





Estrogens have the potential to afford atheroprotection, to prevent excess adiposity and its metabolic complications including insulin resistance, and to lessen hepatic steatosis. Cellular responses to estrogens occur through gene regulation by nuclear estrogen receptors (ERs), and through signal initiation by plasma membrane-associated ER. Leveraging the potentially favorable cardiometabolic actions of estrogens has been challenging, because their reproductive tract and cancer-promoting effects adversely impact the risk to benefit ratio of the therapy. In previous works, we discovered that an estrogen dendrimer conjugate (EDC) comprised of ethinyl-estradiol (E2) molecules linked to a poly(amido)amine dendrimer selectively activates nonnuclear ER, and in mice, EDC does not invoke a uterotrophic response or support ER-positive breast cancer growth. In the present investigation, we employed EDC to determine how selective nonnuclear ER activation impacts atherosclerosis, adiposity, glucose homeostasis, and hepatic steatosis in female mice. In contrast to E2, EDC did not blunt atherosclerosis in hypercholesterolemic apoE−/− mice. Also in contrast to E2, EDC did not prevent the increase in adiposity caused by Western diet feeding in wild-type mice, and it did not affect Western diet-induced glucose intolerance. However, E2 and EDC had comparable favorable effect on diet-induced hepatic steatosis, and this was related to down-regulation of fatty acid and triglyceride synthesis genes in the liver. Predictably, only E2 caused a uterotrophic response. Thus, although nonnuclear ER activation does not prevent atherosclerosis or diet-induced obesity or glucose intolerance, it may provide a potential new strategy to combat hepatic steatosis without impacting the female reproductive tract or increasing cancer risk.
Estrogens have the potential to provide atheroprotection via the activation of estrogen receptors (ERs) in vascular cell types including endothelium, vascular smooth muscle (VSM), and monocytes/macrophages (1–3). The Women’s Health Initiative and other studies provided support of this concept if hormone replacement therapy with estrogens is initiated before the development of advanced atherosclerosis (4, 5). Estrogens also have multiple metabolic actions that occur via ER in numerous target tissues. In pancreatic β-cells, estrogens and their receptors influence insulin secretion and cell survival, and in the hypothalamus, estrogens and ER modulate food intake, energy expenditure and white adipose tissue (WAT) deposition. Estrogens and ERs also govern key processes influencing glucose homeostasis in the liver, adipose tissue, and skeletal muscle (6). Estrogen modulation of body weight, adiposity, and insulin sensitivity is apparent in women, with the risks for weight gain, increasing adiposity and type 2 diabetes rising with the decline in the levels of estrogens that occurs at menopause (7). Hormone replacement therapy with conjugated estrogen and medroxyprogesterone acetate in postmenopausal women causes a 21%–35% decrease in diabetes occurrence (8, 9). Actions of estrogens are additionally critical to maintaining hepatic lipid homeostasis, and these mechanisms are relevant to hepatic steatosis, which entails lipid accumulation in the liver, and nonalcoholic steatohepatitis, in which there is additionally liver inflammation and fibrosis. Hepatic steatosis and steatohepatitis are common complications of obesity and frequent complications of breast cancer treatment with the ER antagonist tamoxifen (10–14). However, leveraging the potentially favorable cardiovascular and metabolic actions of the hormone has been challenging because treatments involving estrogens are associated with uterotrophic and cancer-promoting effects that adversely impact the risk to benefit ratio of the therapy (15–17).
Although ERs, of which there are 2 subtypes, ERα and ERβ, classically function as transcription factors (18, 19), we and others previously discovered that plasma membrane subpopulations of ER mediate a number of nonnuclear processes (20–22). The development of an estrogen dendrimer conjugate (EDC) has made it possible to provide selective activation of nonnuclear ER. In EDC, approximately 20 ethinyl-estradiol (E2) molecules are attached to a large, positively charged nondegradable poly(amido)amine (PAMAM) dendrimer via hydrolytically stable linkages, thereby excluding EDC from the nucleus. Importantly, the nature of the chemical linkage of the PAMAM to E2 is such that the affinity of EDC-bound E2 for ER is similar to that of free E2 (23, 24). EDC has been administered systemically to mice, and it has been demonstrated that the agent promotes endothelial monolayer repair, it attenuates neointima formation invoked by endothelial injury, and it prevents cortical bone loss after ovariectomy without invoking a uterotrophic response or promoting breast cancer xenograft growth (25, 26). How selective nonnuclear ER activation impacts atherosclerosis and metabolic health and disease is currently unknown.
