasmic pH homoeostasis in H. pylori at the extreme pH of 2.5. The response regulator for the acid response is unknown: FlgS belongs to the FlgRS TCS, which regulates flagellar gene expression via FlgR, which, however, is not implicated in the response to acidity (Fig. 2). FlgS regulates several pH homoeostatic genes overlapping with ArsS (Wen et al., 2009). Thus, in H. pylori, the two sensor kinases are required to respond to different degrees of growth medium acidity: ArsS for milder and FlgS for stronger (Scott et al., 2010). Notably, at acidic pH, ArsS and FlgS are both responsible for recruitment of urease to the inner membrane in association with UreI, the urea channel, thereby accelerating the periplasmic buffering (Scott et al., 2010; Marcus et al., 2012).
The CadC proteins of E. coli and Salmonella
CadC is an example of a one-component system, where the sensory and transcriptional regulation mechanisms are incorporated in a single integral membrane protein. In the presence of lysine and at slightly acidic pH, E. coli CadC activates the expression of the two genes of the AR4 system: cadA and cadB (Watson et al., 1992). It has also been shown to act as a negative regulator of the AR3 system and often to be absent in Shigella and enteroinvasive E. coli strains (Casalino et al., 2010). CadC alone is responsible for the detection of low pH, while the lysine permease LysP is needed for the detection of lysine (Tetsch et al., 2008). Mutants of E. coli CadC that failed to respond normally to low pH (in this case, 5.8) were shown to map in its periplasmic domain (Dell et al., 1994). The subsequent solution of the structure of this domain enabled a directed mutagenesis approach aimed at dissecting how CadC detects low pH (Haneburger et al., 2011). These experiments showed that a negative patch of the protein was crucial for pH detection. Mutation of the acidic residues within this patch tended to perturb the function of CadC, making it either not responsive to low pH or stuck in a permanent on-state even at neutral pH.
Interestingly, similar phenotypes were also seen for some mutations of residues that cannot be protonated, showing that (as with PhoQ in Salmonella) it is likely to be the overall structure of this patch, not merely the protonation state of some of the residues within it, which enables the detection of low pH by CadC. Many of these residues are at the dimer interface of the CadC periplasmic domain, so a change in the orientation of the two monomers in a CadC dimer may be responsible for the transduction of the detection signal across the membrane to where it can affect the binding of CadC to the target promoters (Haneburger et al., 2011). Support for this model comes from two recent reports. One showed that CadC contains two periplasmic cysteine residues that form a disulphide bond at pH 7.6, but not at 5.8. Mutations that disrupt this disulphide bond lead to constitutive activation of the cadA and cadB genes at pH 7.6 if lysine is also present and remove the dependency on lysine at pH 5.8. These data are completely consistent with the model where the relative orientation of the two periplasmic domains in the dimeric CadC determines its activity (Tetsch et al., 2011). The second showed that periplasmic cadaverine, the decarboxylated product of lysine, inhibits the activity of CadC by binding to two sites in CadC, one of which overlaps with the cluster of amino acids which are thought to sense pH, and a model was proposed whereby the binding of cadaverine to both sites stabilised the inactive conformation of the CadC dimer (Haneburger et al., 2012).
A further aspect of the mechanism of CadC activation has been described in S. enterica serovar Typhimurium, where it was shown that activation of CadC requires a post-translational cleavage event that liberates an N-terminal fragment which then localises to the inner membrane and is required for activation of the cadBA operon. Mutants in which this cleavage is blocked fail to show activation (Lee et al., 2008b). Whether or not a similar mechanism operates in E. coli or in other bacteria that possess the CadC system has not been reported; however, as the CadC protein sequences are 100% conserved between these two organisms, it is highly likely to be the case. The mechanism of cleavage remains unknown.
In E. coli, expression of CadC is constitutive, whereas in Salmonella, acidic pH and lysine in the growth medium increase the expression of the regulator, which in addition to cadBA induces also the expression of the genes encoding the porins OmpC and OmpF, while it represses the expression of proteins involved in glycolysis, energy production and stress tolerance (Lee et al., 2007). In addition, in vivo expression studies have shown that cadC and cadB are induced during infection of BALB/c mice and macrophages (Heithoff et al., 1997; Eriksson et al., 2003).
Detection mechanisms in the E. coli AR2 network
Description of the ways in which E. coli induces systems for acid resistance is complicated by the fact that there are several different acid resistance systems involved. The well-characterised AR2 system, discussed in detail below, shows a high level of complexity in its regulation and the ways in which several different detection and response mechanisms overlap.
The EvgS/EvgA TCS sits at the top of the regulatory pathways of the AR2 system, and it is thought that it acts as a primary detector of low pH in E. coli in exponentially growing cells (Fig. 3). The evidence for this is that (1) overproduction of the response regulator EvgA leads to a constitutive acid-resistant phenotype, with activation of many of the genes known to be involved in the AR2 network (Masuda & Church, 2002; Ma et al., 2004); (2) a mutation in the sensor kinase EvgS that leads to constitutive expression of the genes regulated by EvgA also makes cells acid resistance (Itou et al., 2009); and (3) deletion of evgA leads to a complete loss of the normal acid-induced expression of the genes of the AR2 network in exponential phase (Burton et al., 2010).
