





Abstract
Objective: Hypertrophy of cardiomyocytes may play an important role in the pathogenesis of cardiac hypertrophy associated with various cardiovascular diseases such as congestive heart failure. The aim of this study was to investigate whether vasopressin (AVP) induces protein synthesis in cultured neonatal rat cardiomyocytes through its specific receptor and whether YM087, a newly synthesized nonpeptide AVP receptor antagonist, inhibits AVP-induced protein synthesis in vitro. Methods: AVP receptors on cardiomyocytes were characterized using the radioligand [3H]AVP. The effects of AVP and YM087 on intracellular free calcium concentration ([Ca2+]i), mitogen-activated protein (MAP) kinase and [3H]-leucine incorporation were investigated in cultured neonatal rat cardiomyocytes. Results: In cardiomyocytes, Scatchard analysis showed a single population of high-affinity binding sites with the expected AVP V1A receptor subtype profile. YM087 showed high affinity for cardiomyocyte V1A receptors with a Ki value of 0.63 nM. In these same cells, YM087 potently inhibited AVP-induced increases in [Ca2+]i and activation of MAP kinase in a concentration-dependent manner. In addition, AVP concentration-dependently stimulated the synthesis of protein without changing the rate of DNA synthesis, and YM087 prevented AVP-induced protein synthesis in a concentration-dependent manner. Conclusions: These results suggest that AVP directly causes protein synthesis and YM087 is a potent inhibitor of AVP-induced protein synthesis of cardiomyocytes and thus may have beneficial effects in the development and regression of cardiomyocytic hypertrophy.
Time for primary review 22 days.
1 Introduction
Many studies have shown the importance of arginine vasopressin (AVP) in both the regulation of vascular tone by induction of vascular smooth muscle cell contraction and the pathogenesis of hypertension [1, 2]. Despite its pathogenic role, the vascular action of AVP has been studied extensively, while little is known concerning the effect of this hormone on the cardiomyocytes. Several studies have demonstrated the presence of receptors for vasoactive peptides such as angiotensin II and endothelin-1 in cardiomyocytes [3, 4]. Recently, binding and cellular calcium mobilization studies indicated that cardiomyocytes possessed AVP receptors (V1A subtype), the activation of which led to intracellular Ca2+ mobilization [5]. The intracellular mechanism of AVP via V1A receptors is coupled through a pertussis toxin-insensitive G protein to phosphatidylinositol-specific phospholipase C (PLC). PLC-mediated hydrolysis of polyphosphoinositides generates two second messengers, inositol 1,4,5-triphosphate, which induces an increase of free intracellular Ca2+ ([Ca2+]i) via a release of Ca2+ from the endoplasmic reticulum, and diacylglycerol, which activates protein kinase C [6, 7].
AVP produces a positive inotropic effect at lower concentrations and negative inotropic action at higher concentrations in perfused isolated rat heart in which coronary artery flow was kept constant [8]. This biphasic effect of AVP was abolished by the inclusion of a V1A receptor antagonist. In addition, AVP stimulated the release of prostacyclin and atrial natriuretic peptide in cultured neonatal rat cardiomyocytes [7]. However, the pathophysiologic role of AVP in cardiomyocytes, especially its possible involvement in cardiac hypertrophy, remains open to question. A number of peptide AVP receptor antagonists such as d(CH2)5Tyr(Me)AVP, a specific V1A receptor antagonist, have been designed [9, 10]. However, their lack of oral bioavailability and their short biological half-life have severely limited their clinical and therapeutical applications. Recently, orally effective nonpeptide V1A receptor specific antagonists, OPC-21268 [11, 12]and SR 49059 [13], and V2 receptor specific antagonists, OPC-31260 [14], SR 121463A [15]and WAY-VPA-985 [16], have become available. Using these antagonists, it is possible to examine the pure V1A and V2 receptor-mediated physiologic actions of AVP in several target tissues such as vascular smooth muscle cells, Swiss 3T3 fibroblasts, cardiac myocytes and renal cortical collecting duct [17–22]. More recently, a nonpeptide V1A/V2 receptor antagonist, YM087 which potently and totally blocked the physiologic and pharmacologic effects of AVP was identified [23, 24].