In the present work, experiments were designed in hypercholesterolemic female mice to interrogate how specific nonnuclear ER stimulation with EDC influences atherogenesis, and in high-fat/high-cholesterol Western diet-fed female mice to assess how such intervention impacts body weight, adiposity, glucose intolerance, and hepatic steatosis. In contrast to E2, EDC did not blunt atherosclerosis, it did not attenuate the increase in body weight or adiposity that results from Western diet feeding, and it did not prevent Western diet-induced glucose intolerance. However, E2 and EDC had comparable beneficial effect on diet-induced hepatic steatosis and predictably only E2 invoked a uterotrophic response. Thus, although nonnuclear ER activation is insufficient to prevent atherosclerosis and a number of metabolic abnormalities, it may provide a potential new strategy to improve hepatic lipid homeostasis without impacting the female reproductive tract.
Materials and Methods
Animal models
All experiments were performed in female mice on C57BL/6 background. Studies of atherosclerosis were done in apoE−/− mice on standard chow (4.3% fat, 0.02% cholesterol). Ovariectomies were performed at 5 weeks of age, at which time, 6-week duration ip osmotic minipumps (Alzet) were placed to deliver vehicle control, E2 at 6 μg/d, empty dendrimer control, or an estrogen equivalent amount of EDC as previously described. The strategy employed yields stable plasma E2 levels of 12nM–49nM and plasma EDC estrogen equivalent levels of 14nM–36nM, which cause equal stimulation of carotid artery reendothelialization mediated by ERα (25). The first minipumps were replaced after 6 weeks in order to provide a second 6 weeks of treatment. Atherosclerotic lesions were evaluated at the end of a total of 12 weeks of treatment (17 wk of age).
To evaluate the impact of nonnuclear ER activation on body composition and insulin sensitivity, wild-type C57BL/6 female mice were placed on either control chow (Harlan Teklad 2016, 12% calories from fat), or a high-fat/high-cholesterol Western diet (Harlan Teklad TD88137, 42% calories from fat, 0.2% cholesterol) beginning at the time of weaning at 4 weeks of age. Ovariectomies were performed at 6 weeks of age, at which time, the first ip minipumps were placed to deliver either vehicle control, E2, dendrimer control, or EDC. The minipumps were replaced after 6 weeks in order to continue the treatments. Studies of adiposity, glucose tolerance, insulin sensitivity, and related parameters were performed at 17 weeks of age. The Western diet-fed mice were also employed to evaluate the impact of nonnuclear ER activation on hepatic steatosis. All animal studies were approved by the Institutional Animal Care and Use Committee at University of Texas Southwestern.
Plasma lipid and liver tissue analyses
Plasma total cholesterol was evaluated as previously described (27). Plasma triglyceride levels were measured by colorimetric enzymatic assay (Roche Diagnostics). Plasma lipid profiles were obtained by column fractionation and measurement of fraction cholesterol content (28). Free fatty acids were measured in freshly collected serum using the VITROS 250 Chemistry System (Ortho-Clinical Diagnostics). Liver histology was evaluated in hematoxylin and eosin-stained sections after paraformaldehyde fixation. Images were captured using a Nikon TE2000-E microscope equipped with a Photometric Coolsnap HQ2CCD camera (×10 magnification). Liver triglyceride content was measured by the Folch method (29). Weighed tissue samples were homogenized in methanol/choloform, and after overnight extraction, 0.7% sodium chloride was added. The aqueous layer was aspirated and duplicate aliquots of the chloroform/lipid layer were dried under nitrogen gas. The lipid was reconstituted in isopropyl alcohol and assayed for triglyceride spectrophotometrically with enzymatic reagents from Thermo DMA (30)
Assessment of atherosclerosis
Atherosclerotic lesions were evaluated in a blinded fashion using our previously reported approach (28, 31). Mice were anesthetized with avertin and killed, blood was obtained for analyses, and perfusion fixation was performed. The heart and aorta were removed, the heart and proximal aorta were embedded in optimal cutting temperature compound, serial frozen sections (10 μm) of the aortic root were obtained, and 4–6 sections per aorta were processed. Images were captured and areas were determined using Image Pro Plus software (32, 33).
Evaluation of adiposity
In the studies of insulin resistance, fat mass and lean body mass was determined by NMR (Minispec NMR Analyzer; Bruker). At the time of euthanasia, sc WAT was quantified by determining the weight of the inguinal fat pad, and visceral WAT was quantified by determining the weights of the uterine, mesenteric, and gonadal fat pads. The abundance of brown adipose tissue (BAT) was also assessed by harvesting and weighing the intrascapular fat pad.
Glucose and insulin homeostasis
For glucose tolerance tests (GTTs), mice were fasted for 4–6 hours and injected ip with D-glucose (1-g/kg body weight). Tail vein blood samples were obtained at the indicated times for plasma glucose measurement by glucometer (ONE TOUCH Ultra2; Johnson & Johnson). Fasting plasma insulin concentrations were determined by ELISA (Crystal Chem, Inc) (34). Homeostasis model assessment-insulin resistance (HOMA-IR) was calculated to assess differences in insulin sensitivity (35, 36).