Schematic representation of detection of low pH and activation of AR2 in Escherichia coli. The activation of the EvgSA TCS (red), occurring at low pH, triggers the expression of the ydeP gene and safA-ydeO operon. YdeO (green) activates the expression of gadE. The protein connector SafA (dark green) activates the PhoPQ TCS (violet) which exerts a negative control on ydeP and safA-ydeO and induces the expression of iraM. The anti-adaptors IraM (pink) inhibits the activity of RssB (grey), which otherwise directs RpoS (yellow) to the degradation through the cellular protease ClpXP. RpoS, freed from RssB, and GadE (blue), together with RcsB (orange), directly activate the expression of genes encoding the components of AR2 system (gadA, gadBC and hdeAB).
EvgS has a large predicted periplasmic domain consisting of two tandem venus flytrap domains. These domains, characterised by two subdomains with a solute-binding cavity or cleft between them (Quiocho & Ledvina, 1996), are often found in periplasmic proteins which have to bind and then import small molecules, and these may constitute the part of the protein that senses low pH in the periplasm.
Most periplasmic domains in the sensor of TCSs are anchored by transmembrane helices on both sides of the domain, but the evidence for a transmembrane helix at the N-terminus of EvgS is not strong, and it may be that this protein has only a single transmembrane helix. Immediately after the predicted membrane-spanning helix, there is a PAS domain. Such domains are common in sensory molecules, but it is not clear whether the PAS domain in EvgS is also involved in some way in sensing acid or whether it has a simpler structural role in the protein. The constitutively active mutant of evgS described above maps in this domain (Itou et al., 2009).
Most sensor kinases of TCSs have a kinase domain which phosphorylates an internal histidine residue on activation of the kinase activity, the phosphate being subsequently transferred to an aspartate residue on the response regulator. EvgS is one of only five unorthodox sensor kinases in E. coli which has an internal phosphorelay where the phosphate is transferred to an internal aspartate and then to a second histidine before finally being transferred to a histidine residue in EvgA. The reason for this complexity is not clear but theoretical studies suggest that kinases with internal phosphorelays may be more sensitive in their response to external stimuli, while also be less affected by noise (Kim & Cho, 2006). It seems likely that EvgS responds directly to periplasmic acidification, but it has been shown in vitro that the kinase activity of a soluble cytoplasmic domain of the protein is inhibited by oxidised ubiquinone, and this inhibition is decreased by some mutations in the PAS domain. Thus, there may be multiple inputs into the activity of EvgS (Bock & Gross, 2002).
Interestingly, the closest homologue to EvgS in E. coli is the redox sensor ArcB, which is regulated by the redox state of the ubiquinone pool, and is also an unorthodox sensor kinase (Georgellis et al., 2001). ArcB is regulated by the reversible oxidation of a cytoplasmic disulphide bond (Malpica et al., 2004), but one of the cysteine residues involved is not found in EvgS. It has been reported recently that ArcA (the response regulator of the ArcAB system) regulates expression of gadE under anaerobic conditions, primarily by antagonising HNS-mediated repression of gadE (Deng et al., 2013), but whether this is affected by acid has not been reported.
Among the targets of activated EvgA are ydeP and the safA-ydeO operon (Fig. 3). The function of YdeP is obscure, although it is known to have a role in acid resistance (Masuda & Church, 2002). YdeO is an activator protein that itself activates gadE, the gene encoding GadE, a central regulator of the AR2 network. SafA (also known as b1500), the small protein encoded by the gene immediately upstream from ydeO, is a connector protein which mediates crosstalk between TCSs. EvgA-induced expression of SafA activates the PhoPQ system, even in the presence of high concentrations of Mg2+ which usually inhibit PhoPQ. This is due to direct interaction between the periplasmic domain of SafA and the sensor domain of PhoQ (Eguchi et al., 2007, 2011, 2012). PhoQ is itself directly activated by low pH (as described above), particularly in the presence of low Mg2+, but in E. coli, this activity is further stimulated by SafA.
XAR in exponential phase via the AR2 system in E. coli is thus partly caused by direct activation of the AR2 genes via YdeO and GadE, and partly via the SafA-mediated activation of PhoPQ (Fig. 3). Activation of the PhoPQ system causes increased acid resistance by regulation of RpoS levels. This alternative sigma factor (known also as σ38 or σS) is present at high levels in stationary phase cells and also increases in its cellular levels and activity following many different stresses, including low pH (reviewed by Hengge-Aronis, 2002; Hengge, 2009). RpoS-regulated genes are important in making cells more resistant to a wide variety of stresses, including acid, in many different organisms. In E. coli, the levels of RpoS are known to vary significantly between different strains (Ferenci et al., 2011) and mutants of E. coli O157:H7 lacking rpoS are less competitive than the wild-type strain in animal models of infection due to their lower acid resistance (Price et al., 2000).
