





Abstract
It has been suggested that mitochondrial reactive oxygen species (ROS), Akt and Erk1/2 and more recently the mitochondrial permeability transition pore (mPTP) may act as mediators of ischaemic preconditioning (IPC), although the actual interplay between these mediators is unclear. The aim of the present study is to determine whether the cyclophilin-D (CYPD) component of the mPTP is required by IPC to generate mitochondrial ROS and subsequently activate Akt and Erk1/2.
Mice lacking CYPD (CYPD−/−) and B6Sv129 wild-type (WT) mice were used throughout. We have demonstrated that under basal conditions, non-pathological mPTP opening occurs (indicated by the percent reduction in mitochondrial calcein fluorescence). This effect was greater in WT cardiomyocytes compared with CYPD−/− ones (53 ± 2% WT vs. 17 ± 3% CYPD−/−; P < 0.01) and was augmented by hypoxic preconditioning (HPC) (70 ± 9% WT vs. 56 ± 1% CYPD−/−; P < 0.01). HPC reduced cell death following simulated ischaemia–reperfusion injury in WT (23.2 ± 3.5% HPC vs. 43.7 ± 3.2% WT; P < 0.05) but not CYPD−/− cardiomyocytes (19.6 ± 1.4% HPC vs. 24.4 ± 2.6% control; P > 0.05). HPC generated mitochondrial ROS in WT (four-fold increase; P < 0.05) but not CYPD−/− cardiomyocytes. HPC induced significant Akt phosphorylation in WT cardiomyocytes (two-fold increase; P < 0.05), an effect which was abrogated by ciclosporin-A (a CYPD inhibitor) and N-2-mercaptopropionyl glycine (a ROS scavenger). Finally, in vivo IPC of adult murine hearts resulted in significant phosphorylation of Akt and Erk1/2 in WT but not CYPD−/− hearts.
The CYPD component of the mPTP is required by IPC to generate mitochondrial ROS and phosphorylate Akt and Erk1/2, major steps in the IPC signalling pathway.
1. Introduction
Mitochondria play a critical role in deciding the fate of cardiomyocytes subjected to acute ischaemia–reperfusion injury. It is well established that the mitochondrial dysfunction resulting from the irreversible pathological opening of the mitochondrial permeability transition pore (mPTP) at the onset of myocardial reperfusion is a major determinant of cardiomyocyte death following an acute myocardial infarction.1,2 The molecular composition of the mPTP remains unclear, although mitochondrial cyclophilin-D (CYPD) has been reported to be a critical regulatory component of the mPTP.3–5 It has been shown that preventing the pathological form of mPTP opening at the onset of myocardial reperfusion underlies the infarct-limiting effects of ischaemic preconditioning (IPC),6–9 a phenomenon in which one or more brief episodes of ischaemia and reperfusion render the myocardium more resistant to a subsequent acute myocardial infarction.10 The signal transduction pathways recruited by IPC prior to myocardial ischaemia have been demonstrated to converge on mitochondria resulting in the generation of mitochondrial reactive oxygen species (ROS), which are released into the cytosol where they activate important signalling protein kinases such as PKC, Akt, and Erk1/2, critical to IPC protection.11,12 The mechanism through which IPC induces mitochondrial ROS production and whether this ROS is actually responsible for activating these protein kinases remains to be demonstrated directly.
There is accumulating evidence to suggest that the mPTP can open reversibly under basal conditions without causing cell death.13–17 The significance of this form of reversible non-pathological mPTP opening is unresolved, although previous studies have suggested that it may contribute to the cardioprotection elicited by IPC via a mechanism that is not as clear yet.18,19 In the present study, we sought to determine whether reversible non-pathological CYPD-dependent mPTP opening is induced by preconditioning, and whether this form of mPTP opening is required by IPC to generate mitochondrial ROS, which in turn are needed to activate the pro-survival protein kinases, Akt, and Erk1/2, critical mediators of the IPC signalling pathway.
2. Methods
2.1 Animals
Experiments using animals were carried out in accordance with the United Kingdom Home Office Guide on the Operation of Animal (Scientific Procedures) Act of 1986. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. B6Sv129 wild-type (WT) mice were obtained from Harlan (UK) and cyclophilin-D knockout mice (CYPD−/−) mice were bred from pairs originally provided by Molkentin's laboratory.3,20 WT and CYPD−/− male mice (8–10 weeks, 20–30 g) were used throughout the study.