Quantitative RT-PCR
Transcript abundance in liver samples was evaluated by quantitative RT-PCR. Total RNA was isolated using RNAzol RT reagent (Sigma) according to the manufacturer’s instructions, and cDNA was generated from 2-μg total RNA using the High Capacity RNA-to-cDNA kit (Applied Biosystems). Inflammation and fibrosis genes were amplified by quantitative RT-PCR using TaqMan gene assays and the ABI HT7900 Real Time PCR System (Applied Biosystems). Fatty acid and triglyceride synthesis-related gene expression was monitored using SYBR green real-time PCR mix from Roche. Primer sequences were obtained from https://pga.mgh.harvard.edu/primerbank/. Hprt or 36B4 expression was evaluated in parallel to indicate total RNA abundance, and relative expression was determined by the 2−ΔΔCT method and is reported normalized to the abundance of the transcript in control group liver.
Statistical analysis
All data are expressed as mean ± SEM. Two-tailed Student’s t test or ANOVA was used to assess differences between 2 groups or among more than 2 groups, respectively, with Newman-Keuls post hoc testing after ANOVA. P < .05 was considered significant.
Results
Impact of EDC on atherosclerosis
Previous studies with EDC have demonstrated that selective activation of nonnuclear ER promotes endothelial nitric oxide (NO) production and endothelial repair (25). Recognizing that NO is an important atheroprotective molecule and that the integrity of the endothelial monolayer is also critical to atheroprotection (37), the impact of nonnuclear ER activation on atherosclerosis severity was determined using EDC. Figure 1, A–D, displays representative aortic root sections in ovariectomized female apoE−/− mice administered control vehicle, E2, empty dendrimer control, or an estrogen equivalent amount of EDC, respectively. Lesions were comparable in the vehicle and dendrimer controls, and as expected, E2 caused a decrease in lesion size. In contrast, EDC had no effect on aortic root lesions. Summary data indicate that although E2 caused a 34% decline in lesion size, the amount of atherosclerosis was unaltered by EDC treatment (Figure 1E). Neither E2 nor EDC affected total circulating cholesterol or triglyceride levels compared with their control treatments (Supplemental Figure 1). Subcutaneously placed minipumps delivering E2 yielded an identical 34% diminution in lesion size as was observed with ip minipumps (Supplemental Figure 2), indicating that the route of delivery does not influence the efficacy of the intervention. Thus, although treatment with E2, which activates both nonnuclear and nuclear ER, blunts atherogenesis, the selective stimulation of nonnuclear ER has no impact on the severity of the disease.
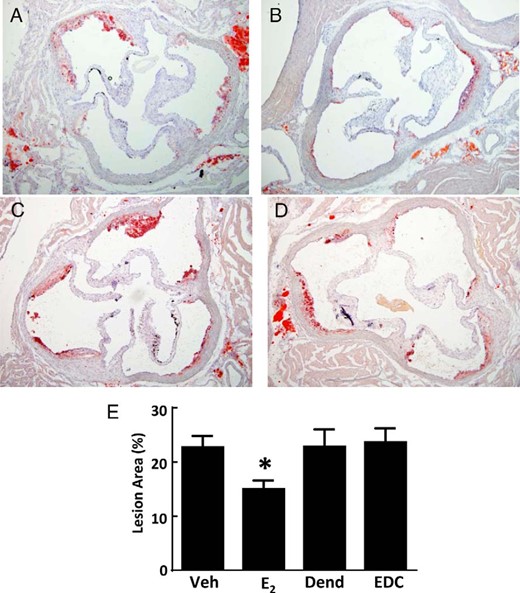
Atherosclerosis in apoE−/− ovariectomized female mice is blunted by E2 but not by EDC. A–D, Representative aortic root sections in ovariectomized female mice treated for 12 weeks with control vehicle (Veh) (A), E2 (B), empty dendrimer control (Dend) (C), or an estrogen equivalent amount of EDC (D) by ip osmotic minipump. E, Summary data for lesion size. Values are mean ± SEM, n = 11–19; *, P < .05 vs vehicle control.
Impact of EDC on body weight and composition
Estrogens acting primarily via ERα contribute to the regulation of body weight and body composition (6), and there is evidence that nongenomic actions of estrogen may be involved (38). To evaluate the impact of selective nonnuclear ER activation on metabolism, studies with control vehicle, E2, control dendrimer, and EDC were performed in control chow-fed and Western diet-fed ovariectomized female wild-type mice. Compared with control treatment, neither E2 nor EDC affected weight gain over 12 weeks in standard chow-fed mice (Figure 2A). Predictably, E2 lowered body weight in Western diet-fed mice, but EDC had no effect (Figure 2B). In both control chow- and Western diet-fed mice, E2 caused a decrease in fat mass and a corresponding increase in lean body mass, and in contrast, these parameters were unaffected by EDC (Figure 2, C–F).

Adiposity in ovariectomized female mice is blunted by E2 but not by EDC. Wild-type female mice were placed on control chow (n = 8–10) (A, C, and E) or Western diet (n = 16–20) (B, D, and F), treated for 12 weeks with control vehicle (Veh), E2, empty dendrimer control (Dend), or an estrogen equivalent amount of EDC, and body weight at 0 and 12 weeks (A and B) and fat mass (C and D) and lean body mass (E and F) at 12 weeks were determined. Values are mean ± SEM. In A and B, *, P < .05 vs time 0 and †, P < .05 vs vehicle control; in C–F, *,P < .05 vs vehicle control.
Specific effects of E2 vs EDC on different fat depots were also evaluated. The BAT depot in the intrascapular fat pad was similar in size in both control groups and E2 and EDC treatment groups, in control chow-fed as well as in Western diet-fed mice (Figure 3, A and B). Subcutaneous WAT in the inguinal fat pad was decreased by 38% with E2 treatment in control chow-fed mice, and E2 decreased it by 59% in Western diet-fed mice (Figure 3, C and D); in contrast, sc WAT abundance was unaffected by EDC. Visceral WAT in the mesenteric fat pad was similar in all 4 treatment groups, in both control chow-fed and Western diet-fed mice (Figure 3, E and F). However, in visceral WAT in the uterine and gonadal fat depots, E2 decreased fat abundance, causing declines of 47% and 45%, respectively, in chow-fed mice, and declines of 77% and 78%, respectively, in the Western diet-fed mice (Figure 3, G–J). As observed previously (25), although E2 invoked a robust uterotrophic response, there was no effect of EDC on uterine weight (Supplemental Figure 3A). These collective findings indicate that processes by which estrogen influences body weight and body composition are not activated by the selective stimulation of nonnuclear ERs with EDC.
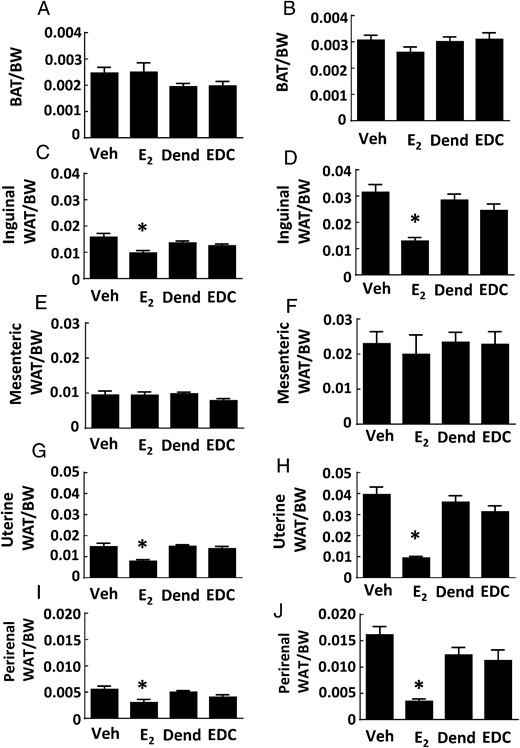
Fat depots in ovariectomized female mice are decreased by E2 but not by EDC. Wild-type ovariectomized female mice were placed on control chow (A, C, E, G, and I) or Western diet (B, D, F, H, and J), treated for 12 weeks with control vehicle (Veh), E2, empty dendrimer control (Dend), or an estrogen equivalent amount of EDC, and multiple fat depots were quantified. The depots were intrascapular BAT (n = 8–10 for control diet and n = 17–21 for high-fat diet) (A and B), inguinal WAT (n = 8–13) (C and D), mesenteric WAT (n = 8–13) (E and F), uterine WAT (n = 8–10 for control diet and n = 16–21 for high-fat diet) (G and H), and gonadal WAT (n = 6–10) (I and J). Values are mean ± SEM; *, P < .05 vs vehicle control.