In the present study the ability of AVP, acting through its specific receptor, to stimulate protein synthesis in cultured neonatal rat cardiomyocytes was examined. The experiments were repeated in the presence of YM087. Finally, as a comparison, the activity of OPC-21268 and OPC-31360 were compared in all assays used.
2 Methods
This investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1985) and the regulations of the Animal Ethical Committee of Yamanouchi Pharmaceutical Co., Ltd.
2.1 Materials
[3H]AVP and [3H]leucine with a specific activity of 80 and 145 Ci/mmol, respectively, were obtained from DuPont-New England Nuclear (Boston, MA). [γ-32P]ATP and [3H]thymidine with a specific activity of 2000 and 83 Ci/mmol, respectively, were obtained from Amersham International (Buckinghamshire, England). AVP and oxytocin were obtained from Peptide Institute Inc., (Osaka, Japan) and d(CH2)5Tyr(Me)AVP (SKF-100273) was from Sigma Chemical Co. (St. Louis, MO). YM087, OPC-21268 and OPC-31260 were synthesized at Yamanouchi Pharmaceutical Co. (Ibaraki, Japan). These non-peptide antagonists were initially dissolved in dimethyl sulfoxide (DMSO) at 10−2 M and diluted to the desired concentration with the assay buffer. The final concentration of DMSO in the assay buffer did not exceed 0.1%, at which specific [3H]AVP binding and [3H]leucine/[3H]thymidine incorporation were not affected. Fura 2-AM was obtained from Dojindo Laboratories (Kumamoto, Japan) and EGTA and ionomycin were from Wako Pure Chemicals (Osaka, Japan). Myelin basic protein (MBP) from bovine brain was obtained from Sigma Chemical Co. (St. Louis, MO). Eagle's minimum essential medium (EMEM), phosphate-buffered saline (PBS) and fetal calf serum (FCS) were from Gibco (Grand Island, NY). Bovine serum albumin (BSA) type V was purchased from Nacalai Tesque Inc. (Kyoto, Japan). Reagents for protein assay were purchased from Bio-Rad Laboratories (Richmond, CA).
2.2 Cell culture
Primary cultures of neonatal rat cardiomyocytes were prepared as originally described by Simpson and Savion with minor modifications [25]. Briefly, 50–100 newborn (2–3 day old) Wistar rat pups were anesthetized with ether in batches of five at a time. Under aseptic conditions, the lower (apical) two-thirds of each heart was excised from decapitated rats and placed in a small Petri dish containing calcium and bicarbonate-free Hanks solution buffered with HEPES (CBFHH) containing 100 IU/ml penicillin and 10 μg/ml streptomycin. The tissues were washed with CBFHH, cut into 1 mm pieces and enzymatically digested for 15 min with 10 ml of 0.1% collagenase type I (Sigma Chemical Co.) at 37°C in a 50 ml conical tube subjected to constant stirring. The supernatant from the first incubation was discarded, 7 ml of fresh enzyme solution was added and the incubation procedure was sequentially repeated until the tissue was totally digested. Starting from the second incubation, supernatants were collected into EMEM containing 20% FCS and centrifuged at 175 g for 10 min. The resulting cell pellets were resuspended and transferred to a 100 mm culture dish (1–2×106 cells/dish) and allowed to remain for 60 min at 37°C in 5% CO2. The fibroblast and endothelial cell culture contaminants adhered to the surface, whereas the cardiomyocytes remained in the supernatant. Nonattached viable cells were collected and seeded into 12-well culture plates (2×105 cells/well). Cardiomyocytes were incubated in EMEM supplemented with 5% FCS for 48 h and then replaced with serum-free medium 24 h before each experiment. In all experiments, cultures of almost pure cardiomyocytes were prepared by use of cytosine arabinoside (10−5 M) during the first 24 to 36 h which preferentially reduces the proportion of non-myocytes as previously described [26].
