





Abstract
The complement and coagulation systems are two related protein cascades in plasma that serve important roles in host defense and hemostasis, respectively. Complement activation on bacteria supports cellular immune responses and leads to direct killing of bacteria via assembly of the Membrane Attack Complex (MAC). Recent studies have indicated that the coagulation system also contributes to mammalian innate defense since coagulation factors can entrap bacteria inside clots and generate small antibacterial peptides. In this review, we will provide detailed insights into the molecular interplay between these protein cascades and bacteria. We take a closer look at how these pathways are activated on bacterial surfaces and discuss the mechanisms by which they directly cause stress to bacterial cells. The poorly understood mechanism for bacterial killing by the MAC will be reevaluated in light of recent structural insights. Finally, we highlight the strategies used by pathogenic bacteria to modulate these protein networks. Overall, these insights will contribute to a better understanding of the host defense roles of complement and coagulation against bacteria.
We take a closer look at how complement and coagulation induce stress to bacterial cells.
Introduction
The complement and coagulation systems are two ancient proteolytic cascades in plasma that play important roles in host defense and hemostasis, respectively. Both systems are more than 400 million years old and probably evolved from a common ancestor, explaining the significant overlap in protein domains (Krem et al., 2000). Complement and coagulation proteins circulate in the blood as inactive precursors, but are activated upon contact with target structures. The resulting proteolytic cascade generates multiple protein cleavage products that trigger numerous events important for inflammation and hemostasis.
Complement is an integral part of the innate immune system, our primary host defense barrier against invading bacteria (Walport, 2001; Ricklin et al., 2010). Whereas the complement reaction exclusively takes place on the bacterial surface, the resulting cleavage products are either linked to the surface or released in fluid-phase. While the released chemotactic peptides attract phagocytes from the blood to the site of infection (Bardoel & Strijp, 2011), the massively deposited C3 fragments enhance bacterial recognition by phagocytes and trigger phagocytosis. Finally, the pore-forming complex (Membrane Attack Complex or MAC) that inserts into the membrane (Müller-Eberhard, 1986) results in direct bacterial killing.
The primary function of the coagulation system is to respond to damage of blood vessels via formation of fibrin, which seals wounds and stops bleeding (hemostasis). Although clotting is a well-known immune defense mechanism in invertebrates, its role in mammalian host defense is largely unknown. However, multiple lines of evidence now suggest that coagulation is important for the early innate immune response since it supports the inflammatory response, generates antimicrobial peptides and induces local entrapment of bacteria in fibrin clots (Frick et al., 2007; Nickel & Renné, 2012).
In this review, we aim to provide detailed insights into how complement and coagulation contribute to host clearance of bacterial infections. In particular, we will focus on the recognition mechanisms that drive activation of these cascades on bacteria and discuss how these systems directly induce stress to bacterial cells. The comparison of these related proteolytic cascades as innate sensors provides us a unique opportunity to discuss the relatively unknown role of coagulation as part of the mammalian innate immune system. Finally, we also highlight how bacterial pathogens block or use these proteolytic cascades to overcome immune clearance. Since both complement and coagulation are pathological during systemic infections, detailed insights into their activation mechanisms and modulatory strategies developed by bacteria could stimulate research into new treatment strategies for infectious diseases.
Bacteria under stress by complement
Complement activation is a step-wise process in which protein binding and cleavage events occur in a well-defined order (Gros et al., 2008; Ricklin et al., 2010): first, the recognition molecules of the three complement pathways (classical, lectin, alternative) bind to the bacterial cell surface (Fig. 1); second, they activate associated serine proteases that form C3 convertase enzymes (Fig. 2); third, C3 convertases cleave the central complement protein C3 to label the target surface with C3b and amplify the labeling process (Fig. 2b); fourth, C5 convertases convert C5 into C5a and C5b; fifth, C5b generates the MAC (C5b-9) (Fig. 2a, Fig. 3). The recognition phase is important for the specific activation of complement on target cells. Recognition molecules generally bind evolutionarily conserved structures that are only present on microbes and absent on host cells. In the past years, our knowledge on these recognition mechanisms of bacteria has increased significantly and below we will provide an overview of bacterial structures that are recognized by complement.
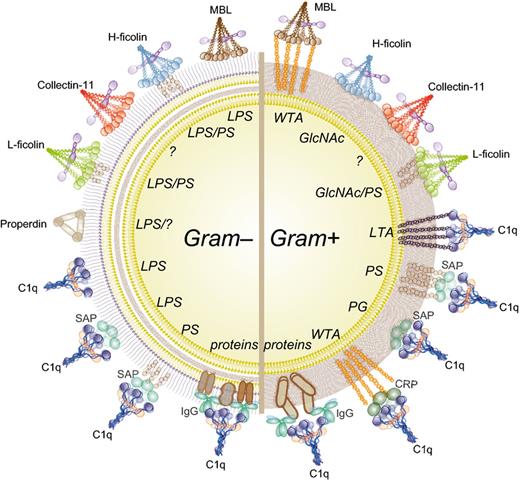
How bacteria are recognized by complement. Complement recognition molecules of the CP (C1q), LP (MBL, ficolins, collectin-11), and AP (properdin) bind to various structures on Gram-positive and Gram-negative bacteria (recognition motifs in italics). Next to direct binding, C1q also indirectly recognizes bacteria via antibodies, CRP or SAP. MBL, mannose-binding lectin; SAP, serum amyloid P; CRP, C-reactive protein; IgG, immunoglobulin G; LPS, lipopolysaccharide; PS, polysaccharides; WTA, Wall Teichoic acid; GlcNAc, N-acetyl glucosamine; LTA, Lipoteichoic acid.
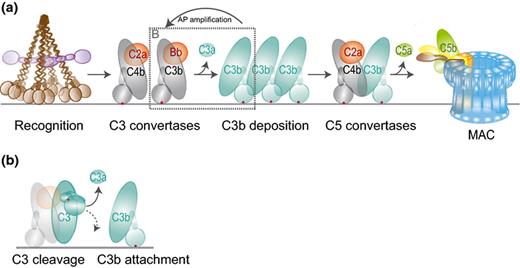
The complement reaction on bacteria. (a) Recognition molecules initiate the complement cascade by their associated serine proteases (C1s and MASP-2, purple) that cleave C4 and C2 to generate a C4b2a complex on the bacterial surface. This complex is a C3 convertase that cleaves C3 into the released anaphylatoxin C3a and C3b, which binds covalently to the bacterial surface. C3b molecules initiate formation of another C3 convertase (C3bBb) that amplifies the C3b labeling process (AP amplification). C3b molecules also generate C5 convertases by binding onto or near the C3 convertases. This initiates a substrate switch, which causes convertases to cleave C5 into soluble C5a and C5b that forms a complex with C6-9 to generate the MAC. This figure was partly modified from Gros et al., 2008. (b) Conversion of C3 by the C3 convertase. C3 holds a reactive thioester bond (red dot) that is hidden within the C3 molecule but exposed upon its cleavage into C3b. The acyl group of the thioester moiety forms a covalent bond with a hydroxyl or amine group, which allows covalent attachment of C3b to the target surface.
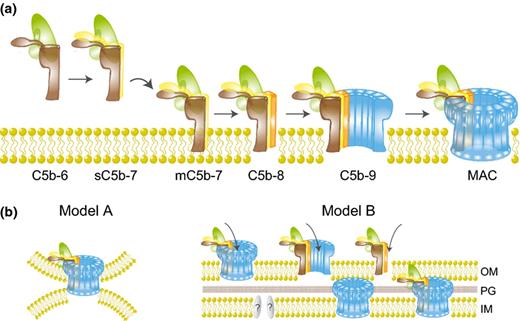
Assembly and bactericidal mechanisms of the MAC. (a) Newly formed C5b reacts with C6 to form the stable C5b6 complex. Binding of C7 results in a hydrophobic complex that targets the membrane (mC5b-7). Membrane insertion is initiated upon binding of C8 (C5b-8) after which 12–18 copies of C9 polymerize to form the pore-forming ring structure (MAC). (b) Several hypotheses for MAC-mediated killing of Gram-negative bacteria. Model A: the MAC simultaneously disrupts the OM and IM via insertion into e.g. Bayer's junctions. Model B: the MAC or its precursors (C5b-8 or C5b-9n) rupture the OM, which allows passage of C5b-9 components (creating another MAC), C9 alone (or fragments of C9), or other factors into the periplasm to disrupt IM integrity. This process may require additional enzymes to breakdown the PG layer.
Recognition of bacteria by the complement system
Bacteria are roughly divided into Gram-positive and Gram-negative bacteria, determined by their cell wall characteristics (Fig. 1). Peptidoglycan (PG) is the common trait in the cell envelope of almost all bacteria and important for cell integrity. It is composed of polysaccharide strands of alternating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acids (MurNAc) that are linked by short peptides (reviewed in Vollmer et al., 2008). Gram-positive bacteria possess one phospholipid membrane covered by a thick PG layer (20–80 nm) (reviewed in Silhavy et al., 2010) that commonly contains complex polysaccharides and teichoic acids linked to the peptidoglycan (wall teichoic acids, WTAs) or the membrane (lipoteichoic acids, LTAs) (Reichmann & Gründling, 2011). In contrast, Gram-negative bacteria contain two membranes (the inner (IM) and outer membrane (OM)) with a thin PG layer (1–7 nm) in between. The lipid composition of the OM is heterogeneous and contains glycolipids called lipopolysaccharide (LPS) (Silhavy et al., 2010).
It has long been recognized that isolated PG, which is unique to bacteria, is a potent activator of the complement system (Verbrugh et al., 1980; Ma et al., 2004). In the context of living bacteria, however, PG is less exposed; it is hidden underneath the outer membrane (OM) of Gram-negative bacteria or shielded by PG-anchored polysaccharides and teichoic acids in Gram-positives. Therefore, complement also recognizes other conserved bacterial structures (Fig. 1). The recognition molecules of the lectin pathway (LP) are the mannose binding lectin (MBL), ficolins (Ficolin-1, Ficolin-2, and Ficolin-3) and collectin-11 (Matsushita & Fujita, 2001; Holmskov et al., 2003; Fujita et al., 2004; Ma et al., 2013). These large multimeric complexes resemble a ‘bunch of tulips’ since they hold a collagen-like domain build up from three identical polypeptide chains that assemble in a coiled-coil structure at the base and multiple specific carbohydrate-binding domains at the top. MBL and collectin-11 contain a C-type carbohydrate-recognition domain (CRD) (Holmskov et al., 1994) that allows binding to carbohydrate and glycoconjugate structures on many different microbes (reviewed in Jack et al., 2001b). The main bacterial targets of MBL are LPS in Gram-negative and WTA in Gram-positive bacteria (Polotsky et al., 1996; Jack et al., 2001a; Park et al., 2010). Collectin-11 was recently identified and described to bind Escherichia coli, Pseudomonas aeruginosa (Hansen et al., 2010), and Streptococcus pneumoniae (Ali et al., 2012). In ficolins, the CRD consists of a fibrinogen-like domain that recognizes both sugar motifs (Matsushita et al., 2013) and acetylated groups present in for instance GlcNAc and GalNAc (Matsushita & Fujita, 2001). The binding to GlcNAc, a major constituent of PG, is believed to be important for the binding to Gram-positive bacteria (Ma et al., 2004).