2.2 Isolating adult murine ventricular cardiomyocytes
Adult ventricular cardiomyocytes were isolated from WT and CYPD−/− mice using collagenase digestion according to a previously established method.21 The isolated cardiomyocytes were then seeded onto sterilized laminin-coated coverslips in M199 medium containing 2 g/L BSA, 0.62 g/L taurine, 0.32 g/L carnitine, 10 units/mL penicillin, 10 mg/L streptomycin, and 25 µmol/L blebbistatin (Calbiochem, USA) before use on the same day of isolation.
2.3 Adult murine ventricular cardiomyocyte model for detecting mPTP opening
To determine whether non-pathological opening of the CYPD-dependent mPTP occurs under basal conditions and is augmented by hypoxic preconditioning (HPC), a well-established and reproducible technique for detecting mPTP opening in the isolated adult cardiomyocyte was used with some modifications.15,18,19,22,23 Briefly, freshly isolated cardiomyocytes were loaded with 1.0 µmol/L calcein-AM + 1.0 mmol/L CoCl2 in order to selectively load the mitochondria with calcein by quenching the cytosolic calcein signal with CoCl2. The loss of mitochondrial calcein fluorescence under basal conditions can then be used as a measure of non-pathological mPTP opening.15,18,19,22,23
WT and CYPD−/− cardiomyocytes were randomized to receive either a standard HPC protocol comprising 10 min simulated ischaemia followed by 10 min simulated reperfusion or control. Simulated ischaemia was induced in a custom-made airtight hypoxic chamber using a ischaemic buffer containing (in mmol/L): 1.0 KH2PO4, 10.0 NaHCO3, 1.2 MgCl2·6H20, 25.0 Na(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES), 74.0 NaCl, 16.0 KCl, 1.2 CaCl2, and 20.0 Na lactate, pH 6.2, bubbled with 95% nitrogen/5% CO2. Simulated reperfusion was achieved by replacing the buffer with M199 medium containing 2 g/L BSA, 0.62 g/L taurine, 0.32 g/L carnitine, 10 units/mL penicillin, 10 mg/L streptomycin. Using a confocal microscope to visualize the cardiomyocytes, the loss of mitochondrial calcein fluorescence was measured over the course of 30 min in the same cells. The cells were imaged at time 0, 10, and 30 min in order to minimize laser-induced oxidative stress on the cells. For each time-point, the mean calcein fluorescence was expressed as a percentage of the initial value. In between imaging, the cells were returned to the incubator and kept at 37.0°C. For the cells that were randomized to receive HPC, after the baseline images at time 0 had been taken, the cells were placed in an ischaemic buffer and oxygen was removed by replacing it with nitrogen gas. Following this, the cells were returned to normoxic buffer (see later) and the same cells were imaged at 10 and 30 min. For each treatment group, 10 cells were counted. This experiment was repeated on three separate occasions giving a total of 30 cells per treatment group.
A Leica confocal microscope (SP5) was used for these experiments. For the mPTP experiments 1024 × 1024, images of cells were acquired using low power (2%) 488 nm argon laser (line averaging of 4, zoom 2.2). The gain was adjusted to achieve maximum signal intensity without saturation.
2.4 Adult murine ventricular cardiomyocyte model for assessing mitochondrial ROS production
To determine whether CYPD is required to generate mitochondrial ROS in response to HPC, WT and CYPD−/− cardiomyocytes were loaded with reduced Mitotracker Red CM-H2XRos (Molecular Probes/Invitrogen) 1.0 µM for 20 min at 37.0°C in the incubator and randomized to the following treatment protocols, following which Mitotracker Red fluorescence, was measured using a fluorescence microscope in order to quantify the mitochondrial ROS production: (i) control, DMSO (0.1%) for 30 min; (ii) HPC, comprising 15 min simulated ischaemia and 15 min simulated reperfusion; (iii) HPC+MPG, HPC in the presence of the ROS scavenger, N-2-mercaptopropionyl glycine (MPG, 300 µmol/L); (iv) HPC+CsA, HPC in the presence of CsA (0.4 µmol/L); (v) MPG alone was given for 30 min; and (vi) CsA alone was given for 30 min. For each treatment group, at least 25 cells were counted, taken from three to four randomly selected fields of view. This experiment was repeated on three separate occasions giving a total of >75 cells per treatment group.