Impact of EDC on glucose homeostasis
Along with their effects on atherosclerosis and body weight and composition, estrogens and ERs contribute to the regulation of glucose homeostasis. In female rodents and primates, ovariectomy results in impaired insulin sensitivity and glucose homeostasis, and these effects are reversed by E2 (39, 40). We employed EDC to evaluate how the activation of nonnuclear ER impacts glucose regulation and did so in both normal chow-fed ovariectomized female mice and mice with diet-induced obesity. On either diet, a uterotrophic response was observed with E2 but not with EDC (Supplemental Figure 3, B and C). In nonobese mice, fasting glucose was not altered by either E2 or EDC (Figure 4A). In mice with diet-induced obesity, treatment with E2 caused a decline in fasting glucose and in contrast EDC had no effect (Figure 4B). Fasting insulin was unaffected by E2 or EDC in normal chow-fed mice (Figure 4C). In Western diet-fed, vehicle-treated mice, there was fasting hyperinsulinemia compared with values in the chow-fed group (P < .05), and this was normalized by E2 (Figure 4D). In contrast, in the dendrimer control and EDC-treated groups fed the Western diet, the fasting insulin levels were similar. In nonobese mice, GTTs were improved with E2 administration (Figure 5A), but GTTs were unaffected by EDC compared with control empty dendrimer (Figure 5B). In mice with diet-induced obesity, there was also a normalization of GTT with E2 treatment (Figure 5C), and EDC had no impact (Figure 5D). HOMA-IR calculations indicated that insulin sensitivity was increased in control chow-fed ovariectomized female mice treated with E2, but EDC had no effect (Figure 5E). Insulin sensitivity assessed with HOMA-IR was even more favorably impacted by E2 in the Western diet-fed mice, whereas EDC continued to cause no change (Figure 5F). Thus, although glucose homeostasis was favorable affected by E2 as has been previously observed (30), the selective activation of nonnuclear ER with EDC did not alter glucose regulation.
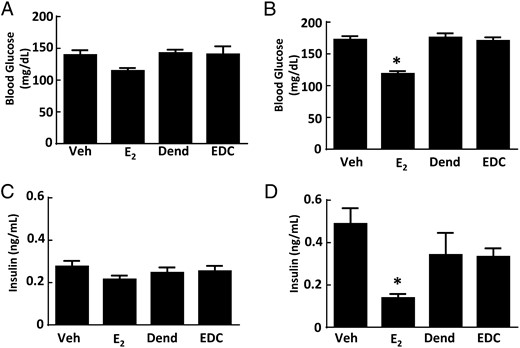
Diet-induced fasting hyperglycemia and fasting hyperinsulinemia are improved by E2 but not by EDC. Wild-type ovariectomized female mice were placed on control chow (A and C) or Western diet (B and D), treated for 12 weeks with control vehicle (Veh), E2, empty dendrimer control (Dend), or an estrogen equivalent amount of EDC, and fasting plasma glucose and insulin were measured. Values are mean ± SEM, n = 8–10 for control chow groups and n = 16–21 for Western diet groups; *, P < .05 vs vehicle control.
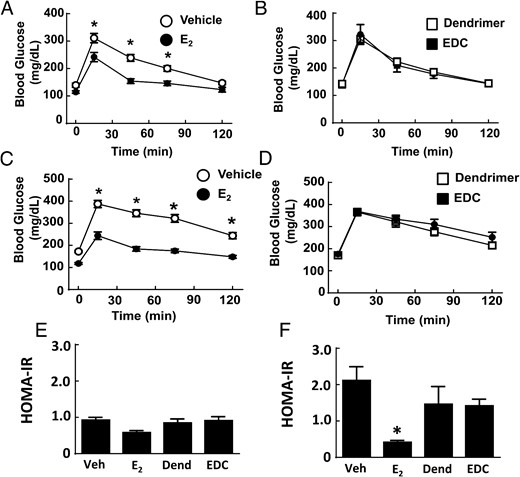
Glucose tolerance is enhanced by E2 but not by EDC. Wild-type ovariectomized female mice were placed on control chow (A, B, and E) or Western diet (C, D, and F), treated for 12 weeks with control vehicle (Veh), E2, empty dendrimer control, or an estrogen equivalent amount of EDC, and GTTs (A–D) and HOMA-IR were evaluated (E and F). Values are mean ± SEM, n = 8–10 for control chow groups and n = 16–21 for Western diet groups; *, P < .05 vs vehicle control.
Impact of EDC on hepatic steatosis
Recognizing that estrogens participate in hepatic lipid homeostasis (11), we compared the effects of E2 and EDC on the hepatic steatosis that accompanies receipt of a Western diet in mice. In the livers of control vehicle- or dendrimer-treated mice, there was considerable lipid deposition noted both histologically (Figure 6, A and C) and by the quantification of liver triglyceride content (Figure 6E). To determine whether the intake of the Western diet employed also causes hepatic inflammation and fibrosis, quantitative RT-PCR was done assessing the expression of F4/80, TNFα and IL-6, and TGFβ, tissue inhibitor of metalloproteinase metallopeptidase inhibitor 1, and collagen, type I, α1, respectively, in livers from control diet- vs Western diet-fed mice (Supplemental Figure 4). None of the indicators of inflammation or fibrosis were elevated in the livers of the Western diet-fed mice. As such, the model employed entails fatty liver but not the development of liver fibrosis or inflammation. Regarding possible modulation of hepatic steatosis by nonnuclear ER activation, E2 and EDC caused comparable declines in liver fat accumulation (Figure 6, B and D), with triglyceride abundance decreased by half (Figure 6E).
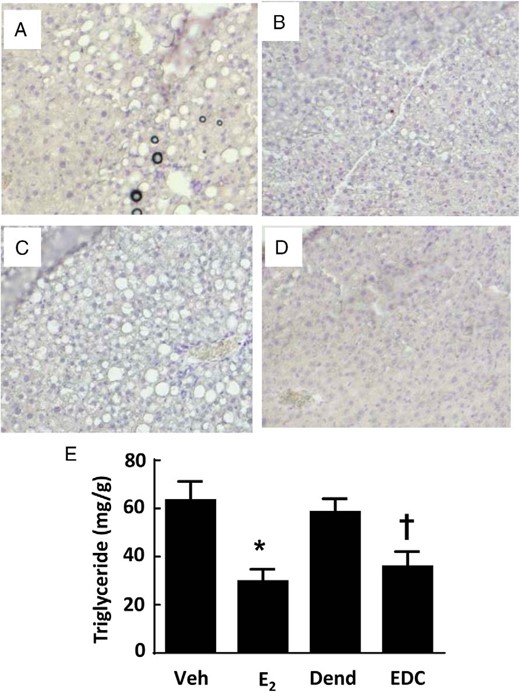
Hepatic steatosis is blunted by E2 and EDC. Wild-type ovariectomized female mice were placed on Western diet and treated for 12 weeks with control vehicle (Veh) (A), E2 (B), empty dendrimer control (Dend) (C), or an estrogen equivalent amount of EDC (D), and liver histology (×10 magnification) (A–D) and triglyceride content (E) were evaluated. In E, values are mean ± SEM, n = 10–13; *, P < .05 vs vehicle control; †, P < .05 vs dendrimer.
To determine the basis for the protection from hepatic steatosis afforded by E2 and EDC, plasma free fatty acids were measured and plasma lipid profiles were performed (Supplemental Figure 5). Compared with the vehicle-treated and dendrimer-treated control groups, neither E2 nor EDC had an effect on plasma free fatty acids, and plasma lipid profiles were also unaffected. These findings suggest that changes in systemic lipid or cholesterol homeostasis do not explain the reversal of hepatic steatosis by E2 and EDC. To further evaluate the underlying mechanisms, transcript abundance for fatty acid and triglyceride synthesis genes in the liver was assessed (Figure 7). Regarding fatty acid synthesis, relative to the vehicle and dendrimer control treatments, both E2 and EDC caused down-regulation of stearoyl-coenzyme A desaturase 1 (Scd1) and sterol regulatory element binding transcription factor 1 (Srebf1), and fatty acid synthase (Fasn) was down-regulated by E2 but not by EDC. As for triglyceride synthesis, peroxisome proliferator activated receptor (PPAR)γ was down-regulated by both E2 and EDC, diacylglycerol O-acyltransferase 1 (Dgat1) was down-regulated by E2, and there was a parallel directional change with EDC (P = .052), and liver X receptor α (LXRα) expression was unaffected by either E2 or EDC. Thus, the selective activation of nonnuclear ER is sufficient to cause the attenuation of hepatic lipid accumulation, and this is related to the down-regulation of genes involved in the regulation of fatty acid and triglyceride synthesis.
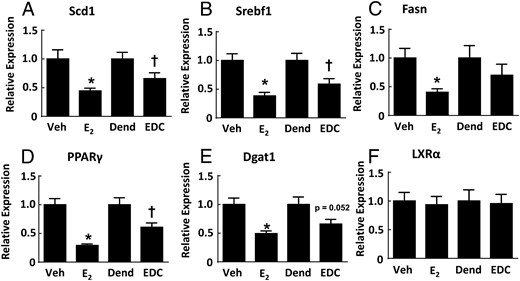
Genes regulating fatty acid and triglyceride synthesis in the liver are down-regulated by E2 and EDC. Wild-type ovariectomized female mice were placed on Western diet and treated for 12 weeks with control vehicle (Veh), E2, empty dendrimer control (Dend), or an estrogen equivalent amount of EDC, and liver samples were obtained. Quantitative RT-PCR was then performed to evaluate transcript levels for Scd1 (A), Srebf1 (B), Fasn (C), PPARγ (D), Dgat1 (E), and LXRα (F). Values are mean ± SEM, n = 9–13; *, P < .05 vs vehicle control; †, P < .05 vs dendrimer.
Discussion
Actions of estrogens mediated by ER have the potential to afford atheroprotection as well as prevent adiposity and obesity-related glucose intolerance and blunt hepatic steatosis (6, 11, 12, 41). In the current investigation, we employed E2 and EDC to evaluate the cardiometabolic responses to selective nonnuclear ER activation in hypercholesterolemic apoE−/− mice and in Western diet-fed wild-type mice. In contrast to E2, EDC did not blunt atherosclerosis in apoE−/− mice, it did not affect the increase in adiposity that resulted from Western diet feeding of wild-type mice, and it did not prevent Western diet-induced glucose intolerance. However, hepatic steatosis invoked by Western diet was similarly improved by E2 and EDC, and predictably only E2 caused a uterotrophic response. Previous studies revealed that in contrast to E2, EDC does not support the growth of estrogen-responsive cancers (23, 25). Thus, nonnuclear ER activation may provide a potential new strategy to combat hepatic steatosis without impacting the female reproductive tract or increasing the risk of cancer.
Nonnuclear actions of ER have been demonstrated previously in vascular cell types involved in modulating atherosclerosis severity, particularly in endothelial and VSM cells. In endothelium, plasma membrane-associated ERα as well as plasma membrane subpopulations of ERβ stimulate endothelial NO synthase enzymatic activity (42, 43), thereby increasing the production of the atheroprotective signaling molecule NO. Plasma membrane ERαs are also necessary and sufficient for the capacity of estrogens to promote endothelial cell growth and migration, which are critical to maintaining the integrity of the endothelial monolayer in vivo (25). In VSM cells, nonnuclear ERα are responsible for the inhibition of cell proliferation by estrogens (3). However, the current studies demonstrated that selective nonnuclear ER activation is insufficient to attenuate atherosclerosis development. This finding is consistent with the observations made in a previous report in which E2-induced atheroprotection was lost in a knock-in mouse in which activation function (AF)-2 in the ligand binding domain of ERα was disrupted by omission of amino acids 543–549 (44). In addition, it has been reported that although the naturally occurring estrogen estetrol does not stimulate the nonnuclear activation of endothelial NO synthase, it activates the 2 ER activation functions AF-1 and AF-2, and promotes the recruitment of steroid receptor coactivator-3 and effectively blunts atherogenesis in LDLR−/− mice (45). Thus, when considered along with these previous findings, the inability of EDC to impact atherosclerosis development in the present investigation indicates that ER-related atheroprotection is primarily driven by nuclear functions of ERα. Furthermore, from a therapeutic standpoint, the current findings with EDC indicate that although reproductive tract and hormone-responsive cancers are unaffected, the selective activation of nonnuclear ER actions does not provide atheroprotection.
Along with their vascular actions, estrogens potently impact body weight and body composition, and these responses are mediated centrally in hypothalamic nuclei and peripherally in adipose tissue. In the hypothalamus estrogen actions via ERα in arcuate nucleus proopiomelanocortin neurons attenuate food intake (46), and through ERα in ventromedial hypothalamic steroidogenic factor-1-expressing neurons the hormone enhances physical activity and energy expenditure and regulates visceral fat distribution (47, 48). Nonnuclear functions may contribute to metabolic regulation by hypothalamic ER, because central E2 treatment causes a rapid increase in excitatory synapses on proopiomelanocortin neurons (46), in immortalized hypothalamic neurons E2 attenuates neuropeptide Y secretion via a membrane form of ERα (49), and there is evidence that anorectic actions of E2 and its modulation of body temperature occur via a Gq-coupled membrane ER (50, 51). In cultured adipocytes, E2 inhibits adipogenesis and lipogenesis (52), and the selective deletion of ERα from adipocytes results in an increase in adiposity in both male and female mice (53). Nonnuclear responses to E2 have been demonstrated in cultured adipocytes (54), but whether they influence adipocyte development or function remains unknown. In the present investigation, in contrast to E2, EDC had no effect on body weight or adiposity, suggesting that selective nonnuclear ER activation does not impact body composition in vivo. However, a potential limitation of the systemic use of EDC is that most likely related to blood-brain-barrier exclusion, there is negligible distribution of PAMAM dendrimer to the brain compared with peripheral tissues; as such, systemically delivered EDC probably has limited access to the central nervous system (55). Studies of centrally administered EDC are now warranted to better understand the involvement of nonnuclear ER functions in hypothalamic regulation of energy homeostasis and body composition.
Besides influencing body weight and composition, estrogens impact glucose homeostasis through processes in numerous tissues and cell types, and nonnuclear ER actions have been suggested to be operative in some of these mechanisms. In skeletal muscle, E2 promotes insulin-stimulated glucose uptake, there is associated activation of Akt and AMP-activated protein kinase (AMPK) (56, 57), and in vitro studies with myotubes have demonstrated that there is enhanced insulin action after short-term E2 treatment (58). Considering the numerous functions of Akt and AMPK in skeletal muscle, roles for nonnuclear ER signaling in skeletal muscle insulin sensitivity would then be predicted to be demonstrable in vivo. There is also data suggesting that nonnuclear ERs are important to pancreatic islet function. In islets, ERs are found primarily outside the nucleus, and E2 attenuates pancreatic islet fatty acid and glycerolipid synthesis to attenuate B cell failure during type 2 diabetes (59). A role for nonnuclear ER is further suggested by the observation that in INS-1 cells E2 activates AMPK, and via AMPK E2 attenuates the expression of Srebf1, a key transcriptional regulator of Fasn (60). E2-ERα stimulation of insulin synthesis also appears to be mediated by nonnuclear ER, with EDC increasing preproinsulin expression in cultured pancreatic islets (61). However, the current findings indicate that in the context of obesity-induced insulin resistance exacerbated by ovariectomy in mice, the selective activation of nonnuclear ER with EDC has negligible impact. As such, nuclear ER actions are likely required for estrogens to have substantive influence on the processes governing glucose homeostasis in vivo.
Although EDC did not impact atherosclerosis severity or diet-induced changes in body weight, body composition or glucose tolerance/insulin sensitivity, the agent had a favorable effect on hepatic steatosis that was comparable with that observed with E2. The improvement in steatosis was not related to changes in systemic lipid or cholesterol homeostasis, and alternatively a number of hepatic genes involved in fatty acid and triglyceride synthesis were down-regulated by both E2 and EDC. These included Srebf1, which is a transcription factor that promotes the expression of numerous genes that participate in lipid synthesis (62). Srebf1 was previously observed to be down-regulated in the liver by the ERα-selective ligand propyl-pyrazole-triol in standard chow-fed mice expressing wild-type ERα or a form of the receptor consisting of the ERα ligand binding domain directed only to plasma membrane, but not in mice deficient in ERα. Related studies employing kinase inhibitors in insulin-stimulated isolated hepatocytes showed that AMPK and the ERK-MAPK pathway participated in membrane ERα down-regulation of Srebf1 (63). In the present work, EDC also down-regulated Scd1, PPARγ, and Dgat1, which were previously not known to be target genes of nonnuclear ER activation in the liver. Consistent with previous studies of E2 impact on hepatic steatosis, the favorable effects of EDC on Srebf1 and liver triglyceride occurred independent of a change in liver LXRα expression (64). Thus, the current findings with EDC reveal that nonnuclear actions of ER underlie beneficial effects of estrogens on hepatic steatosis. The current findings also provide the rationale for future studies of EDC and the additional liver inflammation and fibrosis that characterize nonalcoholic steatohepatitis.
From a therapeutic risk-benefit perspective, the present studies indicate that although actions of estrogens that provide protection from atherosclerosis, obesity or its related insulin resistance may not be effectively captured by the selective stimulation of nonnuclear ER action, agents like EDC may offer a novel means to combat hepatic steatosis without adversely impacting the reproductive tract or cancer risk. The lack of impact on the female reproductive tract or ER-positive breast cancer cells also suggests that their use in males may be feasible (25), and previous studies with E2 in mice administered high-fat diet plus ethanol further suggest that agents like EDC may benefit both nonalcohol-related and alcohol-related fatty liver disease (65). Combining the findings for steatosis with the previous discoveries that nonnuclear ER activation promotes vascular repair and prevents cortical bone loss (25, 26), the rationale for further development of nonnuclear selective ER modulators is expanding.
Acknowledgments
This work was supported by National Institutes of Health Grants HL087564 (to P.W.S.), DK015556 (to J.A.K.), HL098040 (to P.W.S.), and P50 AT006268 (to B.S.K. and J.A.K.) and by the Associates First Capital Corporation Distinguished Chair in Pediatrics (P.W.S.).
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- AF
activation function
- AMPK
AMP-activated protein kinase
- BAT
brown adipose tissue
- Dgat1
diacylglycerol O-acyltransferase 1
- E2
estradiol
- EDC
estrogen dendrimer conjugate
- ER
estrogen receptor
- Fasn
fatty acid synthase
- GTT
glucose tolerance test
- HOMA-IR
homeostasis model assessment-insulin resistance
- LXRα
liver X receptor α
- NO
nitric oxide
- PAMAM
poly(amido)amine
- PPAR
peroxisome proliferator activated receptor
- Scd1
stearoyl-coenzyme A desaturase 1
- Srebf1
sterol regulatory element binding transcription factor 1
- VSM
vascular smooth muscle
- WAT
white adipose tissue.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}