The C1q molecule, which is structurally similar to MBL and ficolins in the LP, mediates activation of the classical pathway (CP). C1q is classically known to bind to Fc regions of IgG or IgM antibodies on the microbial surface. For some bacteria like Neisseria meningitidis, antibodies are essential for an effective complement response (Granoff, 2009). Furthermore, C1q binds bacteria via pentraxins, evolutionarily conserved plasma proteins that recognize foreign antigens and altered-self ligands. The major pentraxins in human plasma are the short pentraxins C-reactive protein (CRP) and serum amyloid P (SAP) (reviewed in Du Clos, 2013) and the long pentraxin 3 (PTX3) (Ma et al., 2011). CRP interacts with phosphocholine (PC) that is present in a number of bacterial structures including the teichoic acid of S. pneumoniae (Volanakis, 2001). SAP binds a variety of bacterial cell wall structures such as carbohydrates, LPS, and peptidoglycan (Hind et al., 1985; Noursadeghi et al., 2000; An et al., 2013). Alternatively, C1q can bind directly to the bacterial surface to activate the CP. C1q was found to bind to a variety of microbial structures including the lipid A region of LPS (Cooper & Morrison, 1978; Tan et al., 2011) and different outer membrane proteins on Gram-negative bacteria (Loos & Clas, 1987; Albertí et al., 1993; Mintz et al., 1995; Merino et al., 1998), and LTA on Gram-positive bacteria (Loos et al., 1986). Moreover, C1q can bind to various carbohydrates (Païdassi et al., 2008), which potentially mediates direct binding to bacteria.
Recently, it was shown that the alternative pathway (AP), generally viewed as an amplification mechanism of the CP and LP, also directly recognizes bacteria via the properdin molecule. It has been reported that properdin, a stabilizing protein for the convertases of the AP, functions as a pattern recognition receptor for bacteria (Spitzer et al., 2007; Kemper et al., 2010). Properdin binds to LPS of certain Gram-negative bacteria but also to LPS-defective E. coli and Salmonella typhimurium (Spitzer et al., 2007). Also, properdin was reported to bind and activate complement on Chlamydia pneumoniae (Cortes et al., 2011). However, since other studies have questioned these data, the direct recognition of bacteria via the AP is still under debate (Harboe et al., 2012). Of note, activation of the AP can also occur at a low-level via the spontaneous ‘tick-over’ process in which hydrolysis of C3 creates the C3b-like molecule C3H2O (Pangburn et al., 1981; Ricklin et al., 2010). This activation process is down regulated on host cells but quickly amplified on bacterial cells.
Thanks to the large variety of recognition mechanisms, almost all bacteria will be recognized by the complement system. Many studies have shown that the relative importance of each pathway may be different for every bacterium. Importantly, the contribution of each pathway may change during an active infection due to upregulation of the acute phase response (CRP and MBL) and the (increased) production of antibodies. Recently, an interesting study reported that binding of MBL to WTA of Staphylococcus aureus occurs exclusively in infants with low levels of anti-WTA antibodies. In adults, higher levels of anti-WTA antibodies compete with MBL for WTA binding and trigger activation of the classical pathway (Park et al., 2010). Whether such a pathway-shift results in a more effective complement response is presently unknown.
Complement reaction on bacteria
Whereas the initial phase of complement recognition is strictly dependent on the composition of the bacterial surface, the next steps of the complement pathway occur similarly on all target surfaces (Fig. 2a). The recognition molecules do not possess enzyme activity but circulate in complex with serine proteases responsible for activation of the proteolytic cascade. The recognition molecules of the LP (MBL, Ficolins, and Collectin-11) circulate in complex with the MBL-associated serine proteases (MASPs) while C1q is complexed with the C1r and C1s proteases. The homologous MASP-2 and C1s proteases can both cleave C4 and C2 to generate a C3 convertase enzyme that consists of surface-bound C4b and protease C2a (Fig. 2a). This convertase catalyzes the covalent deposition of C3b onto the bacterial surface and release of the small anaphylatoxin C3a. The covalent binding of C3b, a critical step in the complement response, is mediated by its reactive thioester. Thanks to the crystal structures of C3 and C3b (Janssen et al., 2005, 2006), we now understand how this thioester is hidden within C3 but becomes exposed in C3b as a result of extensive conformational changes (Fig. 2b). Although the acyl group of the thioester is thought to form a covalent bond with any hydroxyl or amine group (Law & Dodds, 1997; Janssen & Gros, 2007), this reaction is probably less random than generally believed (Law et al., 1981, 1984; Law & Dodds, 1997). Most C3b is linked by an ester bond, which indicates that it reacts better with hydroxyl groups than amine groups (Law et al., 1984). Binding occurs preferentially to hydroxylated sugar groups, but also to amino acids in proteins of which tyrosine, threonine, and serine show the highest reactivity (Tyr>Thr>Ser) (Sahu & Pangburn, 1995).
The deposited C3b molecules initiate the alternative pathway (AP) amplification loop because C3b itself forms the basis of the AP C3 convertase enzyme (C3bBb). Surface-bound C3b forms a complex with Factor B, which is then converted by Factor D (Gros et al., 2008). The resulting C3bBb enzyme converts new C3 molecules into C3b (Fig. 2a and b) and thus rapidly amplifies the number of deposited C3b molecules. This is crucial for initiation of the terminal pathway, since high densities of C3b are required to change the affinity of the convertase for C5 (Rawal & Pangburn, 2001; Pangburn & Rawal, 2002), which can then cleave C5 into the chemo-attractant C5a and soluble C5b that initiates assembly of the MAC.
Host defense functions of complement on bacteria
Complement activation products can exert stress to bacterial cells, either indirectly by supporting cellular immune responses or directly by damaging bacterial membranes. Before we zoom into the direct antibacterial mechanisms, which are the focus of our review, we will shortly summarize the indirect actions of complement. A very important role of complement is to facilitate phagocytosis of bacteria by labeling (or opsonizing) their surface with C3b and its degradation product iC3b, which are recognized by complement receptors 1 (CR1), 3 (CR3), 4 (CR4), and CRIg on phagocytic cells (Vik & Fearon, 1985; Krych-Goldberg & Atkinson, 2001; Helmy et al., 2006; Bajic et al., 2013). Also, the release of anaphylatoxins C5a and C3a is important to trigger a pro-inflammatory status. Moreover, neutrophils and monocytes (C5a), but also eosinophils and mast cells, are attracted to the site of infection through interaction with the C5a (C5aR) or C3a receptor (C3aR) (Haas & Strijp, 2007). Moreover, complement activation can orchestrate adaptive immunity by enhancing antigen presentation via interaction of the C3b degradation product C3d with CR2 on B cells and follicular dendritic cells (reviewed in Carroll & Isenman, 2012). In addition, complement activation products influence T cell immunity via stimulation of antigen presenting cells (APCs) and T cells (for a complete overview see Kemper & Atkinson, 2007; Dunkelberger & Song, 2010).
Direct stress: the Membrane Attack Complex
Direct stress of complement to bacterial cells is mainly exerted through formation of the MAC that kills Gram-negative bacteria. Although the fluid-phase anaphylatoxin C3a and its derivative C3ades-Arg were also reported to exhibit direct antibacterial activity (Nordahl et al., 2004), we will focus on the molecular mechanisms of MAC assembly and lysis of bacteria.
Assembly
The MAC is a multiprotein complex that comprised single copies of C5b, C6, C7, and C8 together with 12–18 copies of C9 (Podack et al., 1980; Müller-Eberhard, 1985). MAC formation is initiated by the cleavage of C5 into C5b by the C5 convertase (Fig. 2a). In contrast to C3 and C4, C5 does not hold a reactive thioester for covalent linkage to the bacterial surface (Cooper & Müller-Eberhard, 1970). Newly formed C5b has an unstable binding site with a half-life of c. 2.5 min (Cooper & Müller-Eberhard, 1970) and needs to react with C6 to form the stable C5b6 complex (Fig. 3a) (DiScipio et al., 1983). Subsequent binding of C7 renders the complex lipophilic and mediates binding to the target membrane (Preissner et al., 1985). Initial membrane insertion is mediated by the binding of C8 to the C5b-7 complex (Monahan & Sodetz, 1981; Müller-Eberhard, 1986; Stewart et al., 1987) with the C8 domain inserting into the lipid bilayer (Steckel et al., 1983). The C5b-8 complex then facilitates recruitment of C9 and catalyzes its polymerization into a pore-forming ring structure that inserts into the target membrane (Fig. 3a) (Podack & Tschopp, 1982b; Tschopp, 1984; Müller-Eberhard, 1985; Tschopp et al., 1985).
Structure and pore-formation
The MAC is a SDS-stable ring-structure (Podack & Tschopp, 1982a) with an inner diameter of c. 100 Å, an outer diameter c. 200 Å, and a length of c. 160 Å (Podack & Tschopp, 1982b; DiScipio & Hugli, 1985; Müller-Eberhard, 1985). Components C6-C9 are all homologous and share a 40 kDa Membrane Attack Complex Perforin (MACPF) domain (Tschopp et al., 1986; Rosado et al., 2008), which is flanked by different regulatory domains at the C- and N-termini (Esser, 1994). This MACPF domain, which is essential for membrane insertion of C8α, C8β, and C9 (Steckel et al., 1983), is also found in perforin (Tschopp et al., 1986), a cytolytic protein released by cytotoxic T lymphocytes or natural killer cells to form pores in target cells (e.g. virus-infected host cells) (Bolitho et al., 2007). The structures of C8 and C8α have recently provided new insights into formation and composition of the MAC pore (Hadders et al., 2007; Lovelace et al., 2011). Since the MACPF domain of C8α is structurally similar to bacterial cholesterol-dependent cytolysins (CDCs), MACPF proteins are believed to use a similar mechanism for membrane insertion as CDC. Like CDC monomers, MACPF proteins contain domains that refold from soluble into transmembrane β-hairpins (TMH) that oligomerize to form a circular pore (Tweten, 2005; Hadders et al., 2007; Lovelace et al., 2011). In the MAC, C9 is the main component making up the pore although the other MAC proteins insert partially in the membrane with C8 as the predominant protein (Hu et al., 1981; Podack et al., 1981; Steckel et al., 1983). Not all MACPF proteins function as lytic toxins: molecules like C6 and the β domain of C8 do not insert into membranes to form lytic pores (Rosado et al., 2008), they rather have a supporting role in formation of polymeric C9 within the MAC.
Bactericidal mechanisms of the MAC
Although the mode of assembly and structural organization of the MAC are well characterized, the exact mechanism by which the MAC kills bacterial cells remains poorly understood. The MAC is reported to kill Gram-negative bacteria while Gram-positive bacteria are resistant (Joiner et al., 1984). However, the most commonly used model systems for MAC lysis are erythrocyte membranes and artificial lipid bilayers. These single-membrane ‘cells’ are much more sensitive to lysis by pore-forming proteins than Gram-negative bacteria that possess two membranes separated by a periplasmic space containing peptidoglycan. Furthermore, bacteria also have membrane repair mechanisms (García et al., 2006). Below, we discuss the proposed bactericidal mechanisms of the MAC and evaluate these hypothetical models in light of the makeup of a Gram-negative cell.
Molecular composition of the bactericidal MAC
While the complete ring-structured C5b-9 complex is generally considered the active ‘killer’ component, many reports have shown that the premature C5b-8 complex also has membranolytic activity. Although there is no direct evidence for C5b-8 to assemble into a pore, this complex can cause leakage of synthetic lipid vesicles (Zalman & Muller-Eberhard, 1990). Also in lipid bilayers and erythrocytes, C5b-8 can increase ion conductance (Shiver et al., 1991) or cause cell lysis, respectively (Tamura et al., 1972). However, the C5b-8 complex does not efficiently kill Gram-negative bacteria, again underlining the difference between lysis of bacteria vs. erythrocytes and lipid bilayers. Interestingly, although C9 is absolutely required for bacterial killing, its polymerization into a SDS-stable ring does not seem essential [observed for both E. coli (Joiner et al., 1985) and S. typhimurium (Joiner et al., 1989)].
Hypothetical models for MAC insertion into the Gram-negative cell envelope
Since damage to the OM alone is unlikely to cause bacterial death (Wright & Levine, 1981a; Tomlinson et al., 1990), the MAC should be able to disturb the IM as well. Currently, a few hypotheses exist on how the MAC gains access to the IM (Fig. 3b). With recent information on the exact dimensions of the MAC (Aleshin et al., 2012; Hadders et al., 2012), we can now further contemplate the likelihood of the proposed events. Both the inner and outer membranes of Gram-negatives are c. 75 Å thick and they are separated by a c. 100 Å periplasmic space, creating a total cell envelope of c. 250 Å (DiRienzo et al., 1978). The first hypothesis considers that one C5b-9 complex would span both membranes (transmural pore) (Born & Bhakdi, 1986). However, with its length of c. 160 Å, it is physically impossible for the MAC to cross the entire cell envelope of Gram-negative bacteria. In addition, only the lower part of the MAC (containing the hydrophobic TMHs) is believed to insert the membrane, making it even less likely for the MAC to span the entire cell envelope. Bayer et al. proposed that the Gram-negative cell wall contains regions, Bayer's junctions, where the IM and OM are in close contact (Bayer, 1968), creating hotspots for MAC perturbation (Fig. 3b, Model A) (Wright & Levine, 1981a,b). Although today the existence of these sites remains controversial (Kellenberger, 1990; Ruiz et al., 2006), they have been proposed as sites where the MAC inserts into the cell envelope. Wright and Levine postulated simultaneous disturbance of the inner and outer membrane by the MAC at these junctions (Wright & Levine, 1981a,b). The second hypothesis proposes that the MAC disrupts the OM but that the lethal activity is executed at the IM (Wright & Levine, 1981b; Dankert & Esser, 1987; Esser, 1994) by allowing access of MAC or other membrane-damaging proteins to the periplasmic space and eventually the IM (Fig. 3b, Model B). Such a mechanism would require these components to pass through the PG layer that covers the inner membrane. It was previously believed that the natural pores in the PG matrix were c. 2 nm, allowing proteins of maximum c. 25 kDa to diffuse (Demchick & Koch, 1996). However, a recent study indicated the presence of PG pore sizes in the range of 10 nm in E. coli (Turner et al., 2013) allowing diffusion of proteins up to c. 64 kDa. Still, the MAC proteins are all larger (C5b c. 179 kDa, C6 c. 102 kDa, C7 c. 97 kDa, C8 c. 150 kDa, and C9 c. 73 kDa), making it plausible that PG degrading enzymes are essential for these components to reach the IM. In analogy, lysis of E. coli via bacteriophage holins can also exclusively occur in the presence of phage endolysins that break the PG layer (Catalão et al., 2013). Many studies were performed analyzing the role of lysozyme (muramidase), an enzyme abundantly present in body fluids including the serum, in the bactericidal reaction of the MAC (reviewed in Taylor, 1983). Initial damage of the OM might allow access of serum-derived lysozyme to the periplasmic space where it can degrade the peptidoglycan layer (Wright & Levine, 1981a). Lysozyme was found to accelerate the bacteriolytic effects of the MAC, although blocking of lysozyme did not abrogate the bactericidal process completely indicating that the enzyme is not absolutely required for MAC-mediated killing of bacteria (Martinez & Carroll, 1980). Even though the C9 molecule without C5b-8 has no toxic effect on bacteria, studies introducing C9 in the periplasm of Gram-negative bacteria showed that the C9 molecule can be lethal by itself when it has access to the inner membrane (Dankert & Esser, 1987; Wang et al., 2000). This hypothesis was strengthened by experiments in which plasmid-derived full-length C9, targeted to the periplasm, led to killing of Gram-negative bacteria whereas C9 expression in the cytoplasm did not have this effect (Wang et al., 2000). It is not known whether C9 causes lysis of the inner membrane in its native conformation. The authors considered the possibility that C9 might be converted into a more toxic form by enzymes present in the periplasmic space (Dankert & Esser, 1986; Wang et al., 2000).
The lethal event
Following pore-formation, it is believed that bacteria are killed due to reduction of the membrane potential (Wright & Levine, 1981a), analogous to pore-forming colicins, a group of toxins secreted by some strains of E. coli (Wright & Levine, 1981a; Cramer et al., 1995; Cascales et al., 2007). Alternatively, bacteria might participate actively in their loss of viability by metabolic events (Kroll et al., 1983). Incubation of bacteria with active complement in the presence of an uncoupler of oxidative phosphorylation completely prevented MAC-mediated killing (Taylor & Kroll, 1983). In line with this, another hypothesis that has not been suggested before is that upon MAC formation, the bacteria might activate a self-death pathway like previously described for mammalian peptidoglycan recognition proteins (PGRPs) (Kashyap et al., 2011).
The MAC on Gram-positive bacteria
For long it has been known that Gram-positive bacteria are protected from killing by the MAC, supposedly due to their thick PG layer that prevents MAC insertion into the membrane (Joiner et al., 1984). Intriguingly, we recently found that the MAC deposits on several Gram-positive bacteria as SDS-stable polymeric C9 structures (Berends et al., 2013). To our surprise, these complexes were located at specific regions on the bacterial cell. For instance, on Streptococcus pyogenes (group A streptococcus, GAS), C5b-9 deposited near the division septum, whereas on Bacillus subtilis the complex was located at the poles (Berends et al., 2013). This specific C5b-9 deposition did not affect bacterial viability. Although this remains to be elucidated, it might indicate that the MAC has a non-classical role in immune defense against these microbes.
Bacteria under stress by coagulation
The coagulation cascade can be activated via different pathways that all converge in a common pathway for clot formation (Sun, 2006). Two pathways can trigger coagulation: the contact system (intrinsic pathway) and the tissue factor (extrinsic) pathway. The tissue factor pathway responds to blood vessel damage via exposure of tissue factor, a subendothelial protein that is normally shielded from contact with blood components. Since this pathway plays no clear role in host defenses, it will not be further discussed here. Microbe-specific activation of coagulation was suggested to occur via the FXII-dependent contact-driven pathway, which results in formation of the pro-inflammatory peptide Bradykinin (BK) (Fig. 4a) and clotting via activation of factor XI (Fig. 4b).
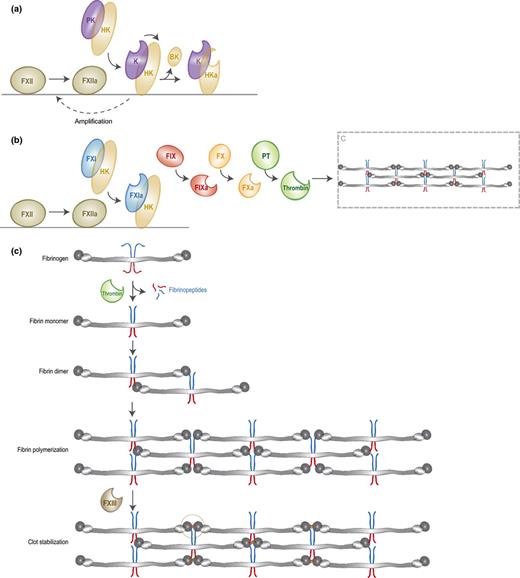
Activation of coagulation on bacteria. The contact activation pathway is initiated by the binding of FXII to negatively charged bacterial structures. FXII is then auto-activated into FXIIa, which either triggers the kallikrein-kinin system (a) or the clotting cascade (b). (a) FXIIa converts PK, which circulates in complex with HK, into active kallikrein. Kallikrein cleaves HK to release BK and activates FXII to amplify the contact system. (b) FXIIa cleaves FXI, which also circulates in complex with HK. FXIa subsequently converts FIX into FIXa. FIXa will then trigger the common pathway of coagulation by converting FX into FXa, which leads to conversion of prothrombin into thrombin that cleaves fibrinogen. (c) Formation of fibrin clots. Activated thrombin cleaves fibrinogen into fibrin monomers that are capable of interacting with adjacent fibrin molecules and thereby form long fibers and finally a fibrin clot. This clot can be stabilized by FXIII or thrombin-activated FXIIIa.
Contact activation on bacteria
The contact system is activated upon binding of FXII to negatively charged structures, as was demonstrated in coagulation assays using dextran sulfate or silica (Citarella et al., 1997; Yoshida et al., 2013). Since these structures are abundantly present on bacteria, FXII can bind to bacterial surfaces (Ben Nasr et al., 1996; Herwald et al., 1998). Target-binding of FXII results in low-level auto-activation into FXIIa, which then converts prekallikrein (PK) into active plasma kallikrein (Fig. 4a) (Wuepper & Cochrane, 1972; Samuel et al., 1992; Citarella et al., 1997). Kallikrein can cleave the associated high molecular weight kininogen (HK) to release the pro-inflammatory peptide bradykinin (BK). Furthermore, kallikrein amplifies the contact system by cleavage of FXII (Schmaier & McCrae, 2007). At least 75–90% of PK circulates in an equimolar complex with HK (Mandle et al., 1976; Scott & Colman, 1980), which is important for effective cleavage of PK by FXIIa (Colman & Schmaier, 1997). The activated form of HK (HKa) has an increased capacity to bind negatively charged surfaces and provides a surface for further activation of the cascade (Scott & Silver, 1984; Reddigari & Kaplan, 1988). Escherichia coli curli and S. typhimurium fimbriae can activate the contact system due to interactions with FXII and HK. All contact factors were assembled on these fibrous structures and mutant strains lacking either curli or fimbriae did not activate the cascade (Ben Nasr et al., 1996; Herwald et al., 1998). Also LPS was described to provide a surface for contact activation by binding to domain 5 in HK, which is responsible for binding to negatively charged surfaces (Perkins et al., 2008). For S. aureus it was proposed that negatively charged teichoic acids induce contact activation (Kalter et al., 1983; Mattsson et al., 2001). Apart from surface-associated molecules, fluid-phase bacterial products can also initiate the contact system. For instance, LPS is released from the bacterial surface during cell growth and can then activate the contact system by binding HK in solution (Perkins et al., 2008). Also, it has been described that excreted bacterial polyphosphates can trigger coagulation through the contact pathway by binding to FXII and to a lesser extent to HK (Smith et al., 2006), depending on the length of the polymer chains (Smith et al., 2010). In a murine infection model, it was shown that bacterial polyP induces vascular leakage through release of BK in a FXII-dependent manner (Müller et al., 2009).
Next to its role in cleavage of PK and HK, contact activation also results in clotting (Fig. 4b). Surface-activated FXIIa can also cleave coagulation factor XI, a homolog of PK that circulates in complex with HK as well. As explained in Fig. 4b, activated FXI (FXIa) triggers the common pathway of coagulation in which subsequent activation of FIX, FX, and prothrombin results in thrombin-catalyzed cleavage of fibrinogen (Fig. 4b and c). Fibrinogen is a highly abundant (4 mg mL−1) and large plasma protein (340 kD) that consists of two sets of three polypeptide chains (Aα, Bβ, and Cγ) organized in three distinct domains: two outer D-domains and a central E-domain connected by coiled coil segments (Kollman et al., 2009). Thrombin drives fibrin formation by cleavage of peptide bonds in both the N-terminal Aα chain and the N-terminal Bβ-chain that are located in the E-domain (Fig. 4c) (Blombäck et al., 1978). Cleavage of the Aα-chain results in release of fibrinopeptide A and exposure of a polymerization site known as EA. This newly exposed EA-site subsequently interacts with the constitutively exposed Da-site on the β-chain segment of the D-domain of adjacent fibrin(ogen) molecules, leading to formation of a staggered overlapping double-stranded fibrin fibril. Cleavage of the Bβ-chain by thrombin and the associated release of fibrinopeptide B is much slower and leads to exposure of the EB polymerization site that interacts with constitutive Db-sites on the β-chain region of the D-domain. This interaction induces a conformational change in the D-domain that allows lateral association of fibrin fibers (Yang et al., 2000). Fibrin fibers are subsequently stabilized by factor XIII or thrombin-activated FXIIIa (Siebenlist et al., 2001), a transglutaminase that introduces covalent intermolecular bonds between C-terminal lysine and glutamine residues of adjacent γ-chains (Chen & Doolittle, 1971). Recently, it was shown that contact activation by bacterial fimbriae and curli indeed results in cleavage of fibrinogen and subsequent fibrin formation (Persson et al., 2003).
The contact system: an innate recognition pathway?
Due to its redundant function for efficient hemostasis, the contact system has been proposed to be part of the innate immune system. Interestingly, the recognition mechanisms of the contact system seem less specific than those of the complement system and innate immunity in general. In order to respond to a large group of microbes, the innate immune system uses a variety of proteins that recognize evolutionarily conserved structures on non-self surfaces. For example, toll-like receptor 4 (TLR4) recognizes Gram-negative bacteria by binding to LPS, the formylated peptide receptor (FPR) responds to all living bacteria by sensing formylated peptides (exclusively produced by bacteria) (Bardoel & Strijp, 2011) and the complement system uses a variety of lectins to bind conserved bacterial sugars or teichoic acids. In contrast, the contact system is not activated by conserved bacterial structures but recognizes bacteria via their negatively charged surface. Still there appears to be some specificity since contact activation does not normally occur in the circulation and seems to be limited to foreign surfaces or human cells in a procoagulant state.
Host defense functions of coagulation on bacteria
The coagulation system is recognized to be important to the immune response of invertebrates. Several invertebrate species contain a coagulation cascade that senses invading microorganisms and neutralizes them via clotting (Osaki & Kawabata, 2004). Horseshoe crab clotting factors are stored in the granules of hemocytes (phagocytic cells) that are released upon sensing bacterial components like LPS. This triggers a proteolytic cascade that eventually leads to cleavage and polymerization of the clottable protein coagulogen that embeds bacteria in insoluble aggregates (Osaki & Kawabata, 2004). In addition to clottable proteins, the immune system of invertebrates uses transglutaminases that are highly homologous to FXIII to stabilize clots containing bacteria. In Drosophila melanogaster, a lack of transglutaminases led to an immune defect since microbes were no longer entrapped in clots (Wang et al., 2010).
In contrast to invertebrates, the mammalian coagulation system was long considered to be exclusively important for hemostasis. However, several studies now indicate that coagulation factors also contribute to the effective elimination of bacteria in mammals. Activation of coagulation on bacteria triggers multiple biological effects important for host defense. While some of these effectors induce direct stress on the bacteria, others support the cellular immune response. Before we zoom in to the direct antimicrobial effects of coagulation, we will briefly summarize the indirect immune functions of coagulation. One important consequence of coagulation at the site of infection is the recruitment of innate immune cells via BK that binds kinin receptors (B1R and B2R) on endothelial, smooth muscle, and innate immune cells (Frick et al., 2007). BK induces vascular leakage and interacts with macrophages to release chemo-attractants. Furthermore, a number of immune cells express protease-activated receptors (PARs) that are cleaved by coagulation proteases (thrombin and FXa) to initiate inflammation (Shpacovitch et al., 2008). Finally, activation of fibrinogen is accompanied by the release of the fibrinopeptides A and B, which are also chemo-attractants (Skogen et al., 1988).
Fibrin formation
The best-defined direct action of coagulation on bacteria is the formation of fibrin at the site of infection. When bacteria become immobilized inside the fibrin network, they are prevented from spreading into the surrounding tissues. It has been known for decades that this local entrapment reduces bacterial dissemination into the bloodstream (Rotstein, 1992). Mice deficient in fibrinogen are therefore more susceptible to infections with GAS due to decreased fibrin production (Sun et al., 2009). For some bacteria like S. aureus and E. coli, it was found that this immobilization inside a fibrin clot is FXIII dependent (Wang et al., 2010) and that bacteria are directly attached to the fibrin fibers via multiple binding sites cross-linked by FXIII. In the absence of FXIII, they appear to be loosely assembled within the clot without direct interaction with the fibers. Furthermore, it has been described that FXIII triggers entrapment of GAS at the site of infection, which subsequently leads to killing of the bacteria due to generation of plasma-derived antimicrobial peptides (Loof et al., 2011). In a murine skin infection model, these bacteria were also shown to cluster within fibrin networks in the presence of FXIII, whereas in the absence of FXIII they are distributed throughout the infection site. This leads to a higher influx of neutrophils and thereby increased inflammation. Furthermore, a systemic response, and therefore bacterial dissemination, was induced when FXIII was absent (Loof et al., 2011). Furthermore, fibrin polymerization mediates clearance of E. coli and S. aureus from the peritoneal cavity and limits the growth of Listeria monocytogenes in the liver (Ahrenholz & Simmons, 1980; Flick et al., 2004; Mullarky et al., 2005).
Antimicrobial peptides
Another direct action of coagulation in host defense against bacteria is the production of antimicrobial peptides (AMPs). First of all, BK itself was found to exhibit antimicrobial activity in vitro (Kowalska et al., 2002) although it has been debated if this is relevant in vivo (Frick et al., 2006). Also, coagulation proteins are sensitive to cleavage by neutrophil-derived proteases resulting in peptides with antimicrobial activity (Nordahl et al., 2005). For instance, thrombin-derived C-terminal peptides (TCP) were produced when fibrin clots or human plasma was incubated with neutrophil elastase, which could induce lysis of microbial membranes (Papareddy et al., 2010). TCPs were found to be antibacterial against the Gram-positive S. aureus, as well as to Gram-negative species P. aeruginosa and E. coli. Membrane permeabilization by the TCP GKY25 was confirmed by electron microscopy (Papareddy et al., 2010). Although it was shown that GKY25 acts on the bacterial membrane, the precise mechanism remains to be elucidated. HKH20, a peptide derived from domain 5 of HK, also displays antibacterial activity against both Gram-positive (S. aureus and Enterococcus faecalis) and -negative species (E. coli and P. aeruginosa) (Nordahl et al., 2005; Ringstad et al., 2007). Antibacterial activity of this peptide and its truncated variants was correlated to the ability to cause leakage in model lipid bilayers, indicating that disruption of membrane integrity is the mechanism that leads to HKH20-mediated bacterial killing (Ringstad et al., 2007). The effect of HKH20 is similar to that of the HK domain 3 derived peptide, NAT26. This peptide kills bacteria that activate the contact system, GAS AP1, S. aureus Cowan I, and S. typhimurium SR11B (Frick et al., 2006) and their cell walls were disintegrated when analyzed by electron microscopy. Furthermore, also this peptide causes leakage of anionic liposomes by disrupting the membrane (Frick et al., 2006). Finally, the fibrinogen-derived peptide GHR28 also exhibits antibacterial activity against the Gram-positive species Streptococcus agalactiae (GBS) and S. aureus, but not against E. faecalis and the Gram-negative bacterium E. coli (Påhlman et al., 2013).
Crosstalk between complement and coagulation
There is a significant amount of crosstalk between the complement and coagulation systems and this has been reviewed in a number of excellent papers (Markiewski et al., 2007; Oikonomopoulou et al., 2012). Here, we highlight a few of these interactions that, in our opinion, are of special interest in light of host defenses against bacterial infections. First, it was reported that several complement proteases activate the final stage of coagulation to form fibrin clots. A major role has been suggested for the MASPs, which circulate in complex with MBL or ficolins and are thus activated on bacterial surfaces via pattern recognition. Next to the role of MASP-2 in activation of complement, this protease was also found to cleave prothrombin into thrombin thereby promoting fibrin clot formation (Krarup et al., 2007). Although the prothrombin turnover rate was only 5% compared to the cleavage by FXa, activation occurred at physiological concentrations of prothrombin, which seemed specific since MASP homologues did not mediate this cleavage. Furthermore, MBL-MASP complexes bound to S. aureus could generate fibrin that was covalently linked to the bacterial surface via FXIII. This suggests that MBL-MASP may lead to localized fibrin formation. In addition to the action of MASP-2 on coagulation, MASP-1 is also involved in clotting. MASP-1 has thrombin-like activity and activates FXIII and fibrinogen to release fibrinopeptides and to induce clot formation (Krarup et al., 2008). Although it is tempting to speculate that the two MASPs might act together to cause localized formation of fibrin, a study comparing both actions indicated that clotting via MBL/ficolin-MASP is predominantly catalyzed by MASP-2 cleavage of prothrombin (Gulla et al., 2010). MASP-1 and MASP-2 can also cleave HK, but release of BK is only shown for MASP-1 (Dobó et al., 2011). Although the physiological importance of MASPs in coagulation clearly needs further investigations, the fact that these proteases are targeted to bacterial surfaces via pattern recognition molecules might trigger a more specific and localized clotting reaction compared to ‘contact’ activation.
Vice versa, coagulation proteases can activate the complement system. A number of coagulation proteases, such as thrombin, FXa, FIXa, and FXIa, directly cleave the central complement components C3 and C5 into their bioactive fragments (Huber-Lang et al., 2006; Amara et al., 2010). In addition, it has been shown that the CP can be directly initiated by activation of C1r by FXIIa (Ghebrehiwet et al., 1981; Kaplan & Ghebrehiwet, 2010) and that kallikrein has Factor-D-like activity since it converts Factor B to form the AP convertase (Hiemstra et al., 1985). Similar to MASP-mediated coagulation activation, the relevance of these findings in vivo is presently unknown. However, it is very intriguing that some of these cleavage reactions generate alternate complement activation products that have different biological activities. In particular, thrombin was reported to cleave both the MAC components C5 and C9 into alternative products. First, thrombin cleaves C9 into a 37 kDa C9b molecule (Biesecker et al., 1982) that theoretically can cross the peptidoglycan layer. In contrast to native C9, this carboxyl-terminal fragment of C9 is able to disturb the membrane potential of membrane vesicles in absence of C5b-8 (Dankert & Esser, 1986). Second, thrombin efficiently cleaves C5 at a different cleavage site than the C5 convertase, generating C5T (Krisinger et al., 2012). This cleavage does not directly lead to release of C5a or C5b, but after cleavage of C5T by the complement C5 convertase, a different C5b molecule was formed that seemed to form a more potent MAC than the conventional C5b-9 complex.