2.5 Adult murine ventricular cardiomyocyte model of simulated ischaemia–reperfusion injury
To determine whether the CYPD is required for cardioprotection elicited by HPC, WT and CYPD−/− cardiomyocytes were randomized to the following treatment protocols prior to subjecting them to simulated ischaemia–reperfusion injury: (i) control, DMSO (0.1%) for 30 min; (ii) HPC, comprising 15 min simulated ischaemia and 15 min simulated reperfusion; (iii) HPC+MPG, HPC in the presence of the ROS scavenger, MPG (300 µmol/L); (iv) HPC+CsA, HPC in the presence of CsA (0.4 µmol/L); (v) MPG alone for 30 min; and (vi) CsA alone for 30 min. The cells were then subjected to 45 min of simulated ischaemia followed by 30 min of simulated reperfusion at the end of which cell death was quantified by the number of cells staining positive for propidium iodide (3 µmol/L). For each treatment group, approximately 150 cells were counted. This experiment was repeated on three separate occasions giving a total of ∼450 cells per treatment group.
2.6 Adult murine ventricular cardiomyocyte model of kinase phosphorylation
To determine whether the HPC-induced phosphorylation of Akt is mediated by ROS and is dependent on CYPD, WT adult ventricular cardiomyocytes were randomized to the following treatment protocols: (i) control, containing DMSO (0.1%); (ii) HPC, comprising 15 min simulated ischaemia and 15 min simulated reperfusion; (iii) HPC+MPG, HPC in the presence of the ROS scavenger, MPG (300 µmol/L); (iv) HPC+CsA, HPC in the presence of the CYPD inhibitor, CsA (0.4 µmol/L); (v) MPG alone; (vi) CsA alone; and (vii) insulin (300 units/L) (this treatment was used as a positive control for Akt phosphorylation), following which the cardiomyocytes were lysed for subsequent western blotting analysis for Akt phosphorylation.
2.7 In vivo adult murine model of myocardial ischaemic preconditioning
To determine whether CYPD is required by IPC to activate Akt and Erk1/2 in vivo, mice were anesthetized by intraperitoneal injection with a combination of ketamine, xylazine and atropine (0.01 mL/g, final concentration of ketamine, xylazine and atropine were 10, 2, and 0.06 g/L, respectively) and body temperature was maintained at 37.0 ± 0.5°C. A tracheotomy was performed for artificial respiration at 120 strokes/min and 220 µL stroke volume using a rodent Minivent (type 845, Harvard Apparatus, Kent, UK) and supplemental oxygen was supplied. A limb lead I electrocardiogram was recorded. A left anterior thoracotomy and a chest retractor were used to expose the heart. Ligation of the left anterior descending coronary artery was performed at ∼2 mm below the tip of the left atrium using a 8/0 prolene monofilament polypropylene suture.21
WT and CYPD−/− mice were randomized to receive either a standard IPC protocol or control (n = 4 per treatment group). IPC comprised of 5 min regional myocardial ischaemia followed by 5 min myocardial reperfusion following which myocardial tissue taken from the preconditioned myocardium was snap-frozen in liquid nitrogen for subsequent western blotting analysis. For the control hearts, myocardial tissue was harvested at an equivalent time-point of the IPC-treated hearts.
2.8 Western blotting analysis for Akt and Erk1/2
A total of 30 µg of protein (from myocardial tissue) or 30 µL of protein (from isolated cardiomyocytes) of each sample was loaded into 12.5% acrylamide gels. The phosphorylation states of Akt (phospho-Akt and Ser-308) and Erk1/2 (p42/p44) protein levels were determined for each of the treated groups (n = 4–6 per group). All the antibodies were obtained from Cell Signalling (Hitchin, Kent) and used in accordance with the manufacturer's instructions. Equal protein loading was confirmed by β-actin (for myocardial tissue) or α-tubulin (for isolated cardiomyocytes) probing of membranes (Abcam Ltd, UK). Relative densitometry was determined using a computerized software package NIH Image 1.63.
2.9 Statistical analysis
Values are mean ± SEM. Data were analysed using one-way analysis of variance (ANOVA), followed by Tukey's multiple comparison post hoc test. Results from the calcein experiments were analysed by two-way ANOVA with post-test analysis using Bonferroni correction. P < 0.05 was considered significant.
3. Results
3.1 Non-pathological mPTP opening occurs under basal conditions and is augmented by hypoxic preconditioning
Over the course of 30 min, there was a significant reduction in mitochondrial calcein fluorescence (expressed as the percentage reduction of baseline fluorescence) indicating that non-pathological mPTP opening was occurring under basal conditions. This effect was greater in WT cardiomyocytes when compared with those lacking CYPD (53 ± 2% in WT vs. 17 ± 3% in CYPD−/−; P < 0.01) (Figure 1A and B), indicating that non-pathological mPTP opening is dependent on CYPD.

(A) Representative confocal images depicting non-pathological mitochondrial permeability transition pore (mPTP) opening occurring under basal conditions (as indicated by the redistribution of calcein from the mitochondria into the cytosol), a phenomenon which occurs to a greater extent in wild-type (WT) adult murine cardiomyocytes compared with those lacking the cyclophilin-D component (CYPD−/−) of the mPTP. (B) Non-pathological mitochondrial permeability transition pore (mPTP) opening occurred under basal conditions (as indicated by the loss of mitochondrial calcein fluorescence expressed as a percentage of the baseline fluorescence), and this phenomenon occurred to a greater extent in wild-type (WT) adult murine cardiomyocytes compared with those lacking the cyclophilin-D component (CYPD−/−) component of the mPTP. Hypoxic preconditioning (HPC) augmented this form of mPTP opening as indicated by a further loss of mitochondrial calcein fluorescence, an effect which again occurred to a greater extent in WT cardiomyocytes compared with those lacking CYPD. n = 3. *P < 0.05 when compared to WT control.
Importantly, HPC was demonstrated to induce a further loss of mitochondrial calcein fluorescence after 30 min which was more marked in the WT cardiomyocytes when compared with those lacking CYPD (70 ± 9% in WT vs. 56 ± 1% in CYPD−/−; P < 0.01) (Figure 1B), indicating that HPC itself can induce non-pathological mPTP opening over and above that occurring under basal conditions. Again, this HPC-induced mPTP opening was dependent on CYPD.
3.2 CYPD is required by hypoxic preconditioning to generate mitochondrial ROS
HPC increased the fluorescence of reduced Mitotracker Red in the mitochondria of WT cardiomyocytes by almost 4-fold (see Figure 2A for representative fluorescence images) following a standard HPC protocol (Figure 2B), indicating that HPC induced the production of mitochondrial ROS. The HPC-induced increase in reduced Mitotracker Red fluorescence was abolished in the presence of the ROS-scavenger, MPG, confirming that the increase in fluorescence reflected mitochondrial ROS production (Figure 2B). Interestingly, the HPC-induced increase in reduced Mitotracker Red fluorescence was also abolished in the presence of the CYPD inhibitor, CsA, and in cardiomyocytes lacking CYPD, HPC failed to increase the fluorescence of reduced Mitotracker Red, suggesting that CYPD is required to generate mitochondrial ROS in response to HPC (Figure 2B). MPG or CsA when given alone did not influence reduced Mitotracker Red fluorescence significantly (Figure 2B).
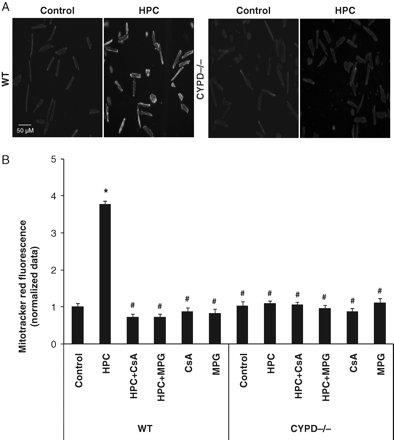
(A) Representative fluorescent images depicting an increase in reduced Mitotracker Red fluorescence (indicating enhanced mitochondrial ROS production) in WT and CYPD−/− adult murine cardiomyocytes in control and following a standard hypoxic preconditioning (HPC) protocol. (B) Hypoxic preconditioning (HPC) induced mitochondrial ROS production (as measured by an increase in reduced Mitotracker Red fluorescence) in wild-type (WT) cardiomyocytes but not CYPD−/− cardiomyocytes. This HPC-mediated increase in reduced Mitotracker Red fluorescence was abrogated in the presence of a ROS scavenger, N-2-mercaptopropionyl glycine (MPG, 300 µM) and mPTP inhibitor, Ciclosporin-A (CsA, 0.4 µM). Data are normalized to the control WT cardiomyocyte group. n = 3. *P < 0.05 vs. WT control and #P < 0.05 vs. WT HPC.