2.3 Radioligand binding studies
The binding of [3H]AVP was studied in confluent cells grown in 12-well culture plates. The assay buffer consisted of 10 mM HEPES (pH 7.4), 125 mM NaCl, 2.6 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 1 mM CaCl2, 5 mM glucose and 0.1% BSA. Incubation was performed at 37°C for 30 min in 250 μl assay buffer containing [3H]AVP with or without various concentrations of the unlabeled compounds. The reaction was terminated by removal of the assay buffer and three rapid washings with 2 ml ice-cold assay buffer free of BSA. Nonspecific binding was determined in the presence of an excess of AVP (1 μM). After solubilization of the monolayer in 0.5 N NaOH, the radioactivity associated with the cells was estimated by a Packard Tricarb scintillation counter (Packard Instrument Co., Inc., Meriden, CT). The concentration of test compounds that caused 50% inhibition (IC50) of the specific binding of [3H]AVP was determined by regression analysis of displacement curves. The inhibitory constant (Ki) was calculated from the following formula: Ki=IC50/(1+[C]/Kd), where [C] is the concentration of radioligand present in the wells and Kd is the dissociation constant of radioligand obtained from the Scatchard plot [27]. Data were analyzed using GraphPad PRISM software (GraphPAD Software, Inc., San Diego). Protein content was determined by the Coomassie blue method, with BSA as a standard.
2.4 Measurement of [Ca2+]i
Serum-deprived monolayer cultures of cardiomyocytes were grown on coverglasses (13.5 mm diameter) and were assayed one day later. Cell monolayers (2×105 cells/cm2) were loaded in EMEM with fura 2-AM (2 μM per coverglass) for 30 min at 37°C and washed, then transferred to fura 2-free medium and incubated an additional 30 min at 37°C. The loaded monolayers were stored in Krebs–Henseleit–HEPES buffer (containing 130 mM NaCl, 5 mM KCl, 1.25 mM CaCl2, 0.8 mM MgSO4, 5.5 mM glucose, 20 mM HEPES, and 0.1% BSA, pH 7.4). The coverglass was placed into a quartz cuvette containing 2 ml Krebs–Henseleit–HEPES buffer and maintained at 37°C with continuous stirring. When thermal equilibrium was reached, the fluorescence signal was recorded with a CAF-110 spectrofluorometer (Japan Spectrometer Co., Tokyo, Japan) with 340/380 nm excitation and 500 nm emission wavelengths. After recording the baseline signal for 3 min, AVP was added to the cuvette in the presence or absence of antagonists to stimulate the mobilization of intracellular calcium. Fluorescence measurements were converted to [Ca2+]i by determining maximal fluorescence (Rmax) with the nonfluorescent Ca2+ ionophore, ionomycin (25 μM), after which minimal fluorescence (Rmin) was obtained by adding of 3 mM EGTA. From the ratio of fluorescence at 340 and 380 nm, the [Ca2+]i was determined using the following equation: [Ca2+]i(nM)=Kd×[(R−Rmin)/(Rmax−R)]×b. The term b is the ratio of fluorescence of fura 2 at 380 nm in zero and saturating Ca2+. Kd is the dissociation constant of fura 2 for Ca2+, assumed to be 224 nM [28]. Since background and cell autofluorescence were usually about 20 times less than that measured from fura 2 loaded cardiomyocytes, they were considered negligible and no correction was made in the calculation of [Ca2+]i.