Overall, the crosstalk between complement and coagulation cascades may enhance innate immunity by generating more specific, localized and effective antibacterial reactions.
Complement and coagulation in bacterial disease
Whereas complement and coagulation are evolved to act locally at the site of infection and prevent bacterial dissemination, both systems are often pathological during systemic infections. The Systemic Inflammatory Response Syndrome (SIRS), a hallmark of sepsis, is characterized by excessive activation of complement and coagulation (De Jong et al., 2010). Septic patients have high plasma levels of complement activation products (C3a, C4a and C5a) of which especially C5a is believed to cause an overwhelming inflammatory response that plays a role in the outcome of sepsis. Also coagulation markers like soluble tissue factor and thrombin-antithrombin complexes (TATc) are strongly increased while endogenous inhibitors of coagulation and the fibrinolytic system are consumed (van der Poll & Opal, 2008). In severe sepsis, coagulation causes disseminated intravascular coagulation (DIC), a serious condition in which microvascular thrombosis, bleeding and microvascular fibrin deposition lead to multiple organ failure, the most common cause of death. Whereas pathogens can directly activate the coagulation system through the contact system, thrombin generation in DIC is mediated by the extrinsic pathway of coagulation (Taylor et al., 1991; Pixley et al., 1993; Levi et al., 1994). The systemic inflammatory response associated with sepsis disturbs the hemostatic balance, with increased levels of IL-6 leading to tissue factor expression on vascular cells (van der Poll et al., 1994) and release of TNFα leading to impaired fibrinolysis (van der Poll et al., 1991). Several experimental studies are now exploring whether therapeutic intervention with complement and coagulation can reduce the mortality of sepsis. Blocking C5a with antibody treatment reduced mortality of experimental models of Gram-negative sepsis (reviewed in Guo & Ward, 2005). Also in a baboon model of E. coli sepsis, complement inhibition at the level of C3 prevented organ damage and pathological coagulation (Silasi-Mansat et al., 2010). Still, sepsis is a complex disease and not all forms of sepsis are the same. For instance, whereas C5a has a negative role in Gram-negative sepsis, it seems essential for the immune response during S. aureus sepsis (Von Köckritz-Blickwede et al., 2010). In analogy, inhibition of coagulation via natural anticoagulants has led to contrasting results in the treatment of sepsis (Esmon, 2004). Thus, even though knowledge of complement and coagulation provides new avenues for the treatment of sepsis, more insights seem required for the pathophysiology of sepsis caused by different bacteria.
How bacterial pathogens deal with stress invoked by complement and coagulation
Since complement and coagulation cause stress to bacterial cells, bacterial pathogens have developed strategies to counteract these pathways. For the complement system, inhibition can occur at almost every level of the cascade. For example, S. aureus secretes different proteins that block C1q binding (Kang et al., 2013), convertase activity (Rooijakkers et al., 2005; Jongerius et al., 2007), and C5 cleavage (Langley et al., 2005; Bestebroer et al., 2010). Since these mechanisms have been subject of many excellent reviews (Rautemaa & Meri, 1999; Lambris et al., 2008; Zipfel et al., 2013) and ‘direct’ complement actions are the focus of this review, we will here exclusively discuss strategies evolved by Gram-negative bacteria that are aimed at inhibiting the MAC (Table 1). Furthermore, we will discuss how bacterial pathogens block coagulation or degrade fibrin clots and produce virulence factors to specifically induce coagulation (Table 2).
MAC evasion by Gram-negative bacteria
| Microorganism | Microbial structure | Category | Function | Reference |
| Borrelia burgdorferi | CD59-like | Mimick or attract host regulators | Inhibition of MAC assembly | Pausa et al. (2003) |
| CRASP-3, -4, -5 | Mimick or attract host regulators | Binding of CFHR1 | Haupt et al. (2007) | |
| CRASP-1 | Expression of inhibitory surface proteins | Blocks MAC assembly | Hallström et al. (2013) | |
| Borrelia garinii | CRASPs | Mimick or attract host regulators | Binding of CFHR1 | Van Burgel et al. (2010) |
| Escherichia coli | ? | Mimick or attract host regulators | Capture soluble CD59 to inhibit MAC assembly | Rautemaa et al. (1998) |
| TraT | Expression of inhibitory surface proteins | Inhibition of MAC assembly | Pramoonjago et al. (1992) | |
| Haemophilus ducreyi | DrsA | Mimick or attract host regulators | Binding of vitronectin | Leduc et al. (2009) |
| Haemophilus influenza | Hsf | Mimick or attract host regulators | Binding of vitronectin | Hallström et al. (2006) |
| Helicobacter pylori | ? | Mimick or attract host regulators | Capture soluble CD59 to inhibit MAC assembly | Rautemaa et al. (2001) |
| ? | Mimick or attract host regulators | Binding of vitronectin | Ringnér et al. (1992) | |
| Klebsiella pneumoniae | O-chain variation | Surface modification | Protects the OM from MAC assembly to mediates serum-resistance | Merino et al. (1992) |
| Moraxella catarrhalis | UspA2 | Mimick or attract host regulators | Binding of vitronectin | Attia et al. (2006) and Singh et al. (2010a,b) |
| Neisseria gonorrhoeae | OpaA | Mimick or attract host regulators | Binding of vitronectin | Duensing & Putten (1998) |
| Neisseria meningitidis | Msf | Mimick or attract host regulators | Binding of vitronectin | Griffiths et al. (2011) |
| Opc | Mimick or attract host regulators | Binding of vitronectin | Sa E Cunha et al. (2010) | |
| NhhA | Expression of inhibitory surface proteins | Blocks MAC deposition | Sjölinder et al. (2008) | |
| LNT sialylation | Surface modification | Protects from serum-mediated killing | Estabrook et al. (1997) | |
| Capsule expression | Surface modification | Protects the OM from MAC insertion | Schneider et al. (2007) | |
| Porphyromonas gingivalis | APS layer | Surface modification | Protects the OM from MAC insertion | Slaney et al. (2006) |
| Rickettsia conorii | OmpB | Mimick or attract host regulators | Binding of vitronectin | Riley et al. (2013) |
| Salmonella minnesota | O-chain variation | Surface modification | Protects the OM from MAC insertion and mediates serum-resistance | Joiner et al. (1982a,b) |
| Salmonella montevideo | O-chain variation | Surface modification | Protects against MAC-mediated killing | Grossman et al. (1987) |
| Salmonella typhimurium | O-chain variation | Surface modification | Protects against MAC-mediated killing | Murray et al. (2005) |
| Rck | Expression of inhibitory surface proteins | Blocks C9 polymerization | Heffernan et al. (1992) | |
| Shigella flexneri | O-chain variation | Surface modification | Protects against MAC-mediated killing | Hong & Payne (1997) |
| Yersinia enterocolitica | YadA | Expression of inhibitory surface proteins | Inhibits C9 binding, thereby MAC formation | Pilz et al. (1992) |
| Ail | Expression of inhibitory surface proteins | Serum-resistance, potentially blocks C9 polymerization | Bliska & Falkow (1992) |
| Microorganism | Microbial structure | Category | Function | Reference |
| Borrelia burgdorferi | CD59-like | Mimick or attract host regulators | Inhibition of MAC assembly | Pausa et al. (2003) |
| CRASP-3, -4, -5 | Mimick or attract host regulators | Binding of CFHR1 | Haupt et al. (2007) | |
| CRASP-1 | Expression of inhibitory surface proteins | Blocks MAC assembly | Hallström et al. (2013) | |
| Borrelia garinii | CRASPs | Mimick or attract host regulators | Binding of CFHR1 | Van Burgel et al. (2010) |
| Escherichia coli | ? | Mimick or attract host regulators | Capture soluble CD59 to inhibit MAC assembly | Rautemaa et al. (1998) |
| TraT | Expression of inhibitory surface proteins | Inhibition of MAC assembly | Pramoonjago et al. (1992) | |
| Haemophilus ducreyi | DrsA | Mimick or attract host regulators | Binding of vitronectin | Leduc et al. (2009) |
| Haemophilus influenza | Hsf | Mimick or attract host regulators | Binding of vitronectin | Hallström et al. (2006) |
| Helicobacter pylori | ? | Mimick or attract host regulators | Capture soluble CD59 to inhibit MAC assembly | Rautemaa et al. (2001) |
| ? | Mimick or attract host regulators | Binding of vitronectin | Ringnér et al. (1992) | |
| Klebsiella pneumoniae | O-chain variation | Surface modification | Protects the OM from MAC assembly to mediates serum-resistance | Merino et al. (1992) |
| Moraxella catarrhalis | UspA2 | Mimick or attract host regulators | Binding of vitronectin | Attia et al. (2006) and Singh et al. (2010a,b) |
| Neisseria gonorrhoeae | OpaA | Mimick or attract host regulators | Binding of vitronectin | Duensing & Putten (1998) |
| Neisseria meningitidis | Msf | Mimick or attract host regulators | Binding of vitronectin | Griffiths et al. (2011) |
| Opc | Mimick or attract host regulators | Binding of vitronectin | Sa E Cunha et al. (2010) | |
| NhhA | Expression of inhibitory surface proteins | Blocks MAC deposition | Sjölinder et al. (2008) | |
| LNT sialylation | Surface modification | Protects from serum-mediated killing | Estabrook et al. (1997) | |
| Capsule expression | Surface modification | Protects the OM from MAC insertion | Schneider et al. (2007) | |
| Porphyromonas gingivalis | APS layer | Surface modification | Protects the OM from MAC insertion | Slaney et al. (2006) |
| Rickettsia conorii | OmpB | Mimick or attract host regulators | Binding of vitronectin | Riley et al. (2013) |
| Salmonella minnesota | O-chain variation | Surface modification | Protects the OM from MAC insertion and mediates serum-resistance | Joiner et al. (1982a,b) |
| Salmonella montevideo | O-chain variation | Surface modification | Protects against MAC-mediated killing | Grossman et al. (1987) |
| Salmonella typhimurium | O-chain variation | Surface modification | Protects against MAC-mediated killing | Murray et al. (2005) |
| Rck | Expression of inhibitory surface proteins | Blocks C9 polymerization | Heffernan et al. (1992) | |
| Shigella flexneri | O-chain variation | Surface modification | Protects against MAC-mediated killing | Hong & Payne (1997) |
| Yersinia enterocolitica | YadA | Expression of inhibitory surface proteins | Inhibits C9 binding, thereby MAC formation | Pilz et al. (1992) |
| Ail | Expression of inhibitory surface proteins | Serum-resistance, potentially blocks C9 polymerization | Bliska & Falkow (1992) |
MAC evasion by Gram-negative bacteria
| Microorganism | Microbial structure | Category | Function | Reference |
| Borrelia burgdorferi | CD59-like | Mimick or attract host regulators | Inhibition of MAC assembly | Pausa et al. (2003) |
| CRASP-3, -4, -5 | Mimick or attract host regulators | Binding of CFHR1 | Haupt et al. (2007) | |
| CRASP-1 | Expression of inhibitory surface proteins | Blocks MAC assembly | Hallström et al. (2013) | |