3.3 Protection by hypoxic preconditioning requires CYPD
A standard HPC protocol reduced cell death following simulated ischaemia–reperfusion injury in WT cardiomyocytes (23.2 ± 3.5% with HPC vs. 43.7 ± 3.2% in control; P < 0.05) (Figure 3). This cardioprotective effect of HPC was found to be dependent on ROS, given that it was abolished in the presence of the ROS scavenger, MPG (23.2 ± 3.5% with HPC vs. 44.1 ± 2.6% with HPC+MPG; P < 0.05) (Figure 3), confirming the important mediatory role of ROS in IPC protection. MPG when given alone had no significant effect on cell death when compared with control (45.5 ± 6.1% with MPG vs. 43.7 ± 3.2% in control; P > 0.05) (Figure 3).
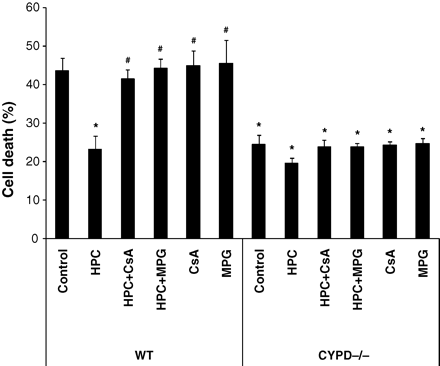
Hypoxic preconditioning (HPC) reduced cell death in wild-type (WT) cardiomyocytes but not CYPD−/− cardiomyocytes following simulated ischaemia–reperfusion injury. This protective effect was abolished in the presence of a ROS scavenger, N-2-mercaptopropionyl glycine (MPG, 300 µM) and mPTP inhibitor, Ciclosporin-A (CsA, 0.4 µM). n = 3. *P < 0.05 vs. WT control and #P < 0.05 vs. WT HPC.
Interestingly, HPC failed to reduce cell death following simulated ischaemia–simulated reperfusion injury in the presence of CsA in WT cardiomyocytes (23.2 ± 3.5% with HPC vs. 41.4 ± 2.6% with HPC+CsA; P < 0.05) and in cardiomyocytes lacking CYPD (19.6 ± 1.4% with HPC vs. 24.4 ± 2.6% in control; P > 0.05), indicating that CYPD is required for the cardioprotective effect elicited by HPC (Figure 3). MPG or CsA when given alone or in the presence of HPC had no significant effect on cell death when compared with control in CYPD−/− cardiomyocytes (23.8 ± 1.1% with HPC+MPG, 23.9 ± 1.8% with HPC+CsA, 24.8 ± 1.3% with MPG, and 23.9 ± 1.8% with CsA vs. 24.4 ± 2.6% in control; P > 0.05) (Figure 3).
3.4 In vivo myocardial ischaemic preconditioning requires the CYPD component of the mPTP to phosphorylate Akt and Erk1/2
Adult murine WT hearts, which were subjected to a standard in vivo myocardial preconditioning protocol,20 demonstrated significantly greater phosphorylation of both Akt and Erk1/2 when compared with control hearts (Figure 4). However, murine hearts lacking CYPD failed to demonstrate a significant increase in Akt or Erk1/2 phosphorylation in response to the IPC stimulus, indicating that IPC-induced kinase phosphorylation is dependent on CYPD (Figure 4).
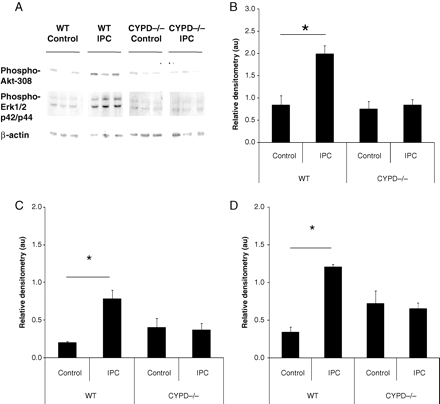
(A) Representative western blot and (B–D) relative densitometries demonstrating that ischaemic preconditioning (IPC) induced the significant phosphorylation of both Akt and Erk1/2 (p42/p44) (normalized to β-actin and measured in arbitrary units, au) in wild-type (WT) but not CYPD−/− (CYPD) adult murine hearts. n = 4 animals/treatment group. *P < 0.05.
3.5 Hypoxic preconditioning requires both CYPD and ROS to phosphorylate Akt
A standard HPC protocol was found to induce significant Akt phosphorylation in WT cardiomyocytes (in arbitrary units: 1.60 ± 0.44 with HPC vs. 0.42 ± 0.15 in control; P < 0.05) (Figure 5). This effect on Akt phosphorylation was abrogated by the ROS scavenger, MPG (1.60 ± 0.44 with HPC vs. 0.50 ± 0.18 with HPC+MPG; P < 0.05) (Figure 5) and the CYPD inhibitor, CsA (1.60 ± 0.44 with HPC vs. 0.65 ± 0.44 with HPC+CsA; P < 0.05) (Figure 5), indicating that HPC-induced phosphorylation of Akt is dependent on both CYPD and ROS. MPG or CsA when given alone had no significant effect on Akt phosphorylation (0.73 ± 0.45 with MPG and 0.41 ± 0.27 with CsA vs. 0.42 ± 0.15 in control; P > 0.05) (Figure 5).
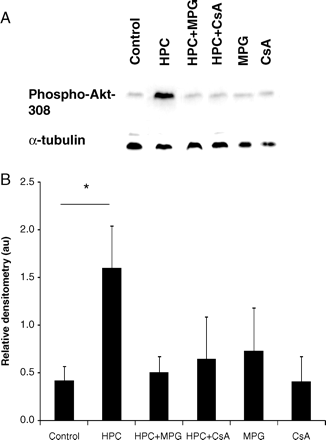
(A) Representative western blots and (B) relative densitometries demonstrating that hypoxic preconditioning (HPC) induced the significant phosphorylation of Akt (normalized to α-tubulin and measured in arbitrary units, au) in wild-type (WT) cardiomyocytes but not in CYPD−/− cardiomyocytes. This effect was abrogated in the presence of ciclosporin-A (CsA, a CYPD inhibitor) and a ROS scavenger, N-2-mercaptopropionyl glycine (MPG, 100 µM). n = 4 animals/treatment group. *P < 0.05.
4. Discussion
The salient findings of the current study are as follows: (i) non-pathological opening of the mPTP occurs under basal conditions and this form of mPTP opening is augmented by hypoxic preconditioning (HPC). This form of mPTP opening occurs to a lesser extent in adult murine cardiomyocytes lacking CYPD; (ii) HPC stimulates mitochondrial ROS production in WT cardiomyocytes but not in those lacking CYPD, suggesting that the generation of ROS is dependent on the CYPD component of the mPTP; (iii) HPC reduces cell death following simulated ischaemia–reperfusion injury in WT cardiomyocytes but not in those lacking CYPD, suggesting that CypD is the likely effector of HPC. (iv) HPC induces Akt phosphorylation in WT cardiomyocytes in a manner which is sensitive to CsA and MPG, suggesting that both CYPD and ROS are required for HPC-induced Akt phosphorylation; and (v) in vivo IPC of the adult murine heart resulted in significant phosphorylation of Akt and Erk1/2 in WT hearts but not in those lacking CYPD, suggesting that the CYP-D component of the mPTP is required for IPC-induced Akt and Erk1/2 phosphorylation. These findings are summarized in a hypothetical scheme in Figure 6.
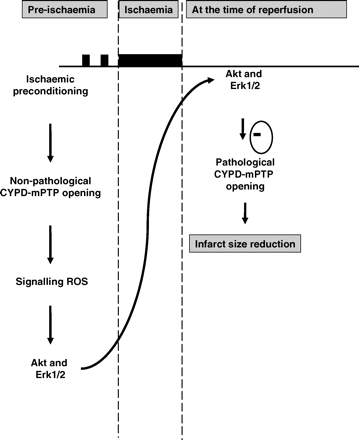
Hypothetical scheme depicting the potential role for non-pathological cyclophilin-D (CYPD) dependent mPTP opening as a mediator of ischaemic preconditioning (IPC) cardioprotection. We postulate that CYPD is required for the mitochondrial release of signalling reactive oxygen species (ROS) into the cytosol in response to an IPC stimulus. The cytosolic signalling ROS then phosphorylate and activate critical pro-survival kinases such as Akt and Erk1/2 which then mediate the inhibition of the pathological form of CYPD-dependent mPTP opening at the onset of myocardial reperfusion.
In the current study, we took advantage of mice lacking the CYPD component of the mPTP to demonstrate that the mPTP is required as a mediator of IPC protection, the generation of mitochondrial ROS and the subsequent activation of the pro-survival kinases Akt and Erk1/2.3–5,20 CYPD is a major regulatory component of the mPTP, and studies have found that its genetic ablation results in resistance to calcium-induced mPTP opening,3,4 a smaller myocardial infarct size,3,4 and resistance to the infarct-limiting effects of IPC.20 Using a well-established model for detecting mPTP opening in adult ventricular cardiomyocytes,15,18,19,22,23 we have demonstrated that under basal conditions, mPTP opening occurs without causing cell death and that a standard HPC protocol can actually augment mPTP opening. Crucially, we have shown that in cardiomyocytes lacking CYPD, this non-pathological mPTP opening is markedly reduced, providing confirmatory evidence that the observed reduction in mitochondrial calcein fluorescence actually represents mPTP opening. Petronilli et al.15 were the first to use this cardiomyocyte model for detecting mPTP opening and they too had observed non-pathological mPTP opening (termed ‘transient’) occurring in adult rat cardiomyocytes under basal conditions as indicated by the CsA-sensitive loss of mitochondrial calcein fluorescence.
In the setting of myocardial ischaemia–reperfusion injury, the prevailing paradigm is that the irreversible pathological opening of the mPTP at the onset of myocardial reperfusion is a critical mediator of cardiomyocyte death, and preventing its opening at this time can dramatically reduce myocardial infarct size.1,2,24 Whether the mPTP plays any role outside the setting of lethal ischaemia–reperfusion injury has been controversial, although accumulating data support the existence of reversible non-pathological mPTP opening occurring under basal conditions. A number of experimental studies have documented ‘flickering’ or ‘oscillations’ in mitochondrial membrane potential that are associated with the production of ROS in isolated mitochondria and cardiomyocytes, a phenomenon which has been attributed to reversible opening of the mPTP under basal conditions.25–28 In contrast, other studies have implicated the opening of other mitochondrial channels as an explanation of this phenomenon.29
A recent experimental study has demonstrated using a variety of cell types including adult rat cardiomyocytes that stochastic superoxide ‘flashes’ occur in individual mitochondria under basal conditions, and that these are associated with mitochondrial membrane depolarization and mitochondrial calcium efflux.17 Crucially, these superoxide flashes were found to be reduced both in the presence of pharmacological inhibitors of mPTP opening (CsA and bongkrekic acid), and in the presence of siRNA to CYPD,3,4 suggesting that these superoxide flashes were the result of non-pathological mPTP opening occurring under basal conditions. Furthermore, in cardiomyocytes subjected to simulated ischaemia–reperfusion injury, widespread mitochondrial superoxide production was associated with the typical pathological mPTP opening that occurs at the onset of reperfusion.17
The significance of this non-pathological form of mPTP opening is unclear, but in the current study we report that it is required to generate ROS, which in turn are needed to activate critical mediators of IPC-induced cardioprotection. In a previous study, we have reported that the pharmacological inhibition of mPTP opening using CsA during a IPC or pharmacological preconditioning protocol abolished the infarct-limiting effects of IPC, suggesting that some form of non-pathological mPTP opening was required during the preconditioning protocol to elicit cardioprotection.18 In a more recent study, Saotome et al.19 have demonstrated that preconditioning with hydrogen peroxide could also be blocked by CsA, suggesting that preconditioning with this agent may be upstream of the CYPD-regulated mPTP. Whether hydrogen peroxide is an upstream activator of the mPTP in the setting of IPC is unknown.