2.5 Mitogen-activated protein kinase activity assay
Cardiomyocytes were seeded into 24-well culture plates at confluency and serum starved for 24 h. Cells were then incubated in EMEM with 0.1% BSA containing the vehicle alone or various concentrations of AVP and/or antagonists for 5 min at 37°C. The assay was stopped by aspirating off the medium and cells were washed with ice-cold PBS and lysed by the addition of lysis buffer (10 mM Tris-HCl, 50 mM β-glycerophosphate, 0.1 mM Na3VO4, 2.0 mM MgCl2, 1.0 mM EGTA, 1.0 mM dithiothreitol, 0.02 mM pepstatin, 0.02 mM leupeptin, 1.0 mM PMSF, 0.5% Triton X-100 and 1000 units/ml aprotinin, pH 7.4). Lysed cells were centrifuged for 10 min at 12 000 g and the pellets were dissolved in 0.1 N NaOH solution containing 1% sodium dodecylsulfate for protein assay. Mitogen-activated protein (MAP) kinase activity was measured using MBP and [γ-32P]ATP as substrates. The assay was performed at 30°C for 10 min in a final volume of 30 μl containing 75 mM β-glycerophosphate, 2 mM dithiothreitol, 6 mM MgCl2, 1 mg/ml MBP and 50 μM [γ-32P]ATP (1 μCi/ml). Reactions were terminated by the addition of 10 μl of 25% trichloroacetic acid. The reaction products were placed on P-81 phospho-cellulose papers (Whatman International Ltd., Maidstone, England) and washed with 1% acetic acid three times. The radioactivity was counted using a liquid scintillation counter and MAP kinase activity was expressed as picomoles of  per minute per milligram of protein.
per minute per milligram of protein.
2.6 Incorporation of [3H]leucine and [3H]thymidine
Cardiomyocytes were seeded into 12-well culture plates and washed with PBS and incubated serum-free EMEM for 24 h. The cultures were then incubated in EMEM with 0.5% FCS and 0.1% BSA containing vehicle alone or various concentrations of AVP and/or antagonists for 24 h and then for 24 h in the same medium containing 2 μCi/ml [3H]leucine or [3H]thymidine. At the end of labeling, cultures were rinsed three times with ice-cold PBS, and incubated with 5% TCA on ice for 20 min. After the cells were washed twice with ice-cold 5% TCA, they were solubilized in 0.5 N NaOH. An aliquot of TCA-insoluble materials was neutralized and radioactivity was determined by a liquid scintillation counter.
2.7 Statistical analysis
Experimental results are expressed as the mean±standard error of the mean (S.E.M.) or the mean with 95% confidence limits. The EC50 and IC50 values were estimated from the concentration–response curves by a nonlinear regression program GraphPad PRISM. Statistical analysis of the data was performed by ANOVA followed by Dunnett's multiple range test.
3 Results
3.1 AVP receptor binding assay
Saturation experiments of increasing concentrations of [3H]AVP binding to cardiomyocytes showed that specific binding was saturable. Scatchard analysis of these data gave linear plots consistent with the presence of a single class of high affinity and low capacity binding site (Fig. 1). The apparent equilibrium dissociation constant (Kd) and the maximal binding capacity (Bmax) were 1.60±0.51 nM and 47.0±8.36 fmol/mg protein, respectively. AVP and several AVP receptor antagonists were tested for their ability to compete for specific [3H]AVP binding (Fig. 2). YM087 inhibited specific binding of [3H]AVP in a concentration-dependent manner with Ki values of 0.63±0.27 nM (Table 1). The Hill coefficients (nH) of YM087 were close to unity, compatible with a single-site competitive model. The affinity of YM087 for V1A receptors was comparable to that of d(CH2)5Tyr(Me)AVP and approximately 20 times higher than that of OPC-21268 with Ki values of 0.54±0.09 nM and 13.0±4.18 nM, respectively. In contrast, OPC-31260 exhibited much lower affinity for V1A receptors with Ki value of 1440±289 nM.
![Displacement of the specific binding of [3H]AVP to cultured neonatal rat cardiomyocytes. Unlabeled compounds added to the binding assay are as follows: ▴, AVP; ○, YM087; □, OPC-21268; ♦, OPC-31260. Specific binding of [3H]AVP is expressed as the percentage of the control binding. