





Abstract
Mutations of GLI-similar 3 (GLIS3) underlie a neonatal diabetes syndrome. Genome-wide association studies revealed that GLIS3 variants are associated with both common type 1 and type 2 diabetes. Global Glis3-deficient (Glis3−/−) mice die of severe diabetes shortly after birth. GLIS3 controls islet differentiation by transactivating neurogenin 3 (Ngn3). To unravel the function of Glis3 in adults, we generated inducible global Glis3-deficient mice (Glis3fl/fl/RosaCreERT2). Tamoxifen (TAM)-treated Glis3fl/fl/RosaCreERT2 mice developed severe diabetes, which was reproduced in TAM-treated β cell–specific Glis3fl/fl/Pdx1CreERT mice, but not in TAM-treated Glis3fl/fl/MipCreERT mice. Furthermore, we generated constitutive β cell– or pancreas-specific Glis3-deficient mice using either RipCre (Glis3fl/fl/RipCre) or Pdx1Cre (Glis3fl/fl/Pdx1Cre) coexpressing mice. We observed that, remarkably, neither type of β cell– or pancreas-specific Glis3-deficient mice phenocopied the lethal neonatal diabetes observed in Glis3−/− mice. All Glis3fl/fl/RipCre mice survived to adulthood with normal glucose tolerance. Thirty percent of Glis3fl/fl/Pdx1Cre mice developed severe diabetes at 3 to 4 weeks of age, whereas 55% of them developed mild diabetes with age. In contrast to the >90% reduction of Ngn3 and near-total absence of insulin (Ins) in the embryonic pancreas of Glis3−/− mice, we found only 75%–80% reduction of Ngn3 and Ins messenger RNA or protein expression in the fetal pancreas of Glis3fl/fl/Pdx1Cre mice. The expression levels of Ngn3 and Ins correlated negatively with the extent of Cre-mediated Glis3 deletion. These mouse models are powerful tools to decipher Glis3 gene dosage effects and the role of GLIS3 mutations/variants in a spectrum of β cell dysfunction in people.
To date, candidate gene and genome-wide association studies have identified >50 susceptibility genes for type 1 diabetes (T1D) and ∼100 susceptibility genes for type 2 diabetes (T2D). Remarkably, among ∼150 loci, only 2, the insulin gene (INS) and the Krüppel-like transcription factor Gli-similar 3 gene (GLIS3), were found to be strongly associated with both T1D and T2D in different populations (1–5). Moreover, a GLIS3 variant has been linked to altered fasting glucose in healthy children and adolescents (6). In this study, we will determine how subtle variations in GLIS3 genotypic expression affect phenotypic presentation by creating a multitude of genetic mouse models targeting the Glis3 gene.
GLIS3 is predominantly expressed in pancreatic β cell, kidney, and thyroid (7–9). Mutations of GLIS3 underlie a rare clinical syndrome, characterized by neonatal diabetes and hypothyroidism (NDH) (7). To gain insight into the function of GLIS3 in neonatal diabetes, 3 groups have independently generated Glis3 global knockout (Glis3−/−) mice using different strategies (10–12). Recapitulating the clinical presentation of NDH patients with GLIS3 mutations, Glis3−/− mice develop neonatal diabetes, hypothyroidism, and polycystic kidney and die within the first few days of life. Mechanistically, GLIS3, synergistically with hepatocyte nuclear factor 6 and forkhead box A2, controls islet differentiation by direct transactivation of neurogenin 3 (Neurog3, also called Ngn3) (11), the pancreatic endocrine lineage-defining transcription factor (13).
Consistent with an autosomal recessive inheritance in humans (7, 14), heterozygous Glis3-mutant mice remained euglycemic while on regular chow diet. However, they developed diabetes in response to high-fat diet feeding because of impaired compensatory β cell proliferation and mass expansion (15). We previously showed that GLIS3 controls obesity-induced β cell proliferation partly by direct regulation of cyclin D2 expression (15). Furthermore, we found that GLIS3 is a potent Ins transactivator in vitro (8), which was reinforced by our in vivo finding that GLIS3 is required for the maintenance of normal insulin expression in adult β cells in mice (15). Moreover, the binding of GLIS3 to the Ins promoter is required for the binding of other Ins gene regulators, such as pancreatic and duodenal homeobox 1, V-maf avian musculoaponeurotic fibrosarcoma oncogene homolog A, and neurogenic differentiation 1, to the Ins promoter (16). In addition, genetic variation of GLIS3 may also affect β cell viability and/or susceptibility to diabetes-associated stress (17, 18).
Rat Ins2 promoter-driven Cre (RipCre) and Pdx1Cre transgenic mice have been widely used to achieve β cell– or pancreas-specific gene targeting, although both promoters were shown to also drive transgene expression in the brain (19). A novel β cell–specific Cre mouse line, mouse Ins1 gene promoter (8.5 Kb)-driven CreERT (MipCreERT), was shown to exhibit Cre activity exclusively in islet β cells, and not in the brain (20). However, the absence of Mip-directed brain expression was recently challenged by an observation that the Mip activity was identified in multiple hypothalamic nutrient-sensing neurons in the same Ins1 gene promoter-driven green fluorescent protein transgenic mice (21).
To gain insight into the role of GLIS3 in pancreatic β cells, we have generated 3 tamoxifen (TAM)-inducible Glis3-deficient mouse lines, including Glis3fl/fl/RosaCreERT2, Glis3fl/fl/Pdx1CreERT, and Glis3fl/fl/MipCreERT mice, as well as 2 constitutive β cell– or pancreas-specific Glis3 knockout mice using RipCre (Glis3fl/fl/RipCre) and Pdx1Cre (Glis3fl/fl/Pdx1Cre) mice. After evaluation of the phenotypes, Cre recombination activity, and potential underlying mechanisms, we found that subtle differences in Cre recombination activity determined the gene dosage effects of Glis3 in the pancreas or β cells, which were directly correlated with the expression levels of its 2 key direct downstream targets such as Ngn3 and Ins in the fetal and newborn pancreata. These results document a key role for GLIS3 as the predominant transactivator of Ngn3 (11) and Ins (8, 15, 16), and minor differences in gene dosage of Glis3 (e.g., 10% versus <1%) produce substantive changes in the expression level of Ngn3 and Ins, culminating in significant differences in phenotype among the multiple Glis3 knockout mouse lines.
Materials and Methods
Generation of global and pancreas- or β cell–specific Glis3-targeted mice
We generated Glis3fl/fl and Glis3−/− mice, as described previously (11). RosaCreERT2 (22), MipCreERT (20), RipCre (23), and Pdx1Cre (24) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). We crossed Glis3fl/fl mice with these Cre transgenic mice to obtain individual Glis3-deficient mice. To activate CreERT nuclear localization and Glis3 deletion globally or selectively in pancreatic progenitors or β cells, we administrated TAM (Sigma-Aldrich, St. Louis, MO; dissolved in peanut oil with 10% ethanol) to 8-week-old male mice intraperitoneally at a dose of 3 mg/mouse/d for 7 consecutive days (15). Animals were considered to have mild diabetes (250 to 350 mg/dL) or severe diabetes (>350 mg/dL) based on random morning blood glucose measurements. For studies in embryos and newborn pups, we collected tissues from both genders. We performed studies on the role of thyroid hormone during pregnancy in female mice and all other studies in male mice. All animal studies were performed using protocols approved by the Institutional Animal Care and Use Committees at Baylor College of Medicine (Houston, TX) and Case Western Reserve University (Cleveland, OH).
Gavage glucose tolerance test and insulin measurement
We fasted mice for 6 hours, delivered d-glucose (1.5 g/kg body weight) into the stomach by a gavage needle, and measured blood glucose levels at 0, 15, 30, 60, and 120 minutes postgavage using Embrace Glucometer (Omnis Health, Nashville, TN), and plasma insulin and pancreatic insulin content (1/3 adult pancreas), as described previously (15).
Immunofluorescence staining and positive cell quantification
We performed immunofluorescence staining on the paraffin-embedded, 5-µm sections of dissected pancreas and fixed them in freshly made 4% paraformaldehyde in phosphate-buffered saline, as described previously (11). Primary antibodies were guinea pig polyclonal insulin antibody (ab7842; Abcam, Cambridge, MA) and glucagon (Gcg) antibody (FL-180) (sc-13091; Santa Cruz, Dallas, TX) (Table 1). The primary antibodies were detected with secondary antibodies conjugated to fluorescein isothiocyanate or rhodamine (Invitrogen, Carlsbad, CA). We used an Axiovert (Zeiss, Thornwood, NY) microscope with Axiovision imaging software to capture fluorescent images and used Adobe Photoshop to process the postacquisition images. We estimated Ins- or Gcg-positive cell area, using ImageJ 1.4 (NIH), on 4 fluorescent sections (approximately every 10th section) processed from 4 independent pancreata.
List of Primary Antibodies Used in This Study
| Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Insulin (human) | Guinea pig polyclonal insulin antibody | ab7842 (Abcam, Cambridge, MA) | Guinea pig polyclonal | 1:200 | |
| Full-length glucagon of human origin | Glucagon antibody | FL-180 (sc-13091; Santa Cruz, Dallas, TX) | Rabbit | 1:200 |
| Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Insulin (human) | Guinea pig polyclonal insulin antibody | ab7842 (Abcam, Cambridge, MA) | Guinea pig polyclonal | 1:200 | |
| Full-length glucagon of human origin | Glucagon antibody | FL-180 (sc-13091; Santa Cruz, Dallas, TX) | Rabbit | 1:200 |
List of Primary Antibodies Used in This Study
| Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Insulin (human) | Guinea pig polyclonal insulin antibody | ab7842 (Abcam, Cambridge, MA) | Guinea pig polyclonal | 1:200 | |
| Full-length glucagon of human origin | Glucagon antibody | FL-180 (sc-13091; Santa Cruz, Dallas, TX) | Rabbit | 1:200 |
| Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| Insulin (human) | Guinea pig polyclonal insulin antibody | ab7842 (Abcam, Cambridge, MA) | Guinea pig polyclonal | 1:200 | |
| Full-length glucagon of human origin | Glucagon antibody | FL-180 (sc-13091; Santa Cruz, Dallas, TX) | Rabbit | 1:200 |
RNA isolation and quantitative polymerase chain reaction
We isolated islets from mouse pancreata using collagenase P (Roche, Branchburg, NJ) digestion. Briefly, we prepared the work solution of collagenase P at 1 mg/mL in Hank's balanced salt solution buffer (Invitrogen) and injected 3 mL collagenase P solution into common bile duct under the microscope with a 30 G 1/2” needle. We then removed the entire pancreas in a 15-mL tube and incubated the organ in 3 mL collagenase P solution at 37°C for 6–7 min. We terminated the digestion by putting the tube on ice and adding 10 mL 1× phosphate-buffered saline with 5% fetal bovine serum, spun down the islets by centrifugation, and picked up individual islets manually. To obtain samples from mouse embryos, we microdissected pancreata from embryonic day (E)15.5 embryos under a microscope. We used TRIzol (Invitrogen) or Mini RNA Isolation I Kit (Zymo Research, Irvine, CA) to isolate total RNA, which was treated with amplification grade DNase I (Invitrogen) to remove genomic DNA contamination. We performed reverse transcription using random primers (New England Biolabs, Ipswich, MA) and quantitative polymerase chain reaction in mouse tissues, as previously described (11), with the following primers: Glis3-F, 5′-ACGTGCTTCTGGACTGGCTG-3′; Glis3-R, 5′-GGATGCTGGCACAAGTATGGC-3′; Ngn3-F, 5′-GTCGGGAGAACTAGGATGGC-3′; Ngn3-R, 5′-GGAGCAGTCCCTAGGTATG-3′; Ins1-F, 5′-CCAGCTATAATCAGAGACCATCA-3′; Ins1-R, 5′-GTTTGACAAAAGCCTGGGTG-3′; Ins2-F, 5′-TCTTCCTCTGGGAGTCCCAC-3′; Ins2-R, 5′-GTCTGAAGGTCACCTGCTCC-3′.
Thyroxine enzyme-linked immunosorbent assay and replacement therapy
We measured serum thyroxine (T4) level in female mice at day 14 of gestation using the T4 enzyme-linked immunosorbent assay kit following the manufacturer’s instructions (Calbiotech, Spring Valley, CA). In l-thyroxine (L-T4) replacement experiment, we dissolved L-T4 (Sigma-Aldrich) in 1 M NaOH to make the stock solution at 100 mg/mL. Neonatal pups received daily subcutaneous injections of L-T4 (0.4 µg/g body weight) (25) or vehicle for up to 10 days.
Statistical analysis
The standard Student’s 2-tailed t test was used for comparisons. Results are presented as the mean ± standard error.
Results
RosaCreERT2-mediated Glis3 inactivation in adult mice produces severe diabetes
The neonatal lethality of Glis3−/− mice (10–12) precludes the use of this model to study the role of GLIS3 in the adult pancreas. To circumvent this limitation, we generated TAM-regulatable Glis3fl/fl/RosaCreERT2 mice by intercrossing Glis3fl/fl mice with RosaCreERT2 mice. These animals reached adulthood without any sign of compromised health. We administered TAM to 8-week-old male Glis3fl/fl/RosaCreERT2 mice to induce ubiquitous deletion of the Glis3 gene. Two weeks after TAM injection, Glis3fl/fl/RosaCreERT2 mice developed diabetes, whereas vehicle-treated Glis3fl/fl/RosaCreERT2 mice remained euglycemic [Fig. 1(A)]. The physical condition of TAM-treated Glis3fl/fl/RosaCreERT2 mice deteriorated with time. Four weeks after TAM, they exhibited significantly lower body weight [Fig. 1(B)] and severe hyperglycemia (>800 mg/dL) [Fig. 1(C)], whereas their plasma insulin [Fig. 1(D)] and pancreatic insulin content [Fig. 1(E)] were almost undetectable. To assess the degree of Cre activation, we isolated islets from Glis3fl/fl/RosaCreERT2 mice 2 weeks after TAM or vehicle treatment. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) showed that the level of Glis3 messenger RNA (mRNA) in the islets of TAM-treated mice was only ∼7% of that in vehicle-treated mice [Fig. 1(F)], indicating >90% RosaCre recombination induced by TAM administration in these mice. Furthermore, immunofluorescent staining revealed relatively intact islet structure but marked reduction [Fig. 2(A) and 2(B)] or near total absence of Ins expression [Fig. 2(C)] at 4 weeks after TAM treatment. In contrast, the expression of Gcg was unchanged between vehicle- and TAM-treated mice (Fig. 2), which is in agreement with a recent report that GLIS3 is not expressed in α cells (26). The physical condition of TAM-treated mice continued to deteriorate rapidly after 4 weeks, and we had to euthanize them.
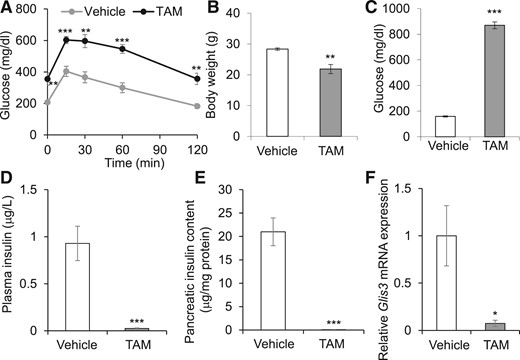
Glis3 ubiquitous inactivation in adult mice leads to fulminant diabetes. (A) Eight-week-old Glis3fl/fl/RosaCreERT2 male mice were treated with TAM or vehicle. Two weeks after treatment, gavage glucose tolerance test was performed after 6-hours fast. Plasma glucose was measured at time 0, 15, 30, 60, and 120 minutes after glucose injection (1.5 g/kg body weight) (N = 6). (B–F) Glis3fl/fl/RosaCreERT2 mice were treated with TAM or vehicle, and 4 weeks after treatment, their body weights (B), blood glucose (C), plasma insulin (D), pancreatic insulin content (E), and Glis3 gene expression in the isolated islets were presented (N = 6). *P < 0.05; **P < 0.01; ***P < 0.001, versus vehicle-treated mice.
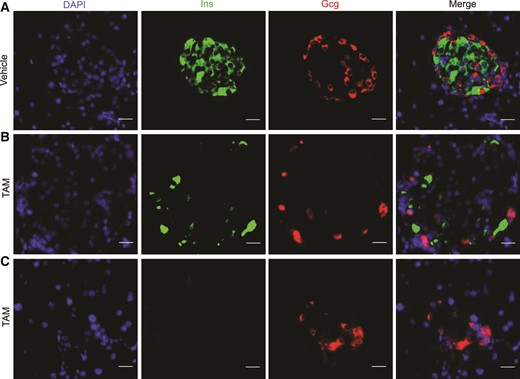
Glis3 inactivation in adult mice drastically reduces insulin expression in the islets. Immunostaining of Ins and Gcg in the pancreas of Glis3fl/fl/RosaCreERT2 mice at 4 weeks after vehicle (A) or TAM (B, C) treatment. Representative images of 4 fluorescent sections (approximately every 10th section) processed from 4 independent pancreata for each genotype. Scale bar, 20 μm.
The phenotypes in Glis3fl/fl/RosaCreERT2 mice were intriguing; however, we could not rule out the possible contribution of Glis3 deletion in non-β cells or other organs given that RosaCreERT2 mediated global Glis3 deletion in these mice. To address this concern, we generated, in parallel, 2 TAM-inducible β cell–specific Glis3 knockout mouse lines using Pdx1CreERT and MipCreERT transgenic mice. We previously observed that TAM-treated Glis3fl/fl/Pdx1CreERT mice phenocopied the Glis3fl/fl/RosaCreERT2 with the same treatment (15). Unexpectedly, however, TAM-treated Glis3fl/fl/MipCreERT mice failed to develop diabetes [Fig. 3(A)]. The mRNA expression of Ins1 and Ins2 in the isolated islets of TAM-treated Glis3fl/fl/MipCreERT mice was ∼50% of that in vehicle-treated mice [Fig. 3(B)]. The recombination efficiency of Pdx1CreERT and MipCreERT in response to TAM administration in vivo was ∼90% and 66%, respectively, as reflected by the Glis3 mRNA expression as determined by qRT-PCR in islets isolated from the treated animals [Fig. 3(C)].
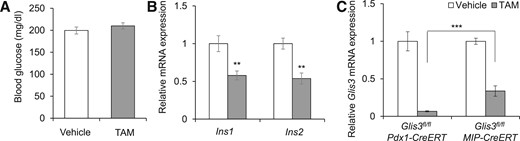
TAM-treated Glis3fl/fl/MipCreERT mice fail to develop diabetes. (A) The morning random blood glucose in Glis3fl/fl/MipCreERT mice at 8 weeks after vehicle or TAM treatment (N = 6). (B) The mRNA expression of Ins1 and Ins2 of islets isolated from Glis3fl/fl/MipCreERT mice at 2 weeks after vehicle or TAM treatment (N = 6 to 8). (C) The mRNA expression of Glis3 in the isolated islets of Glis3fl/fl/Pdx1CreERT at 10 days and of Glis3fl/fl/MipCreERT mice at 2 weeks after vehicle or TAM treatment (N = 6 to 8). **P < 0.01; ***P < 0.001.
Constitutive RipCre-mediated Glis3 deletion does not cause diabetes in mice
Several potential factors, including the tissue specificity of Cre activity, dosage, and action duration of TAM, may affect the CreERT recombination efficiency, causing divergent phenotypes (19, 27). To circumvent the need for TAM injection, we generated constitutive β cell–specific Glis3 knockout mice using RipCre and compared the phenotype of these animals with that in global Glis3−/− mice. As previously reported, Glis3−/− mice die within a few days of life due to severe neonatal diabetes (10–12). In contrast, Glis3fl/fl/RipCre mice displayed normal glucose tolerance test during adulthood [Fig. 4(A)]. qRT-PCR showed that the mRNA level of Glis3, Ins1, and Ins2 in the islets of Glis3fl/fl/RipCre was 47% to 59% of that in Glis3fl/fl mice [Fig. 4(B)], indicating a moderate β cell Cre efficiency (∼50%) in this line.
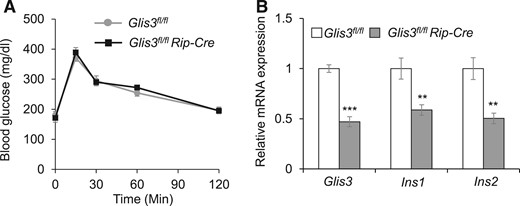
Glis3fl/fl/RipCre mice do not develop diabetes. (A) Gavage glucose tolerance test (GTT) was performed after 6-hours fast in Glis3fl/fl/RipCre and Glis3fl/fl control mice. (B) The mRNA expression of Glis3, Ins1, and Ins2 in the isolated islets from Glis3fl/fl and Glis3fl/fl/RipCre mice. These mice were 10–12 weeks of age (N = 6). **P < 0.01; ***P < 0.001, versusGlis3fl/fl control mice.
Constitutive Pdx1Cre-mediated Glis3 deletion causes variable diabetes in mice
One possible reason for the different phenotypes between Glis3fl/fl/RipCre and Glis3−/− mice is the timing of Cre activity during embryonic development. We used protamine-Cre (28) that deleted Glis3 gene in spermatogenesis to produce Glis3−/− mice. In contrast, RipCre activity peaks at secondary transition, which starts from E13.5 in β cell development (29). Moreover, we previously reported that GLIS3 is required for proper islet differentiation via direct transactivation of Ngn3 (11). RipCre might miss the critical time window for Glis3 deletion prior to islet morphogenesis in the pancreas. To address this concern, we generated a pancreas-specific Glis3 knockout line using constitutive Pdx1Cre transgenic mice to produce Glis3fl/fl/Pdx1Cre mice. Interestingly, Glis3fl/fl/Pdx1Cre mice failed to reproduce the neonatal lethality phenotype observed in Glis3−/− mice (11). In contrast, we found that only 30% of Glis3fl/fl/Pdx1Cre mice developed severe diabetes at 3 to 4 weeks of age, and another 55% of them developed relatively mild diabetes with age; the remaining ∼15% did not develop diabetes up to 25 weeks of age [Fig. 5(A)]. To determine whether the floxed Glis3 per se affects glucose homeostasis, we performed glucose tolerance test and found that they displayed similar degrees of glucose tolerance [Fig. 5(B)]. In contrast to RipCre, which is associated with impaired β cell function (30), multiple lines of evidence suggest that the Pdx1Cre line that we used in the current study (24) does not change the pancreas development and glucose homeostasis (31, 32). At 3 weeks of age (right after weaning), Glis3fl/fl/Pdx1Cre mice displayed a tendency to be glucose intolerant after a standard glucose challenge [Fig. 5(C)]. At 10 weeks old, the subgroup (with random morning blood glucose ≤350 mg/dL) of Glis3fl/fl/Pdx1Cre mice exhibited marked glucose intolerance [Fig. 5(D)].
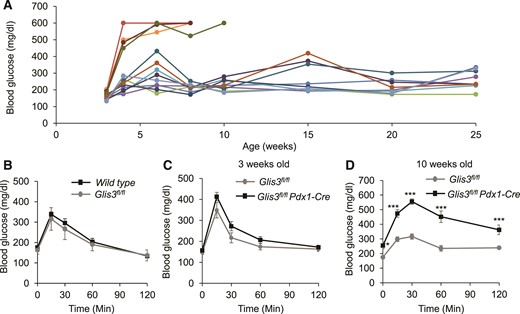
Glis3fl/fl/Pdx1Cre mice develop variable diabetes phenotypes. (A) The morning random blood glucose was monitored in Glis3fl/fl/Pdx1Cre mice at different ages, as indicated. Each line indicates an individual mouse. (B) Gavage glucose tolerance test was performed after 6-hours fast in male Glis3fl/fl and wild-type mice at 12 weeks old (N = 6). (C, D) Gavage GTT was performed after 6-hours fast in male Glis3fl/fl/Pdx1Cre mice at 3 weeks old (C) and 10 weeks old (D) (n = 6). **P < 0.01; ***P < 0.001, versusGlis3fl/fl control mice.
To gain insight into the mechanisms that may underlie the difference between Glis3fl/fl/Pdx1Cre and Glis3−/− mice, we performed histological hematoxylin and eosin staining as well as immunostaining of Ins and Gcg on the newborn pancreas sections. We quantified islet area and hormone-positive cell area normalized to total pancreas area. As shown in Fig. 6, islet area was markedly reduced in both Glis3fl/fl/Pdx1Cre and Glis3−/− mice, compared with that in their control mice. However, the remaining islet area in Glis3−/− mice was significantly lower than that in Glis3fl/fl/Pdx1Cre mice by ∼37% [Fig. 6(A) and 6(B)]. Moreover, small islet remnants were visible in the pancreas of Glis3fl/fl/Pdx1Cre mice [Fig. 6(A)], but essentially nonexistent in Glis3−/− mice [Fig. 6(C)]. Similarly, the numbers of insulin-positive cells and Gcg-positive cells were markedly reduced in both genotypes, but much more so in the Glis3−/− than in the Glis3fl/fl/Pdx1Cre mice [Fig. 6(C–E)]. Notably, the insulin-positive cell area was more substantially reduced than the islet area in Glis3−/− mice (∼75%), compared with that in Glis3fl/fl/Pdx1Cre mice (∼37%) [Fig. 6(C) and 6(D)], which is consistent with the fact that GLIS3 has dual roles in regulating islet differentiation (11, 12) and insulin expression (8, 15, 16). These results indicated substantially more severe defects of islet differentiation and insulin expression in Glis3−/− mice than in Glis3fl/fl/Pdx1Cre mice.
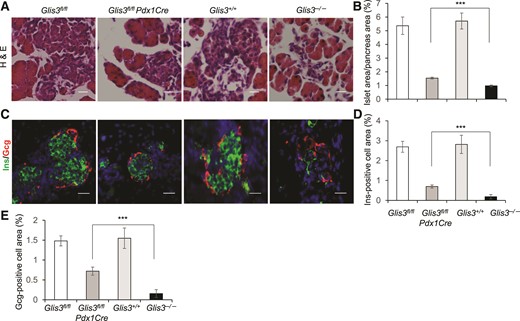
More severe insulin deficiency in the pancreas of Glis3−/− mice than that in Glis3fl/fl/Pdx1Cre mice at birth. (A, B) Representative images and quantification of islet area for each genotype, as indicated. (C–E) Representative images and quantification of Ins and Gcg staining for each group of mice, as indicated. We performed these studies on 4 hematoxylin and eosin (H&E) or fluorescent sections (approximately every 10th section) processed from 4 independent pancreata for each genotype. Scale bar, 40 μm. ***P < 0.001.
Congenital hypothyroidism is not a major determinant of defective islet differentiation and lethality in Glis3−/− mice
One other factor that may underlie the differences in phenotype is that deletion of Glis3 in other organs may contribute to the neonatal lethality in Glis3−/− mice. In addition to diabetes, GLIS3 mutations also cause congenital hypothyroidism (7, 14). There is a potential link between the thyroid and the endocrine pancreas in that thyroid hormone was suggested to play a role in islet development during embryogenesis (33–35) as well as postnatal β cell maturation (36, 37). It is important to note that, to obtain Glis3−/− mice, we had to intercross Glis3+/− mice. Given the fact that maternal thyroid hormones are transported across the placenta during gestation and are essential for normal fetus development (38, 39), we measured the serum level of T4 in the Glis3+/+ and Glis3+/− mothers at day 14 of pregnancy. Consistent with an autosomal recessive inheritance in humans (7, 14), the level of serum T4 in the pregnant Glis3+/− mothers was comparable to that in pregnant Glis3+/+ control mice [Fig. 7(A)]. It is thus not reasonable to attribute the defect of islet development in Glis3−/− mice to fetus thyroid hormone deficiency. Instead, the reduced expression of Ngn3 associated with Glis3 deficiency is the culprit (11). This notion was supported by the observation that Glis3−/− mice displayed no intrauterine growth retardation; they fed normally and were indistinguishable from Glis3+/+ mice at birth (11). Moreover, studies on thyroid transcription factor paired box gene 8 (Pax8) knockout mice also revealed that fetus thyroid hormone deficiency does not affect its own development in utero. Pax8-deficient mice exhibit relatively normal development till term, but display apparent growth retardation 1 week after birth, and die shortly after weaning because of the absence of thyroid follicles (40).
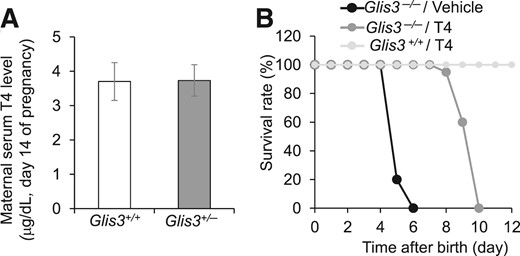
Hypothyroidism is not a major determinant of the severe neonatal diabetes and lethality in Glis3−/− mice. (A) Serum total T4 levels of maternal Glis3+/+ and Glis3+/− mice at midgestation (N = 5 to 8). (B) Survival rate of Glis3+/+ and Glis3−/− mice treated daily with L-T4 (0.4 µg/g body weight) or vehicle. N = 10 to 12 mice for each group.
To examine the contribution of neonatal hypothyroidism to the postnatal lethality, we injected L-T4 at 0.4 μg/g body weight/day subcutaneously to Glis3−/− and Glis3+/+ mice from birth and observed that all vehicle-treated Glis3−/− mice died at postnatal day 4 to 6, as we previously reported (11), whereas L-T4-treated Glis3−/− mice survived up to postnatal day 10 [Fig. 7(B)]. These results suggested that congenital hypothyroidism may have contributed to the early neonatal lethality, but its correction does not prevent early postnatal lethality in Glis3−/− mice.
Differential gene dosage of Glis3 is the main determinant of the phenotype
To determine whether the levels of residual Glis3 mRNA expression underlie the different phenotypes in Glis3−/− and Glis3fl/fl/Pdx1Cre mice, we isolated pancreata from these mice and their corresponding control mice at E15.5 and at birth and examined the expression of Glis3 as well as its 2 key direct downstream targets Ins and Ngn3 by qRT-PCR. We found that the mRNA level of Glis3 and Ins2 was essentially undetectable, whereas the mRNA level of Ngn3 and Ins1 was <10% of their controls in the pancreata of Glis3−/− mice (Fig. 8). In contrast, the Glis3 mRNA in the pancreata of Glis3fl/fl/Pdx1Cre mice was ∼10% of their control Glis3fl/fl mice. The mRNA level of Ngn3, Ins1, and Ins2 in the pancreata of Glis3fl/fl/Pdx1Cre mice was ∼20%–30% of their control Glis3fl/fl mice. It is interesting that, compared with Glis3fl/fl/Pdx1Cre mice, Ngn3 mRNA level was reduced in the islets of Glis3−/− mice at E15.5 [Fig. 8(A)], but not at birth [Fig. 8(B)]. This may be associated with the fact that NGN3 normally peaks at E15.5 during the major wave of endocrine cell genesis and is greatly diminished at birth in mice (41, 42). These data suggest that 10% of normal Glis3 gene dosage is sufficient to produce ∼20% of normal levels of Ngn3 and Ins in Glis3fl/fl/Pdx1Cre mice. Differential dosages of Glis3 related to Cre activity cause significant difference of the expression of Ngn3 and Ins, which may underlie the difference in the diabetes phenotypes exhibited by Glis3fl/fl/Pdx1Cre and Glis3−/− mice.