| Borrelia garinii | CRASPs | Mimick or attract host regulators | Binding of CFHR1 | Van Burgel et al. (2010) |
| Escherichia coli | ? | Mimick or attract host regulators | Capture soluble CD59 to inhibit MAC assembly | Rautemaa et al. (1998) |
| TraT | Expression of inhibitory surface proteins | Inhibition of MAC assembly | Pramoonjago et al. (1992) | |
| Haemophilus ducreyi | DrsA | Mimick or attract host regulators | Binding of vitronectin | Leduc et al. (2009) |
| Haemophilus influenza | Hsf | Mimick or attract host regulators | Binding of vitronectin | Hallström et al. (2006) |
| Helicobacter pylori | ? | Mimick or attract host regulators | Capture soluble CD59 to inhibit MAC assembly | Rautemaa et al. (2001) |
| ? | Mimick or attract host regulators | Binding of vitronectin | Ringnér et al. (1992) | |
| Klebsiella pneumoniae | O-chain variation | Surface modification | Protects the OM from MAC assembly to mediates serum-resistance | Merino et al. (1992) |
| Moraxella catarrhalis | UspA2 | Mimick or attract host regulators | Binding of vitronectin | Attia et al. (2006) and Singh et al. (2010a,b) |
| Neisseria gonorrhoeae | OpaA | Mimick or attract host regulators | Binding of vitronectin | Duensing & Putten (1998) |
| Neisseria meningitidis | Msf | Mimick or attract host regulators | Binding of vitronectin | Griffiths et al. (2011) |
| Opc | Mimick or attract host regulators | Binding of vitronectin | Sa E Cunha et al. (2010) | |
| NhhA | Expression of inhibitory surface proteins | Blocks MAC deposition | Sjölinder et al. (2008) | |
| LNT sialylation | Surface modification | Protects from serum-mediated killing | Estabrook et al. (1997) | |
| Capsule expression | Surface modification | Protects the OM from MAC insertion | Schneider et al. (2007) | |
| Porphyromonas gingivalis | APS layer | Surface modification | Protects the OM from MAC insertion | Slaney et al. (2006) |
| Rickettsia conorii | OmpB | Mimick or attract host regulators | Binding of vitronectin | Riley et al. (2013) |
| Salmonella minnesota | O-chain variation | Surface modification | Protects the OM from MAC insertion and mediates serum-resistance | Joiner et al. (1982a,b) |
| Salmonella montevideo | O-chain variation | Surface modification | Protects against MAC-mediated killing | Grossman et al. (1987) |
| Salmonella typhimurium | O-chain variation | Surface modification | Protects against MAC-mediated killing | Murray et al. (2005) |
| Rck | Expression of inhibitory surface proteins | Blocks C9 polymerization | Heffernan et al. (1992) | |
| Shigella flexneri | O-chain variation | Surface modification | Protects against MAC-mediated killing | Hong & Payne (1997) |
| Yersinia enterocolitica | YadA | Expression of inhibitory surface proteins | Inhibits C9 binding, thereby MAC formation | Pilz et al. (1992) |
| Ail | Expression of inhibitory surface proteins | Serum-resistance, potentially blocks C9 polymerization | Bliska & Falkow (1992) |
| Microorganism | Microbial structure | Category | Function | Reference |
| Borrelia burgdorferi | CD59-like | Mimick or attract host regulators | Inhibition of MAC assembly | Pausa et al. (2003) |
| CRASP-3, -4, -5 | Mimick or attract host regulators | Binding of CFHR1 | Haupt et al. (2007) | |
| CRASP-1 | Expression of inhibitory surface proteins | Blocks MAC assembly | Hallström et al. (2013) | |
| Borrelia garinii | CRASPs | Mimick or attract host regulators | Binding of CFHR1 | Van Burgel et al. (2010) |
| Escherichia coli | ? | Mimick or attract host regulators | Capture soluble CD59 to inhibit MAC assembly | Rautemaa et al. (1998) |
| TraT | Expression of inhibitory surface proteins | Inhibition of MAC assembly | Pramoonjago et al. (1992) | |
| Haemophilus ducreyi | DrsA | Mimick or attract host regulators | Binding of vitronectin | Leduc et al. (2009) |
| Haemophilus influenza | Hsf | Mimick or attract host regulators | Binding of vitronectin | Hallström et al. (2006) |
| Helicobacter pylori | ? | Mimick or attract host regulators | Capture soluble CD59 to inhibit MAC assembly | Rautemaa et al. (2001) |
| ? | Mimick or attract host regulators | Binding of vitronectin | Ringnér et al. (1992) | |
| Klebsiella pneumoniae | O-chain variation | Surface modification | Protects the OM from MAC assembly to mediates serum-resistance | Merino et al. (1992) |
| Moraxella catarrhalis | UspA2 | Mimick or attract host regulators | Binding of vitronectin | Attia et al. (2006) and Singh et al. (2010a,b) |
| Neisseria gonorrhoeae | OpaA | Mimick or attract host regulators | Binding of vitronectin | Duensing & Putten (1998) |
| Neisseria meningitidis | Msf | Mimick or attract host regulators | Binding of vitronectin | Griffiths et al. (2011) |
| Opc | Mimick or attract host regulators | Binding of vitronectin | Sa E Cunha et al. (2010) | |
| NhhA | Expression of inhibitory surface proteins | Blocks MAC deposition | Sjölinder et al. (2008) | |
| LNT sialylation | Surface modification | Protects from serum-mediated killing | Estabrook et al. (1997) | |
| Capsule expression | Surface modification | Protects the OM from MAC insertion | Schneider et al. (2007) | |
| Porphyromonas gingivalis | APS layer | Surface modification | Protects the OM from MAC insertion | Slaney et al. (2006) |
| Rickettsia conorii | OmpB | Mimick or attract host regulators | Binding of vitronectin | Riley et al. (2013) |
| Salmonella minnesota | O-chain variation | Surface modification | Protects the OM from MAC insertion and mediates serum-resistance | Joiner et al. (1982a,b) |
| Salmonella montevideo | O-chain variation | Surface modification | Protects against MAC-mediated killing | Grossman et al. (1987) |
| Salmonella typhimurium | O-chain variation | Surface modification | Protects against MAC-mediated killing | Murray et al. (2005) |
| Rck | Expression of inhibitory surface proteins | Blocks C9 polymerization | Heffernan et al. (1992) | |
| Shigella flexneri | O-chain variation | Surface modification | Protects against MAC-mediated killing | Hong & Payne (1997) |
| Yersinia enterocolitica | YadA | Expression of inhibitory surface proteins | Inhibits C9 binding, thereby MAC formation | Pilz et al. (1992) |
| Ail | Expression of inhibitory surface proteins | Serum-resistance, potentially blocks C9 polymerization | Bliska & Falkow (1992) |
How bacterial pathogens modulate the coagulation system
| Microorganism | Microbial structure | Target | Effect | Reference |
| Staphylococcus aureus | Clumping factor | Fibrinogen | Adherence to host tissue and bacterial aggregation | Fitzgerald et al. (2006) |
| Coagulase | Prothrombin and fibrinogen | Activation of prothrombin to form fibrin clots | Friedrich et al. (2003) | |
| Efb | Fibrinogen and C3b | Inhibition of phagocytosis, neutrophil adherence to fibrinogen and platelet aggregation | Ko et al. (2011, 2013) and Shannon & Flock (2004) | |
| FnbpA | Fibrinogen | Activation and aggregation of platelets | Fitzgerald et al. (2006) | |
| SSL10 | Prothrombin | Delay in clotting times | Itoh et al. (2013) | |
| vWbp | Prothrombin, fibrinogen and FXIII | Activation of prothrombin and FXIII to form and stabilize fibrin clots | Cheng et al. (2010) and Thomer et al. (2013) | |
| Group A streptococci | M protein | Fibrinogen and HK | Inhibition of complement deposition Assembly and activation of contact system | Smeesters et al. (2010) |
| SclA and SclB | TAFI | Inhibition of fibrinolysis | Bengtson et al. (2009) | |
| SIC | HK | Inhibition of HK binding to the bacterial surface and inhibition of antimicrobial peptides of HK | Akesson et al. (2010) | |
| Group G streptococci | FOG | HK | Assembly of contact system and release of BK | Wollein Waldetoft et al. (2012) |
| PG | HK | Assembly of contact system Inhibition of HK-derived antimicrobial peptides | Wollein Waldetoft et al. (2012) | |
| Salmonella typhimurium | Fimbriae | FXII, HK and fibrinogen | Assembly and activation of contact system | Herwald et al. (1998) |
| Escherichia coli | Curli | FXII, HK and fibrinogen | Assembly and activation of contact system | Ben Nasr et al. (1996) and Herwald et al. (1998) |
| Gram negative bacteria | LPS | HK | Activation of contact system | Perkins et al. (2008) |
| Various bacterial species | PolyP | FXII and HK | Activation of contact system and release of BK | Müller et al. (2009) and Smith et al. (2006) |
| FV | Acceleration of thrombin formation and clot stabilization | Morrissey et al. (2012) |
| Microorganism | Microbial structure | Target | Effect | Reference |
| Staphylococcus aureus | Clumping factor | Fibrinogen | Adherence to host tissue and bacterial aggregation | Fitzgerald et al. (2006) |
| Coagulase | Prothrombin and fibrinogen | Activation of prothrombin to form fibrin clots | Friedrich et al. (2003) | |
| Efb | Fibrinogen and C3b | Inhibition of phagocytosis, neutrophil adherence to fibrinogen and platelet aggregation | Ko et al. (2011, 2013) and Shannon & Flock (2004) | |
| FnbpA | Fibrinogen | Activation and aggregation of platelets | Fitzgerald et al. (2006) | |
| SSL10 | Prothrombin | Delay in clotting times | Itoh et al. (2013) | |
| vWbp | Prothrombin, fibrinogen and FXIII | Activation of prothrombin and FXIII to form and stabilize fibrin clots | Cheng et al. (2010) and Thomer et al. (2013) | |
| Group A streptococci | M protein | Fibrinogen and HK | Inhibition of complement deposition Assembly and activation of contact system | Smeesters et al. (2010) |
| SclA and SclB | TAFI | Inhibition of fibrinolysis | Bengtson et al. (2009) | |
| SIC | HK | Inhibition of HK binding to the bacterial surface and inhibition of antimicrobial peptides of HK | Akesson et al. (2010) | |
| Group G streptococci | FOG | HK | Assembly of contact system and release of BK | Wollein Waldetoft et al. (2012) |
| PG | HK | Assembly of contact system Inhibition of HK-derived antimicrobial peptides | Wollein Waldetoft et al. (2012) | |
| Salmonella typhimurium | Fimbriae | FXII, HK and fibrinogen | Assembly and activation of contact system | Herwald et al. (1998) |
| Escherichia coli | Curli | FXII, HK and fibrinogen | Assembly and activation of contact system | Ben Nasr et al. (1996) and Herwald et al. (1998) |
| Gram negative bacteria | LPS | HK | Activation of contact system | Perkins et al. (2008) |
| Various bacterial species | PolyP | FXII and HK | Activation of contact system and release of BK | Müller et al. (2009) and Smith et al. (2006) |
| FV | Acceleration of thrombin formation and clot stabilization | Morrissey et al. (2012) |
How bacterial pathogens modulate the coagulation system
| Microorganism | Microbial structure | Target | Effect | Reference |
| Staphylococcus aureus | Clumping factor | Fibrinogen | Adherence to host tissue and bacterial aggregation | Fitzgerald et al. (2006) |
| Coagulase | Prothrombin and fibrinogen | Activation of prothrombin to form fibrin clots | Friedrich et al. (2003) | |
| Efb | Fibrinogen and C3b | Inhibition of phagocytosis, neutrophil adherence to fibrinogen and platelet aggregation | Ko et al. (2011, 2013) and Shannon & Flock (2004) | |
| FnbpA | Fibrinogen | Activation and aggregation of platelets | Fitzgerald et al. (2006) | |
| SSL10 | Prothrombin | Delay in clotting times | Itoh et al. (2013) | |
| vWbp | Prothrombin, fibrinogen and FXIII | Activation of prothrombin and FXIII to form and stabilize fibrin clots | Cheng et al. (2010) and Thomer et al. (2013) | |
| Group A streptococci | M protein | Fibrinogen and HK | Inhibition of complement deposition Assembly and activation of contact system | Smeesters et al. (2010) |
| SclA and SclB | TAFI | Inhibition of fibrinolysis | Bengtson et al. (2009) | |
| SIC | HK | Inhibition of HK binding to the bacterial surface and inhibition of antimicrobial peptides of HK | Akesson et al. (2010) | |
| Group G streptococci | FOG | HK | Assembly of contact system and release of BK | Wollein Waldetoft et al. (2012) |
| PG | HK | Assembly of contact system Inhibition of HK-derived antimicrobial peptides | Wollein Waldetoft et al. (2012) | |
| Salmonella typhimurium | Fimbriae | FXII, HK and fibrinogen | Assembly and activation of contact system | Herwald et al. (1998) |
| Escherichia coli | Curli | FXII, HK and fibrinogen | Assembly and activation of contact system | Ben Nasr et al. (1996) and Herwald et al. (1998) |
| Gram negative bacteria | LPS | HK | Activation of contact system | Perkins et al. (2008) |
| Various bacterial species | PolyP | FXII and HK | Activation of contact system and release of BK | Müller et al. (2009) and Smith et al. (2006) |
| FV | Acceleration of thrombin formation and clot stabilization | Morrissey et al. (2012) |
| Microorganism | Microbial structure | Target | Effect | Reference |
| Staphylococcus aureus | Clumping factor | Fibrinogen | Adherence to host tissue and bacterial aggregation | Fitzgerald et al. (2006) |
| Coagulase | Prothrombin and fibrinogen | Activation of prothrombin to form fibrin clots | Friedrich et al. (2003) | |
| Efb | Fibrinogen and C3b | Inhibition of phagocytosis, neutrophil adherence to fibrinogen and platelet aggregation | Ko et al. (2011, 2013) and Shannon & Flock (2004) | |
| FnbpA | Fibrinogen | Activation and aggregation of platelets | Fitzgerald et al. (2006) | |
| SSL10 | Prothrombin | Delay in clotting times | Itoh et al. (2013) | |
| vWbp | Prothrombin, fibrinogen and FXIII | Activation of prothrombin and FXIII to form and stabilize fibrin clots | Cheng et al. (2010) and Thomer et al. (2013) | |
| Group A streptococci | M protein | Fibrinogen and HK | Inhibition of complement deposition Assembly and activation of contact system | Smeesters et al. (2010) |
| SclA and SclB | TAFI | Inhibition of fibrinolysis | Bengtson et al. (2009) | |
| SIC | HK | Inhibition of HK binding to the bacterial surface and inhibition of antimicrobial peptides of HK | Akesson et al. (2010) | |
| Group G streptococci | FOG | HK | Assembly of contact system and release of BK | Wollein Waldetoft et al. (2012) |
| PG | HK | Assembly of contact system Inhibition of HK-derived antimicrobial peptides | Wollein Waldetoft et al. (2012) | |
| Salmonella typhimurium | Fimbriae | FXII, HK and fibrinogen | Assembly and activation of contact system | Herwald et al. (1998) |
| Escherichia coli | Curli | FXII, HK and fibrinogen | Assembly and activation of contact system | Ben Nasr et al. (1996) and Herwald et al. (1998) |
| Gram negative bacteria | LPS | HK | Activation of contact system | Perkins et al. (2008) |
| Various bacterial species | PolyP | FXII and HK | Activation of contact system and release of BK | Müller et al. (2009) and Smith et al. (2006) |
| FV | Acceleration of thrombin formation and clot stabilization | Morrissey et al. (2012) |
Bacterial evasion of the MAC
Surface modification
A long known mechanism by which Gram-negative bacteria resist MAC-mediated killing is by modification of the LPS molecules. LPS forms a physical and chemical barrier around the OM and is build up of three regions; the lipid moiety in the outer leaflet of the OM called lipid A; the core oligosaccharide containing 10–12 sugars; and on the exterior a polysaccharide chain of repeating units, the O-chain. Bacteria lacking the O-chain grow in rough-looking colonies, therefore termed ‘rough’ while wild-type species are termed ‘smooth’ (reviewed in Caroff et al., 2002). It has been recognized for decades that the O-chain is involved in complement resistance since smooth-type bacteria are usually serum-resistant whereas the rough bacteria are serum-sensitive. The O-chain does not prevent complement activation or C5 cleavage but probably sheds the MAC from the OM and/or sterically hinders its insertion into the OM (Joiner et al., 1982a,b). For several bacterial species, it was shown that bacteria with longer O-chains are better protected against the MAC. Furthermore, a higher density of O-chains increases MAC resistance (Grossman et al., 1987; Merino et al., 1992; Hong & Payne, 1997; Murray et al., 2005). LPS expressed by N. meningitidis, but also N. gonorrhoeae and Haemophilus influenzae, lack the O-specific side-chain. Alternatively, most disease-associated strains of N. meningitidis produce two distinct short carbohydrate chains, in which a lacto-N-neotetraose (LNT) moiety is present. The LNT epitope serves as the major site for sialylation that reduces immunogenicity and is associated with increased serum-resistance (Estabrook et al., 1997). However, the importance of sialylation remains controversial, indicating that this strategy is of minor importance for MAC evasion by meningococci (Schneider et al., 2007).
Another bacterial mechanism to evade MAC-dependent killing is formation of a capsule. Although a bacterial capsule can prevent complement activation in general, we will solely discuss how a capsule blocks the MAC specifically. Neisseria meningitidis expresses an (α2→8)-linked polysaccharide capsule that also contains sialic acids (Kahler et al., 1998). In most serotypes this capsule did not affect C3b deposition (Vogel et al., 1997), but specifically prevented MAC insertion in the OM (Schneider et al., 2007). Recently it was reported that capsule expression in meningococci is enhanced by increased temperature, which occurs for instance during inflammation when the bacteria need to interfere with immune killing (Loh et al., 2013). Also the outer surface of Porphyromonas gingivalis bears a glycan layer containing anionic polysaccharides (APS) that mediate direct resistance against the MAC. This APS did not inhibit MAC deposition on the bacteria (Slaney et al., 2006) but probably prevents the MAC from inserting into the OM.
Expression of inhibitory surface proteins
Gram-negative bacteria also express surface-localized proteins to inhibit MAC formation. TraT is a plasmid-encoded OM protein of E. coli K12 that mediates MAC-resistance at the level of C5b6. TraT is believed to block C5b6 assembly or structurally alter the complex into an inactive form (Pramoonjago et al., 1992). The OM protein NhhA of N. meningitidis mediates bacterial colonization and was also recognized to be important for complement resistance. NhhA prevents MAC deposition to the bacterial surface although it was not determined at what level in the complement cascade the protein has its effect (Sjölinder et al., 2008). Borrelia burgdorferi, the causative agent of Lyme disease, expresses the OM-localized complement regulator-acquiring surface proteins (CRASP) family. The CRASP-1 (or CspA) protein was recently identified to inhibit MAC attack by binding C7 and C9, thereby inhibiting assembly and membrane insertion of the MAC (Hallström et al., 2013). Yersinia enterocolitica virulence is associated with the expression of an autotransporter protein in the OM, Yersinia adhesin A (YadA). The protein mediates protection against MAC-mediated killing by inhibiting C9 deposition on the bacteria. However, it was not completely elucidated whether inhibition of MAC assembly was not just a result of inhibition at the level of C3 (Pilz et al., 1992). More recently, binding of C3b or iC3b by YadA was described whereby Factor H, normally promoting complement decay on host cells, was attracted to the bacterial surface to enhance bacterial survival (Schindler et al., 2012). Another outer membrane protein expressed by Y. enterocolitica, Ail, also conferred complement resistance when it was expressed in E. coli (Bliska & Falkow, 1992). Potentially, Ail acts by inhibiting C5b-9 assembly. A structurally and functionally related membrane protein expressed by S. typhimurium, Rck, interferes with MAC formation by inhibiting C9 polymerization (Heffernan et al., 1992).
Mimicking or attracting host regulators
To protect the host from self-damage, complement is tightly regulated by multiple fluid-phase and cell-bound proteins (for a review see Zipfel & Skerka, 2009). Bacteria often hijack these complement regulators to resist MAC-mediated killing (reviewed in Blom et al., 2009). CD59 is a membrane-bound glycophosphatidylinositol (GPI) lipid-anchored glycoprotein that protects host cells from the MAC by binding C5b-8 and preventing incorporation of polymeric C9. On the other hand, CD59 interferes with C8 and C9 insertion into the membrane (Zalman et al., 1986; Meri et al., 1990; Rollins & Sims, 1990; Letho & Meri, 1993; Farkas et al., 2002). The OM protein CD59-like of B. burgdorferi is functionally similar to human CD59 since it interacts with C8 and C9 to inhibit MAC assembly (Pausa et al., 2003). Escherichia coli and Helicobacter pylori were found to capture the soluble form of CD59, which is often shed from host cell membranes (Väkevä et al., 1994), to their OM and thereby prevent MAC-mediated killing (Rautemaa et al., 1998, 2001).
Another human regulator of MAC is the abundant serum protein vitronectin (S-protein, 250–450 μg mL−1). This protein, that also regulates coagulation (Preissner, 1991; Felding-Habermann & Cheresh, 1993), can bind fluid-phase C5b67 complexes and prevent formation of an active MAC (Milis et al., 1993; Podack et al., 1977). Bacteria can capture vitronectin to prevent MAC-mediated killing (reviewed in Singh et al., 2010b). Meningococcal surface fibril (Msf) (Griffiths et al., 2011), OM protein Haemophilus surface fibril (Hsf) of H. influenzae (Hallström et al., 2006), surface protein UspA2 of Moraxella catarrhalis (Attia et al., 2006; Singh et al., 2010a), DrsA of Haemophilus ducreyi (Leduc et al., 2009), and autotransporter protein OmpB expressed by Rickettsia conorii (Riley et al., 2013) all mediate recruitment of vitronectin to the bacterial surface, thereby mediating serum resistance. Since bacterial acquisition of vitronectin also fulfills additional roles in pathogenesis like bacterial adherence (Singh et al., 2010b), the role of vitronectin binding proteins in MAC evasion was not always studied [e.g. vitronectin binding to the surface of H. pylori (Ringnér et al., 1992), OpaA of N. gonorrhoeae (Duensing & Putten, 1998), and Opc protein of N. meningitidis (Sa E Cunha et al., 2010)]. However, it cannot be excluded that these bacterial vitronectin-binding proteins also mediate serum resistance.
Finally, bacteria can regulate the MAC by attracting the complement Factor H-related protein 1 (CFHR1), a C5 convertase inhibitor present in the circulation at a concentration of 20–100 μg mL−1 (Timmann et al., 1991; Heinen et al., 2009). CFHR1 is related to Factor H, the C3 convertase regulator that binds C3b (Wu et al., 2009). CFHR1 specifically regulates the AP C5 convertase by binding both C3b and C5 (Skerka et al., 1991; Timmann et al., 1991). Moreover, formation and membrane insertion of the MAC is controlled by CFHR1 (Heinen et al., 2009). The B. burgdorferi CRASP-3, -4, and -5 proteins were all found to attract CFHR1, thereby mediating serum-resistance of B. burgdorferi (Haupt et al., 2007). Some strains of the different genospecies Borrelia garinii express CRASPs that can also bind CFHR1 to mediate MAC escape (Van Burgel et al., 2010).
Polyphosphates
Microorganisms store long polymers of phosphate (polyphosphates or polyP) that are essential to bacterial motility and survival (Rao & Kornberg, 1996; Kim et al., 2002). Recently, it was discovered that polyP, when excreted, specifically inhibit MAC assembly on target surfaces by binding and destabilizing the C5b6 complex (Wat et al., 2014). Accordingly, it was described that mutants of N. meningitidis lacking exopolyphosphatase, that cleaves polyP, were protected from MAC-mediated killing (Zhang et al., 2010). This indicates that the production of long polymers of polyP forms an additional mechanism for bacteria to deal with stress invoked by the MAC, although more proof of this concept has yet to be provided.
Bacterial modulation of coagulation
Mechanisms to block coagulation
One mechanism bacteria use to block coagulation is to prevent contact activation. In GAS, the secreted protein SIC (streptococcal inhibitor of complement) inhibits the binding of HK to the bacterial surface thereby down-regulating the contact system. Furthermore, SIC inhibits the antimicrobial activity of HK-derived peptides (Akesson et al., 2010; Frick et al., 2011). Staphylococcus aureus secretes the staphylococcal superantigen-like protein 10 (SSL10) that delays clotting by binding to prothrombin. This interaction does not inhibit thrombin activity, but blocks the binding of prothrombin to platelets, which is important for its activation (Itoh et al., 2013). No other bacterial inhibitors of contact activation have been described so far but yet to be defined S. aureus surface proteins can also block contact activation (Mattsson et al., 2001).
Degradation of fibrin clots
Many pathogenic bacteria have developed mechanisms to exploit the human fibrinolytic system to escape the physical boundaries of a fibrin clot. Fibrinolysis is normally mediated by the serine protease plasmin, which is formed by the conversion of plasminogen by the plasminogen activators tPA and uPA (Reichel et al., 2012). Many bacteria can attract plasminogen to their surface where it can be converted into active plasmin by host activators to subsequently induce fibrinolysis. Borrelia burgdorferi expresses a variety of plasminogen receptors on their surface, including the outer surface proteins (OspA and OspC) and Erp proteins (ErpA, ErpC, ErpP), to establish plasmin formation during all stages of infection (Brissette et al., 2009). Other examples of bacterial plasminogen receptors include the H. influenzae surface protein E (PE) (Barthel et al., 2012) and surface-bound enolases of several other bacteria (Pancholi & Fischetti, 1998; Bergmann et al., 2001; Chung et al., 2011; Floden et al., 2011). Next to expression of plasminogen-activating molecules at the surface, some bacteria also produce secreted plasminogen activators. Well-known are the streptococcal streptokinase (STK), S. aureus staphylokinase (SAK), and Yersinia pestis Pla. Both STK and SAK form a complex with plasminogen resulting in its autolytic cleavage into a form of plasmin that is less sensitive to inactivation by α2-antiplasmin. They are highly specific for human plasminogen (Sun et al., 2004). Pla does not form a complex but proteolytically converts plasminogen to plasmin. It has been shown that Pla is also capable of cleaving and inactivating α2-antiplasmin, which will result in uncontrolled fibrinolysis (Kukkonen et al., 2001). Since plasmin cleaves a broad range of substrates, including extracellular matrix proteins, surface-associated plasmin activity may have evolved to not only degrade fibrin clots but also facilitate bacterial spread through host tissues (Brissette et al., 2009).
Mechanisms to promote coagulation
In contrast to mechanisms described above, bacteria also induce the coagulation system to either release vasoactive peptides or promote clotting. Enhancement of coagulation can be accomplished via several different mechanisms. A well-characterized mechanism by which S. aureus promotes clotting is via the production of coagulases. Staphylococcus aureus secretes two of these coagulases, named coagulase and von Willebrand factor binding protein (vWbp), which are both able to bind and activate prothrombin (McAdow et al., 2012). The binding of coagulases induces a conformational change in prothrombin that leads to its non-proteolytic activation into thrombin. In this way, coagulases induce the conversion of fibrinogen to fibrin and the formation of a clot (Friedrich et al., 2003). It was recently described that both coagulase and vWbp are necessary for the formation of abscesses in which the bacteria can replicate and are protected from the host immune cells. Furthermore, they have been demonstrated to be important virulence factors in vivo (Cheng et al., 2010) that promote S. aureus agglutination in the bloodstream (McAdow et al., 2011). Finally, a recent study shows that vWbp is also able to bind FXIII and that the vWbp-prothrombin complex first associates with fibrinogen and then with FXIII. This leads to non-proteolytic activation of FXIII and thereby clot stabilization (Thomer et al., 2013). Clotting may be an advantage for bacteria since they can use clots to hide from phagocytes (Guggenberger et al., 2012). For S. aureus, it was suggested that the coagulase-induced fibrin clots are structurally different from conventional clots. Mouse models have shown that these clots are beneficial for escaping phagocytic clearance in the tissues and promote bacterial growth and dissemination through the blood (McAdow et al., 2012).
Next to the secreted coagulases, bacterial pathogens also express fibrinogen-binding proteins that confer bacterial clotting. The S. aureus clumping factor A (ClfA) is a surface-bound protein that binds fibrinogen and causes bacteria to clump. A different S. aureus surface protein that binds fibrinogen is the fibronectin binding protein A (FnbpA). This protein forms fibrinogen bridges between S. aureus and platelets leading to activation and aggregation of platelets, ultimately resulting in clot formation. Since FnbpA and ClfA bind similar regions of fibrinogen, it is expected that ClfA can also activate platelets in a similar way (Fitzgerald et al., 2006).
In addition to induction of clotting, bacteria can also promote activation of the contact pathway. One way to promote contact activation is to enhance the binding of HK to the bacterial surface where it is more susceptible to activation by host proteases. M1 protein of GAS binds HK via domain 5 (for binding to negatively charged surfaces) and domain 6 (for binding to PK). In addition, the fibrinogen-binding protein (FOG) and protein G of group G streptococci (GGS) are capable of binding HK, albeit with distinct functions. FOG, but not protein G, is capable of releasing BK from HK whereas protein G inhibits the specific release of the AMP NAT26 that results from cleavage of HK (Wollein Waldetoft et al., 2012). Furthermore, bacteria can activate the contact system by secreting proteases that can for instance activate FXII and PK or cleave HK to release BK (for a review see Tapper & Herwald, 2000; Nickel & Renné, 2012). Well-known for this activity are the cysteine proteinases staphopain A (ScpA) and staphopain B (SspB) from S. aureus that act directly on HK in human plasma to release BK (Mattsson et al., 2001; Imamura et al., 2005; Wollein Waldetoft et al., 2012). Staphopain A (ScpA) can thereby induce vascular leakage in vivo (Imamura et al., 2005).
Finally, bacteria also promote coagulation by inactivation of endogenous regulators. Thrombin activatable fibrinolysis inhibitor (TAFI) is a negative regulator of fibrinolysis that is activated by both plasmin and thrombin. GAS uses collagen-like surface proteins A and B (SclA and SclB) to recruit TAFI to its surface (Påhlman et al., 2007). GAS can also attract both plasmin and thrombin to its surface and thus locally activate TAFI, which results in inhibition of fibrinolysis (Bengtson et al., 2009).
Misuse of coagulation proteins for immune escape
Finally, bacteria use the coagulation system to modulate immune cells. Recent investigations by our group revealed that the Extracellular fibrinogen binding protein (Efb) of S. aureus covers bacteria with an anti-opsonic shield of fibrinogen that effectively blocks phagocytosis (Ko et al., 2013). Efb uses its C-terminal domain to bind C3b molecules on the surface of S. aureus. At the same time, it can attract fibrinogen (via its N-terminal domain) to the bacterial surface and thereby generate a layer of fibrinogen around the bacterial cell that effectively blocks phagocyte recognition of opsonic antibodies and C3b (Ko et al., 2013). Efb binding to fibrinogen also prevents aggregation of platelets and attachment of neutrophils to fibrinogen (Ko et al., 2011; Shannon & Flock, 2004). Surface-expressed M protein of GAS can also bind fibrinogen to cover the bacterial surface. The M protein-fibrinogen complexes decrease complement deposition and might also avoid antibody binding by steric hindrance (Smeesters et al., 2010). The formation of these complexes was shown to lead to neutrophil activation, which neither M protein nor fibrinogen by itself is capable of. The coiled coil structure within the M1 protein was responsible for the binding and assembly of four fibrinogen molecules, which creates a M1-fibrinogen network, distinct from a conventional fibrin clot (Macheboeuf et al., 2011). Furthermore, it was shown that the virulence of GAS depends both on the presence and functionality of M protein as well as the presence of host fibrinogen, indicating that both host and bacterial factors contribute to virulence (Uchiyama et al., 2013).
Conclusions
Our insights into the molecular interplay between complement, coagulation and bacteria have increased significantly over the past years. Not only do we know more about their modes of recognition but also the way these systems induce stress to bacterial cells has become more lucent. Still, the poorly understood basis for MAC-dependent killing of Gram-negative bacteria remains an important challenge to elucidate in the future. Although the importance of coagulation in clearance of infections is now established, a key question remains to whether coagulation is mainly activated via bystander inflammatory reactions at sites of vascular injury or whether this occurs specifically on bacteria via the contact activation pathway. The evolutionary conservation of this pathway certainly suggests an important function and evidence for its role in innate immunity is steadily accumulating. Still, the pro-coagulatory mechanisms of some bacterial pathogens suggest that coagulation is not always harmful to bacteria. Pathogens seem to induce clot formation to escape the immune system or induce contact activation to release the vasoactive component BK, which could be an advantage since increased vascular permeability promotes bacterial dissemination. Since bacterial modulators of complement and coagulation contribute to bacterial virulence, they may provide new targets for infection control. Overall, the multiple strategies evolved by pathogenic bacteria to modulate complement and coagulation are strong indicators of a constant battle between bacteria and these two protein networks during infections.
Acknowledgements
The authors were supported by grants of the Netherlands Organization for Scientific Research (NWO-Vidi, #91711379) to SHMR&ETMB) and the Dutch Heart Foundation (2010T068) to RTU. We thank Toon Verhoeven and Michiel van Gent for excellent assistance with graphical design and Reindert Nijland and Daphne Stapels for critical reading of the manuscript.
Authors' contribution
E.T.M.B. and A.K. contributed equally to this work.
References
Author notes
Equal contribution
Editor: Jos van Strijp

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}