RpoS levels also increase when E. coli cells growing logarithmically are shifted to pH 5 (Heuveling et al., 2008), and a link between the acid-sensing TCS EvgAS and the levels of RpoS has been proposed (Eguchi et al., 2011). In fact, one of the targets of the activated PhoP response regulator is the iraM gene, encoding one of several ‘anti-adaptors’ that control RpoS levels by inhibiting the activity of the RssB protein, which in turn directs RpoS to the cellular protease ClpXP (Fig. 3). Thus, activation of PhoP leads, via activation of IraM and suppression of RssB, to increased RpoS levels, which can enhance expression of several genes of the AR2 system including the central regulator, GadE.
A further level of complexity linking detection of acid and regulation is superimposed on this system via the RcsB protein (Fig. 3). This protein, itself a response regulator, is required for much of the expression of the AR2 system. It is needed for expression of both GadE-dependent and GadE-independent promoters following acidification, and its absence causes a strong acid-sensitive phenotype (Castanie-Cornet et al., 2007, 2010; Krin et al., 2010; Johnson et al., 2011). RcsB can be activated by many external signals including low pH (Clarke, 2010), and it is implicated in the regulation of several stress response pathways. In addition to its effects on many of the promoters of the AR2 system and on levels of H-NS protein, which also regulate these promoters, RcsB activation also has the effect of increasing cellular RpoS levels via activation of the small RNA RprA, which in turn enhances the translation of RpoS mRNA (Majdalani et al., 2002). Thus, RcsB influences several different aspects of acid resistance and must be considered a key component of XAR mechanisms in E. coli. Again, the precise nature of the way in which whereby pH directly leads to RcsB activation is not known.
Amino acid-dependent XAR in E. coli
In the previous sections we have described the systems that neutralophilic bacteria employ to protect themselves from the deleterious effects of increasing protons level in the cell. However, the extent to which this can be accomplished varies significantly among the different bacteria.
The human stomach is clearly the anatomical site where acid stress is more harsh for bacteria entering in contact with us. While H. pylori uses the urease system to achieve acid acclimation (reviewed by Sachs et al., 2003, 2005; Zanotti & Cendron, 2010), other bacteria which do not possess the urease system mostly rely on the amino acid-dependent XAR systems which are also extremely powerful (Foster, 2004; Kanjee & Houry, 2013). In this section, we will deal in particular with the most potent of them, that is, the E. coli AR2 system, also in light of its potential involvement in pathogenicity.
Chemical and physiological issues
The extreme acidity (pH 1.5–2.5) of the gastric compartment in the mammalian GIT is ascribed to the activity of the gastric H+,K+-ATPase (or ‘proton pump’), an enzyme present in the gastric mucosa from cartilaginous fish to mammals, which exchanges luminal K+ for cytoplasmic H+ (Koelz, 1992; Smolka et al., 1994). Gastric acidity has long been recognised to perform fundamental functions such as (1) modulation of the digestive process, by halting carbohydrate digestion and triggering digestion of proteins via their denaturation and the stimulation of pepsin's proteolytic activity; (2) augmentation of the absorption of calcium and iron; and (3) prevention from gut colonisation and spreading to other sites of the body by potentially harmful ingested bacteria (Giannella et al., 1972; Howden & Hunt, 1987; Tennant et al., 2008). This latter function is of particular importance for human health. In fact, transit through the gastric compartment is always a challenge for orally acquired bacteria, whether they are friends or enemies.
As mentioned in the previous sections, neutralophilic bacteria that during their life face the gastric compartment with a pH lower than 2.5, which is too low to support growth, either succumb or execute survival strategies (see ‘ATR and acid resistance as protective mechanisms’). In the latter case, those bacteria which are poorly armed to control this stress perform a massive attack involving billions of cells, of which only few will survive and reach the intestine (e.g. V. cholerae), whereas others are equipped with potent XAR mechanisms that ensure that most, if not all, ingested cells will pass through the stomach safely (e.g. E. coli, S. flexneri). As a consequence, these latter bacteria have an extremely low infectious dose (it can be as low as 10–100 cells). Therefore, bacterial XAR can be regarded as a fitness and a virulence trait, because it increases the chance of pathogens to reach the gut and cause infection.
Pathogens entering the host via ingestion of contaminated food are typically in stationary phase. Therefore, it is not surprising that XAR systems are maximally expressed in bacteria at this stage of growth, with the AR2 system being a notable exception (see ‘Detection mechanisms in the E. coli AR2 network’ and Ma et al., 2004; Burton et al., 2010). Based on a number of reports (Gorden & Small, 1993; Lin et al., 1995; Hersh et al., 1996; Lin et al., 1996; Riggins et al., 2013), the criteria adopted to define XAR in E. coli, the most thoroughly studied microorganism, are the following: (1) no need for pre-adaptation to a mildly acidic pH prior to extreme acid challenge; (2) robust survival (≥ 10%; that is < 1 log of bacterial loss) upon exposure of 1 h or more (up to 6 h) to a pH ≤ 2.5; and (3) dependence on the addition of specific amino acids in the pH 2.0–2.5 minimal medium used for challenge. The latter point means that the contribution of each of the XAR systems dependent on amino acids, namely the glutamate-, glutamine-, arginine- and lysine-dependent systems, can be distinguished by simply supplementing the minimal medium in which the microorganism is challenged with one of the aforementioned amino acids.
To date, the microorganisms that were shown to possess at least one amino acid XAR system (Table 1) include commensal and pathogenic strains of E. coli, several food-borne pathogens such as S. flexneri, L. monocytogenes and S. enterica serovar Typhimurium (Lin et al., 1995, 1996; Cotter et al., 2001; Waterman & Small, 2003; Kieboom & Abee, 2006; Tennant et al., 2008; Lu et al., 2013) and more recently also a newly discovered species of Brucella, that is, B. microti (Occhialini et al., 2012). For some of these systems, anoxic growth (i.e. no O2) was shown to be essential for the full development of XAR: in S. enterica, the expression and activity of the AR3 system can be measured only under anaerobic conditions and not under the standard assay conditions (i.e. in minimal medium at a pH of 2.5 supplemented with 0.4% glucose and with 10 mM Arg) used for E. coli (Kieboom & Abee, 2006). This finding might provide an explanation for the inability to detect some of the amino acid-dependent XAR systems in bacteria grown under standard laboratory conditions, which typically involve aerobic or microaerobic culturing.
On the contrary, E. coli grown under oxygen levels that fall within the so-called anaerobic transition (i.e. O2 levels in the range 1–10 μM, which are more typical of the stomach and gut epitelium environments) displays a full ability to resist pH 1.6–2.0 even in the absence of gadC and rpoS, indispensable effectors of the AR2 and AR1 systems under aerobic conditions, respectively (Riggins et al., 2013).
A description of the structure and function of the structural components of the amino acid-dependent XAR systems in E. coli is provided in a recent review (Kanjee & Houry, 2013). Herein, based on the available structural and biochemical data, we mainly intend to highlight some special features that characterise these systems, beyond the simple chemistry depicted in Fig. 1. The mechanisms underlying amino acid-dependent XAR systems are becoming more and more clear to us, although there are still some open issues.
By analysing the general mechanism of action of the XAR systems, it is evident that bacteria experiencing extreme acid stress in the stomach must take advantage of substrates which (1) are readily available in the environment (food or GIT); (2) can be transported into the cell with no ATP expense; and (3) either release ammonia or consume protons by their irreversible incorporation into the reaction product (Fig. 4). Equally important is the possibility to use the products of the reaction either to accomplish cellular metabolic needs or for signalling purposes. The latter possibility should be also taken into account given that the products of the reaction are released into the medium and the neutralisation of the extracellular environment, though conceivable, may not be the sole explanation (Planamente et al., 2012).
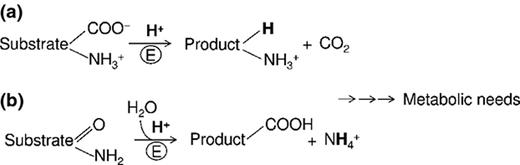
General chemistry of the reactions that use amino acids to neutralise acidity in the intracellular environment. (a) Proton-consuming decarboxylation reaction. (b) Ammonia-releasing amidohydrolase reaction.
Starting from these assumptions, the substrates more apt to fulfil the above requirements are amino acids. In fact, (1) they can undergo proton-consuming decarboxylation reactions (a, in Fig. 4); (2) some possess amido groups which via amidohydrolases can give rise to NH3 (b, in Fig. 4) and can also be sequentially subjected to both reactions; (3) are readily available in the environment and, last but not least; (4) possess a carbon skeleton which can be easily recycled (i.e. no dead-end products).
Recall that in E. coli there are four recognised amino acid-dependent XAR systems which have been investigated in terms of regulation and mechanism of action: they are the glutamate-dependent (AR2), arginine-dependent (AR3), lysine-dependent (AR4) and the newly discovered glutamine-dependent XAR systems (Gong et al., 2003; Iyer et al., 2003; Moreau, 2007; De Biase & Pennacchietti, 2012; Lu et al., 2013). For the latter system, we propose the name AR2_Q, because it shares some regulatory and structural components with the AR2 system (Tucker et al., 2002; Tucker et al., 2003; Weber et al., 2005; Hayes et al., 2006; Lu et al., 2013). The amino acids glutamic acid (Glu), glutamine (Gln), arginine (Arg) and, in part, lysine (Lys) were probably selected by bacteria because they are among the most abundant amino acids in food (as proteins constituents as well as in their free form) and in addition they (or their products) also play crucial physiological roles. The basic principle on which the amino acid-dependent AR systems work is depicted in Fig. 1: they consist of two major structural components, namely a cytoplasmic decarboxylase (in AR2, AR3 and AR4) or an amidohydrolase (in AR2_Q) and an inner membrane antiporter which exchanges the incoming substrate of cognate enzyme for the exported product of the reaction.
Elegant work carried out by Richard and Foster (Richard & Foster, 2004) on the AR2 and AR3 systems of E. coli, the most intensively studied XAR systems, provided evidence that in stationary-phase cells, grown under moderately aerobic conditions, the acid challenge at a pH of 2.5 in the absence of added amino acids causes the abrogation of the negative electrical potential (typically in stationary phase ΔΨ = −50 mV). The loss of the inner membrane integrity, as confirmed also by the drop of the pHi to 3.5, is therefore life threatening for these neutralophilic bacteria. However, following the addition of either glutamate or arginine, the pHi did not fall below pH 4.2 and 4.7 and the electrical potential flipped to + 30 mV and + 80 mV, respectively (Richard & Foster, 2004). The pHi measured in the presence of Glu and Arg nicely fits with the pH optimum of the corresponding inducible amino acid decarboxylases (Shukuya & Schwert, 1960; Blethen et al., 1968).
Thus, besides consuming the protons massively entering into the cell, the intracellular accumulation of the decarboxylation products (GABA or agmatine) is responsible for increasing the number of intracellular positive charges, which in turn will lead to ΔΨ inversion. The major advantage of a positive ΔΨ consists in counteracting further entry of protons, a strategy similarly adopted by acidophiles (Foster, 2004). The recent finding that E. coli cells can be challenged at a pH as low as 1.2 in rich LB medium and still recover viability upon returning to neutral pH suggests that in the absence of protective mechanisms, the pHi can go even lower than 3.5 (Riggins et al., 2013).
The complexity of AR2 system
The AR2 system (and possibly the AR2_Q system) possesses several features that are somehow unique to this XAR system and therefore make it an interesting object of investigation. Besides the very complex transcriptional regulation, which is partially dealt with in this review, the most intriguing aspect for those working on this system is the way it functions. As for any other XAR system, AR2 requires an extracellular supply of Glu to function (Lin et al., 1996; Castanie-Cornet et al., 1999; De Biase et al., 1999) and it is common practice to test for AR2 by adding 0.6–1.5 mM glutamate in the minimal medium supplemented with 0.4% glucose, at pH ≤ 2.5 (i.e. standard assay conditions). The availability of glutamate in the challenge medium was also shown to be the limiting factor to support cell density-dependent survival (Cui et al., 2001). On the other hand, under standard assay conditions, 10 μM extracellular Glu was shown to be sufficient to activate AR2, whereas to detect AR3 and AR4 activity, extracellular Arg and Lys must be at least 0.5 mM (Diez-Gonzalez & Karaibrahimoglu, 2004).
Now, the question is why does the system need such a small amount of extracellular Glu if there is plenty of Glu in the cell? In fact, metabolomic studies on E. coli cells grown to the exponential phase in the presence of various carbon sources (i.e. glucose, glycerol, acetate) have shown that Glu represents the most abundant metabolite, as it is approx. 100 mM, that is, it represents 40% of the total measured intracellular metabolome (Bennett et al., 2009). This means that the intracellular levels of Glu exceed by a factor of 10 000 the extracellular Glu needed for AR2 to operate. Probably, extracellular Glu is required for pH homoeostasis because, based on calculations of intracellular protons in the cell following an extreme acid shock (Foster, 2004), in the absence of an external supply of this amino acid, its intracellular pool will be rapidly depleted. This would be undesirable because high intracellular Glu levels are important for driving forward the transamination reactions of which it is the major nitrogen donor for biomass build up (Bennett et al., 2009). Moreover, Glu plays an important role as major intracellular counter-ion to K+ (McLaggan et al., 1994). Notably, its accumulation in E. coli was shown to be dependent on pH, with GABA replacing Glu during a moderate osmotic shock and at a pH below 8.0 (Ogahara et al., 1995). Similar findings were reported for L. monocytogenes in which two pH-dependent GABA synthesising circuits exist (Karatzas et al., 2012, 2010): the first (operative at mildly acidic pH) involving GABA production and its intracellular accumulation (≥ 100 mM) with no GABA export and the second (operative at pH < 3.5) involving both GABA production and export. Thus, GABA is not just the product of a proton-consuming reaction, but plays additional roles in the cell (and probably between cells). A metabolomic study investigating the effect of inorganic and organic acid stress on pathogenic E. coli O157:H7 provided evidence that at a pH of 3.2, intracellular Glu concentration decreases whereas GABA levels increase (Fletcher, 2012).
Regardless of the molecule which accumulates in the cytosol, that is, Glu or GABA, the high concentrations reported make feasible that both molecules might perform intracellular buffering. In fact with respect to Arg and Lys, Glu and GABA share a unique chemical feature, that is, they both possess a carboxyl group with a pKa close to the pHi measured in acid shocked cells (Richard & Foster, 2004), namely the γ-carboxyl group of Glu (pKa = 4.1) and the carboxyl group of GABA (pKa = 4.0). Therefore, when the bacterial cells are subjected to an extreme acid stress, the intracellular co-presence of two molecules that possess a chemical group capable of buffering at pH of 4.2–5 provides a clear advantage over Arg, Lys and their decarboxylation products, all of which lack a carboxyl group with the ‘right’ pK for buffering the acidified intracellular environment.
The actual protonation status of Glu and GABA when they are taken up and released, respectively, are dealt with by two very recent and independent reports from the groups of Ma et al. (2013) and Tsai et al, (2013). The starting assumptions and the results of their experimental work are depicted in Fig. 5. Basically, both groups reached the conclusion that GadC ‘selects’ for the entrance and the exit only of the species at the desired protonation state, that is, GadC supports exclusively the entrance of Glu0/−1and the exit of GABA+1. These are extremely important results because they eventually provide an answer to an issue which has long being debated in this field (Booth et al., 2002; Foster, 2004; Feehily & Karatzas, 2013) that was already put forward in a recent review (De Biase & Pennacchietti, 2012). In fact, it was always suspected a ‘flaw’ in the logic of how the system works: even though glutamate decarboxylase consumes protons during decarboxylation reaction, still the entry of glutamate itself will exacerbate the acid stress because at a pH lower than 2.5, it should be accompanied by the concomitant entry of protons carried by the fully or partially protonated α-carboxyl group of glutamate (the same was supposed to be caused by the protonated α-carboxyl groups of arginine and lysine of the AR3 and AR4 systems). In addition, GABA was suspected to exist only as GABA0 in the cell and therefore to exit from it in this form. The work of Tsai et al. (2013) and Ma et al. (2013) has shed light on the real mechanism of Glu/GABA antiport. We now can be more confident that GadC works in such a way to avoid not only proton influx, but also a futile proton-neutral antiport and to promote exclusively proton extrusion (Fig. 5). This means that even though GadC is presented with the choice of three different protonated forms of Glu and two protonated forms of GABA (Fig. 5), only Glu0 (or possibly Glu−1) and GABA+1 are effectively exchanged by the cell (Ma et al., 2013; Tsai et al., 2013). Both groups performed in vitro experiments using GadC reconstituted and correctly oriented into proteoliposomes. The problem was attacked by different approaches, although the conclusions were practically overlapping. The finding that GadC (but also AdiC) acts by a ‘logic-gate mechanism’, which consists in recognising the substrates by their net electric charge rather than by the protonation state of the single carboxyl groups, is fascinating and sets the basis for future work, which should implement functional analysis with crystallography by itself not sufficient to reach conclusive answers (Tsai & Miller, 2013; Tsai et al., 2013).
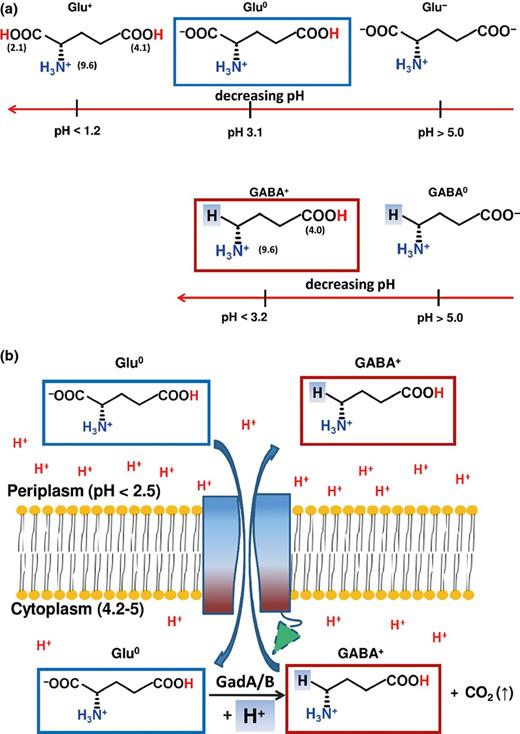
GadC ‘logic-gate mechanism’. (a) Possible protonation states of Glu and GABA and their occurrence at different pHs. The indicated pH is where the species above the value is the most abundant (> 95%). (b) Proposed mechanism for selection of Glu0 and GABA+: following acidic pH activation, the C-plug of GadC (green triangle with a dashed border) is displaced from the substrate transport channel of GadC. The protein (shaded from blue to dark red) will then recognise the substrates via a charge-based mechanism that will allow exclusively the entry of Glu0 (blue border as in A) and the exit of GABA+ (red border as in A).
In both reports on the substrate selectivity of GadC, some of the experiments were carried out with Gln, an amino acid which was shown to be actively transported by GadC in exchange with Glu or GABA (Ma et al., 2012). This amino acid was used because it lacks the γ-carboxyl group, and therefore, it exists only in two protonation states Gln0 and Gln+1. The experiments carried out with Gln helped to establish that the only species which is transported via GadC is Gln0, that is, deprotonated on the α-carboxyl group (Ma et al., 2013; Tsai et al., 2013). Besides being a valuable tool for demonstrating the logic-gate mechanism, the experiments performed with Gln further support the ability of GadC to recognise and transport intracellularly this amino acid, as well as Glu. This may have very important implications because the coupled system AR2/AR2_Q provides several clear advantages to bacteria that employ it. First, a scrutiny of the levels of free glutamine in food suggests that this amino acid is indeed more readily available to bacteria than Glu and Arg (Lu et al., 2013). Second, the entry of Gln in the cell, which occurs only in the Gln0 form (Ma et al., 2013; Tsai et al., 2013), not only is coupled to ammonia release by the acid-inducible glutaminase YbaS, but also acts as an important supply of cellular Glu which, depending on the harshness of the acid stress, can be either accumulated in the cell or converted by GadA and GadB into GABA, with further relief from acid stress by the consumption of additional protons.
The outcome of the biochemical, structural and functional studies on E. coli GadB and GadC, the structural components of the AR2 system, allowed investigators to pinpoint another chemical aspect that deserves to be brought to readers’ attention, that is, the important role that aspartate, glutamate and histidine residues play in modulating a prompt and efficient response to extreme acid stress.
Escherichia coli GadB takes advantage of His465, a residues in the penultimate position of each monomer in the hexamer, to undergo efficient auto-inactivation at pH > 5.5 (Gut et al., 2006). His465 belongs to the C-terminal tail which consists of the last 14 amino acid residues in the polypeptide chain that become ordered and plugs the active site funnel in each subunit of the GadB hexamer. It was demonstrated that His465 is instrumental for the control of GadB activity only at pH > 5.5, that is, when it is in the fully deprotonated form and thus can perform nucleophilic attack over the PLP-Lys276 Schiff base (Pennacchietti et al., 2009). Notably, when this residue is replaced with Ala, closure of the active site is no longer possible and the outcome is an enzyme which is still capable of catalysing the decarboxylation reaction but at pH values close to neutrality. This is clearly a disadvantage for bacteria and this probably explains why a His residue in either penultimate or ultimate position is always present in the C-tail of GadB of many bacteria that employ the AR2 system for acid survival (De Biase & Pennacchietti, 2012). Notably, in the active site of GadB (but also of GadA), there are an aspartate and a glutamate residue, namely Asp86 and Glu89, which depending on the protonation state participate in the binding of the substrate and in the cooperativeness of the system, respectively (Capitani et al., 2003; Pennacchietti et al., 2009; Ho et al., 2013).
The activation of GadB is not the only cooperative event in the AR2 system. The pH-dependent activity profile of GadC was shown in vitro to be a cooperative process which allows full activity of this protein only at pH < 6.0 and in vivo measurement of GABA export have provided clear evidence that GadC is fully active only when the external pH drops to below 3.0 (Richard & Foster, 2004; Ma et al., 2012). Notably, also in GadC, the C-terminal portion, consisting of the last 40 amino acids, plugs the transport channel, thereby effectively restricting the activity of the antiporter only to acidic pH values (Ma et al., 2012). The candidates for an efficient locking were shown to be again His residues. Also, the N-terminal portion of GadB contains several Asp and Glu residues which must become protonated to allow the formation of triple helical bundles that recruit the protein to the membrane compartment where its activity will be even more beneficial to the cell (Capitani et al., 2003; Gut et al., 2006; De Biase & Pennacchietti, 2012). Furthermore, the presence of this bundle accelerates the closure of the active site, thereby indicating that very distant parts of the protein undergoing a conformational change must be connected by a relay mechanism, which is not so obvious on the basis of the crystal structure.
In the section ‘’, the important contribution of His residues in the sensing process was highlighted. A recent example on the role of Asp residues comes from a work on the periplasmic chaperone HdeA (also part of AR2) in which two Asp residues, namely Asp20 and Asp51, once protonated (i.e. when the periplasmic pH suddenly drops to harmful levels) trigger the unfolding of this protein, which is the necessary step to allow HdeA to exert its protective activity from unfolding of periplasmic and membrane proteins (Foit et al., 2013).
Role of AR2 in E. coli pathogenicity
The news media quite often remind us that microbial resistance to common antibiotics, multidrug-resistant bugs and evolution of pathogenic E. coli strains are major health concerns. The remarkable ability of E. coli (commensal and pathogenic strains) to survive a prolonged exposure to extreme acid stress indeed poses a threat, which can become even stronger if one considers that E. coli has an astonishingly plastic genome.
Human pathogenic E. coli cause hundreds of million cases of dysentery and one million deaths per year. The comparison between several E. coli genomes indicates that the ‘core’ genome of this microorganism consists of c. 2200 genes (half of the genome) mostly encoding essential metabolic processes, whereas the E. coli pangenome (the full set of nonorthologous genes among all genomes) consists of over 12 000 genes. Therefore, it is an open genome with the potential of being reshaped into new ‘pathotypes’ by horizontal gene transfer (Rasko et al., 2008; Touchon et al., 2009). Indeed, virulence is the outcome of the acquisition and the loss of genes (Croxen & Finlay, 2010). In a never-ending process, the ‘right’ combination of novel genes contributes to the emergence of pathotypes (Croxen & Finlay, 2010). According to current models, the pathogenic lifestyle leads to greater exposure to host immune defences, which in turn selects for variants that can evade those defences. Selection for such variants results in higher mutation and recombination rates which are responsible of the emergence of epidemic organisms. The severe German outbreak caused by an EAEC (enteroaggregative E. coli) strain O104:H4 in May–July 2011, with more than 4000 cases in 13 European countries and over 50 deaths, is a recent, dramatic example (Frank et al., 2011).
Intestinal E. coli include seven pathovars, and among them are the enteropathogenic E. coli (EPEC), enterohaemorrhagic E. coli (EHEC) and the atypical enteropathogenic E. coli (ATEC). These pathovars harbour the locus of enterocyte effacement (LEE), which contains virulence genes responsible for colonisation and invasion factors, that is, type III secretion system (TTSS), regulators, chaperones and effectors (Schmidt, 2010). In EHEC, EPEC and ATEC, LEE-encoded genes cooperate to cause the development of attaching and effacing (A/E) lesions in the GIT, characterised by the rearrangement of the epithelial cells, effacement of the microvilli and formation of a pedestal-like structure on which bacteria adhere. A/E lesions and AR2 are both associated with the presence of DNA islands acquired by horizontal gene transfer, one for pathogenicity (LEE; 35.6 kb) and the other for acid fitness (AFI, acid fitness island; 14 kb) (Hommais et al., 2004). For pathogenicity to be established, the timely expression of AR2 and the formation of A/E lesions during the various stages of the infection process must be appropriately coordinated, so that some activities are switched either on or off depending on the environmental signal perceived. In this respect, the finding that the expression of AR2 genes and that of the virulence genes in EHEC go in the opposite direction and is under the control of specific regulators and quorum-sensing signalling is of particular relevance. GadE, the central regulator of AR2, downregulates the genes of LEE (Kailasan Vanaja et al., 2009; Tree et al., 2011); on the other hand, Ler, the positive regulator of LEE genes, represses many AR genes, including gadE (Abe et al., 2008). Furthermore, Hughes et al. found that the quorum-sensing signalling molecules acyl homoserine lactones (AHLs), present in the rumen of cattle, activate the AR2 genes via the regulator SdiA, thereby allowing bacterial survival in the acidic stomachs, but also downregulate LEE to avoid untimely activation of the virulence genes in the hostile environment of the acidic stomachs (Hughes et al., 2010). These observations together with the findings that GadX, an additional regulator of AR2, represses transcription of perA (the plasmid-encoded regulator in EPEC; Shin et al., 2001) and that a gadE mutant in EHEC shows an increased adherence to Caco-2 cells (Tatsuno et al., 2003), point towards a definite link between AR2 and E. coli virulence. As a matter of fact, Bhagwat et al. (2005) found that mutations in gadE in some EHEC isolates correlate with a decreased AR2 system activity in vitro and may contribute to their infectious dose and infectivity.
Conclusions
It can be seen from the descriptions above that there are diverse ways by which bacteria can sense acid and respond to it. In most of the cases, some are still imperfectly understood, but the major players have clearly been identified. In general, the pathways that link sensors to genes encoding proteins that act to provide acid resistance can be very complex. The reasons for this are not clear, but it may relate to the fact that bacteria need to integrate multiple signals to express the appropriate resistance genes, particularly in environments where host organisms which they are infecting are trying to kill them, the host stomach and the acidic phagolysosome in macrophages being obvious examples of such environments. In the former, the anatomical site preceding the colonisation site, that is, the gut, most of the bacteria entering into our body must obligatory transit. In this respect, the most efficient systems are those relying on urease and amino acid decarboxylases.
Considerations of acid resistance in the gastric compartment are also important when thinking about the animals that act as reservoirs for pathogens. For example, the spread of E. coli EHEC O157:H7, a major health concern especially in the USA, has been ascribed to the intensive use of corn and other cheap grains (i.e. amino acid-rich food) to feed cattle by beef growers (Diez-Gonzalez et al., 1998; Russell et al., 2000). The grain-based diet, compared to hay and pasture feeding, was shown to increase enormously the numbers of acid-resistant E. coli in the cattle colon. This was attributed to the reduced ability of hay and grass to generate SCFAs compared to grains, such as corn. The consequent acidification of the colon in grain-fed cattle was therefore envisaged the main source for counter-selecting acid-resistant EHEC. It might be difficult to switch back to hay and grass diet, although highly desirable, but our increasing knowledge of the link between AR2 and E. coli pathogenicity might open the way to new antibacterial strategies which do not rely on classical antibiotics as recently suggested (Nguyen & Sperandio, 2012).
We have noted earlier in this review that in several different bacterial species the acid resistance phenotype makes an important contribution to their pathogenesis in humans. If this is the case, we can predict that mutations that reduce acid resistance should reduce pathogenicity, that there should be a good correlation between acid resistance and pathogenicity, and that acid-resistant organisms should have a lower multiplicity of infection than nonacid resistant ones. These predictions are not always easy to test, for several reasons. First, in many cases (including E. coli), a good animal model for human infection does not exist, although colonisation in animals such as cattle and lamb can be studied (Price et al., 2004; Tree et al., 2011). Second, acid resistance is very dependent on choice of acid, growth conditions and growth phase, and it thus may be difficult to know how well acid resistance as measured in the laboratory represents acid resistance as it may occur in the GIT. The presence of multiple resistance systems can make interpretation of results in the laboratory particularly hard to extrapolate to the situation in an infection. Nonetheless, the data discussed earlier suggest an important link in many cases between acid resistance and ability to cause human disease.
Acknowledgement
This work was in part supported by grant from La Sapienza University of Rome and by Fondazione Roma to D.D.B. and by grants from BBSRC and the Darwin Trust of Edinburgh to P.L.
References
Author notes
Editor: Lee Kroos

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}