In the present study, we demonstrate for the first time that a standard HPC protocol augments this form of non-pathological mPTP opening, a finding which is compatible with previous studies that have reported mPTP opening in response to the preconditioning mimetic, diazoxide.18,23 It has been established that in response to an IPC stimulus, mitochondria generate ROS and that this signalling ROS is required to mediate IPC protection, most probably through the activation of pro-survival protein kinases such as PKC, PI3K-Akt and MEK1/2-Erk1/2, although this has not previously been shown directly.11 However, the mechanism for the generation of this mitochondrial ROS is unresolved and has been attributed by some studies to the inhibition of mitochondrial respiration and in others to the opening of the ATP-dependent mitochondrial potassium channel (mitoKATP channel).30 In the current study, we provide evidence that the ROS generated by HPC, and observed in WT adult murine ventricular cardiomyocytes, is dependent on the CYPD component of the mPTP, given that cardiomyocytes lacking CYPD failed to generate mitochondrial ROS in response to HPC. Furthermore, the phosphorylation of Akt by ROS in response to preconditioning was also abrogated by CsA, suggesting that CYPD was upstream of both ROS production and kinase phosphorylation. Whether the mitoKATP channel acts in conjunction with CypD in generating ROS in response to the preconditioning stimulus prior to ischaemia is not clear. However, at myocardial reperfusion and in the context of inhibiting detrimental lethal mPTP opening, recent studies by Garlid and co-workers31 have linked the opening of the mitoKATP channel to the mPTP inhibition.
The mechanism through which non-pathological mPTP opening generates ROS is unclear, although a ROS-induced ROS release coupled with respiratory chain inhibition has been proposed.17,28 In this model, any form of mitochondrial stress, such as that induced by an IPC stimulus, results in partial inhibition of the electron transport chain, which then generates ROS within the mitochondria, triggering non-pathological mPTP opening and resulting in partial mitochondrial membrane depolarization and further inhibition of the electron transport chain and so the cycle continues. One can surmise that under basal conditions, the naturally occurring ROS-scavenging system keeps the system in check. However, in response to an IPC stimulus, the ROS generated by the mitochondria overwhelms the ROS-scavenging capabilities of the mitochondria, the result of which is ROS entering the cytosol where it activates important mediatory kinases implicated in IPC protection.
In this respect, it has been established that the activation of certain pro-survival kinases such as Akt and Erk1/2 in response to the IPC protocol is critical to protection.32 The pharmacological inhibition of the upstream protein kinases PI3K or MEK1/2 has been reported to abolish the infarct-limiting effect of IPC, and a recent experimental study has confirmed that mice lacking PI3K-γ are resistant to IPC protection,33 confirming the critical role of the PI3K-Akt pathway as a mediatory protein kinase cascade in IPC. Interestingly, in the current study, we have shown that IPC did not significantly activate either Akt or Erk1/2 in murine hearts lacking CYPD in vivo, suggesting that some form of mPTP opening is required to activate these mediatory kinases in the setting of IPC. We would speculate that the non-pathological form of mPTP opening which is accentuated by HPC is required to generate sufficient mitochondrial ROS which then enters the cytosol and activates these critical protein kinases. In the current study, we have directly demonstrated that the Akt phosphorylation induced by HPC was dependent on the presence of CYPD and was mediated by ROS. A previous experimental study has reported that the preconditioning mimetic diazoxide results in the activation of Erk1/2 through the mitochondrial generation of ROS, but in this study mPTP opening was not investigated.34 Therefore, the inability to generate ROS in response to a preconditioning stimulus and the lack of Akt and Erk1/2 phosphorylation with preconditioning may explain the lack of a cardioprotective response to IPC in mice lacking a CYPD.20
In summary, we report that the mPTP can reversibly open in adult cardiomyocytes under basal conditions, and that this form of mPTP opening is augmented by hypoxic preconditioning. Crucially, the CYPD component of the mPTP is critical to IPC protection as it is required to generate mitochondrial ROS, which in turn activates the important mediatory protein kinases, Akt, and Erk1/2.
Funding
This work was supported by the British Heart Foundation RG/03/007. S.M.D. is funded by the Medical Research Council (EAA17568) and S.Y.L. is funded by the Wellcome Trust (WT081285). Funding to pay the Open Access publication charges was provided by the Wellcome Trust. This work was undertaken at University College London Hospital/University College London (UCLH/UCL) who received a proportion of funding from the Department of Health's National Institutes of Health Research (NIHR) Biomedical Research Centres funding scheme.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}