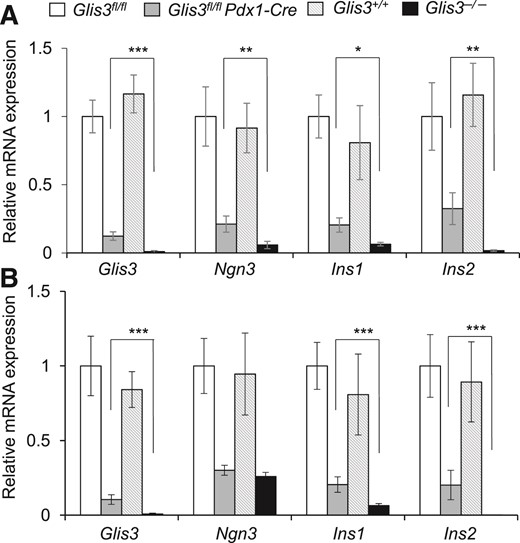
The residual dosage of Glis3 in the knockout mice determines the phenotype. The mRNA expression of Glis3, Ngn3, Ins1, and Ins2 in the E15.5 (A) and newborn (B) pancreata (N = 6), *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Mutations or variants of GLIS3 have been associated with neonatal diabetes (7, 14) as well as both T1D and T2D (1–5). Glis3 global knockout mice die of severe neonatal diabetes within a few days of birth (10–12). In the current study, we found that TAM-inducible global Glis3 deletion in adult Glis3fl/fl/RosaCreERT2 mice led to fulminant diabetes due to drastically reduced insulin expression in the pancreas. This indicates that GLIS3 is required for maintaining insulin expression in adult β cells. Interestingly, this phenotype was reproduced in TAM-treated Pdx1CreERT (15), but not in MipCreERT-mediated Glis3 deletion in adult mice. In contrast to the most widely used Ins2Cre lines (RipCre) (19), MipCreERT is driven by mouse Ins1 promoter and is believed to be a better β cell–specific Cre line due to the absence of leaky Cre recombination activity in the brain in the early study (20); we note, however, that a recent report suggested that Mip activity was detectable in hypothalamic nutrient-sensing neurons (21). Although a number of variables including β cell heterogeneity and nonoverlap of targeted cell populations may affect the consistency of the phenotypes exhibited by these genotypes, we did observe clear differences of Cre efficiency between Pdx1CreERT and MipCreERT lines. The efficiency of Pdx1CreERT line (90%), very similar to that in RosaCreERT2 line (93%), was significantly higher than that in MipCreERT mice (66%). The TAM-induced recombination activity of MipCreERT has been observed in 78%–92% insulin-expressing cells by β-galactosidase staining (20). Notably, the recombination efficiency of MipCreERT in Glis3fl/fl/MipCreERT mice may be underestimated by qRT-PCR because Glis3 is also expressed in non-β islet cells and possibly contaminating ductal cells (7, 26) in the handpicked islets. These results suggest that Cre expression efficiency appears to be the best determinant of the Glis3 gene dosage effects in these bigenic mice.
In these inducible knockout models, another variable in transgene induction and Cre activation is the need for TAM administration to activate CreERT nuclear localization and Glis3 deletion. The dosage and injection timeline of TAM may affect TAM-induced recombination and phenotypic expression (27). To circumvent this variable, we generated 2 constitutive β cell– or pancreas-specific Glis3 knockout mouse lines using Glis3fl/fl crossed with RipCre and Pdx1Cre transgenic mice, respectively. Neither of these bigenic mouse lines phenocopied the Glis3−/− mice. Glis3fl/fl/RipCre mice remained euglycemic from birth to adulthood, whereas Glis3fl/fl/Pdx1Cre mice developed diabetes at different times after the neonatal period. Pdx1-mediated activation of Cre expression and deletion of Glis3 expression are expected to occur in the developing pancreas of Glis3fl/fl/Pdx1Cre mice from embryonic stage E8.5, prior to islet formation (19); and the fact that Glis3fl/fl/Pdx1Cre mice did not display the fulminant neonatal diabetes and lethality observed in Glis3−/− mice was unexpected. We subsequently examined the expression level of residual Glis3 mRNA in the newborn pancreata that reflected the Cre efficiency and found that Pdx1Cre (>90%) displayed much higher efficiency than RipCre (∼50%), although it did not reach that in protamine-Cre in Glis3−/− mice (∼100%). In Glis3fl/fl/Pdx1Cre mice, the presence of only ∼10% of residual Glis3 was sufficient to maintain ∼20% of Ngn3 and Ins in the pancreas, which underlies the differences in phenotype between Glis3fl/fl/Pdx1Cre and Glis3−/− mice.
Using Glis3 inactivation as a tool to study islet developmental biology and diabetes, we must be cognizant of the fact that Glis3 deficiency also leads to hypothyroidism and polycystic kidney (10–12). Mutations in GLIS3 were shown to underlie a rare syndrome of neonatal diabetes and polycystic kidney in clinical syndromes that include NDH (7). Autosomal-dominant polycystic kidney disease is thought to be a risk factor for diabetes following renal transplantation (43). However, there is no direct evidence that polycystic kidney itself impairs islet development or β cell function. In contrast, there is emerging evidence that thyroid hormone may play a role in pancreatic cell fate decisions (33–35) as well as postnatal β cell growth and maturation (36, 37). Our L-T4 replacement experiment indicated that concurrent congenital hypothyroidism is at most a minor accomplice, but not a culprit, in the early neonatal death of Glis3−/− pups. The defect of islet differentiation in the pancreas of Glis3−/− mice was not associated with congenital hypothyroidism because the serum thyroid hormone level in the Glis3+/− mothers during pregnancy was comparable to that of the wild-type controls. These data suggest that loss of Glis3 expression in extrapancreatic tissues plays little role in the fulminant diabetes and early postnatal death in Glis3−/− mice.
We note that there are limitations to the conclusions that can be drawn from our study. One caveat is that we used multiple genetic mouse models to examine the gene dosage effect of Glis3 expression. Theoretically, one potential strategy that circumvents this approach is the use of multiple different doses of TAM to achieve varying levels of Cre recombination and duration (27), and thus the corresponding targeted levels of Glis3 expression. However, it is a major challenge to attain minor differences in Cre activity using graded doses of TAM to the degree of precision, for example, 90% versus 100%, that we have achieved in Glis3fl/fl/Pdx1Cre and Glis3−/− mice, respectively. We further note the lack of a Pdx1Cre-only mouse line as an additional control, although this should not be of major concern because the Pdx1Cre line used in our study (24) had been documented by multiple groups to have no defect in pancreas development and glucose homeostasis (31, 32).
In summary, we have analyzed the Cre efficiency, Glis3 gene dosages, and phenotypes in 2 Glis3 global knockout mice, such as Glis3−/− and TAM-treated Glis3fl/fl/RosaCreERT2 mice, as well as in 4 pancreas- or β cell–specific Glis3 knockout mice generated by intercrossing Glis3fl/fl with MipCreERT, RipCre, Pdx1CreERT2, and Pdx1Cre transgenic mice, respectively. We conclude that the Cre efficiency determines the residual Glis3 gene dosages that cause different phenotypes mainly through the direct transactivation of Ngn3 and Ins (Fig. 9). These mouse models are powerful tools to study Glis3 gene dosage effects in islet differentiation and mature β cells. They provide a unique opportunity to decipher the role of GLIS3 mutations/variants in a spectrum of β cell dysfunction seen in people.
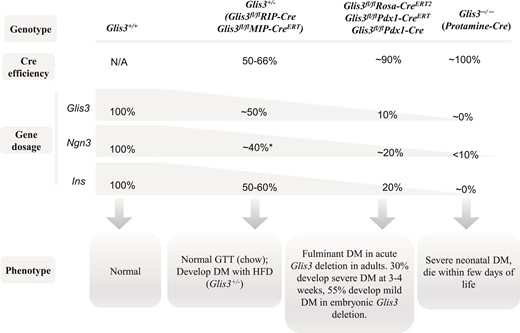
Schematic diagram of Cre efficiency, Glis3 dosage, and phenotype. The recombination efficiency of RipCre and MipCreERT was ∼50% to 66% in islets, causing ∼50% reduction of Glis3 and Ins expression, which is similar to the level in Glis3+/− mice. These mice exhibit normal glucose homeostasis under regular chow condition, and may develop diabetes after high-fat diet feeding (Glis3+/− mice). *Note: The expression of Ngn3 was reduced by ∼40% in fetal Glis3+/− mice, but not changed in Glis3fl/fl/RipCre, and TAM-treated Glis3fl/fl/MipCreERT mice. RosaCreERT2, Pdx1CreERT, and Pdx1Cre display more robust recombination efficiency (∼90%), leading to ∼90% reduction of Glis3. The residual ∼10% of Glis3 is sufficient to transactivate ∼20% of Ngn3 and Ins in the pancreas. These mice develop fulminant diabetes in acute Glis3 deletion in adults (RosaCreERT2, Pdx1CreERT), and they develop variable degrees of diabetes in embryonic Glis3 deletion (Pdx1Cre). Protamine-Cre–mediated Glis3 deletion (Glis3−/−) shows essentially 100% recombination efficiency, resulting in near total absence of Glis3 and Ins, as well as <10% of Ngn3 in the pancreas, culminating in fulminant neonatal diabetes and death.
Abbreviations:
- E
embryonic day
- Gcg
glucagon
- L-T4
l-thyroxine
- mRNA
messenger RNA
- NDH
neonatal diabetes and hypothyroidism
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- T1D
type 1 diabetes
- T2D
type 2 diabetes
- TAM
tamoxifen
Acknowledgments
This work was supported by National Institutes of Health (P30-DK079638) for a Diabetes Endocrinology Research Center at Baylor College of Medicine (T32-HL066991); Juvenile Diabetes Foundation (Award 46-2010-752); American Diabetes Association (Grant 1-14-MN-01); T.T. and W.F. Chao Global Foundation; Cindy and Frank Liu Family Foundation; Cunningham Family Foundation; Betty Rutherford Chair in Diabetes Research at Baylor St. Luke’s Medical Center (to L.C.); American Heart Association Grant 13SDG17090096 (to Y.Y.); and MetroHealth Medical Center, Case Western Reserve University start-up funds (to Y.Y.).
Disclosure Summary: The authors have nothing to disclose.
References
Author notes
Address all correspondence and requests for reprints to: Yisheng Yang, PhD, MD, Division of Endocrinology, Department of Medicine, MetroHealth Medical Center, Case Western Reserve University, 2500 Metrohealth Drive, Cleveland, Ohio 44109. E-mail: [email protected] or Lawrence Chan, Division of Diabetes, Endocrinology and Metabolism, Departments of Medicine and Molecular and Cellular Biology, Baylor College of Medicine, One Baylor Plaza, Houston, Texas 77030. E-mail: [email protected].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}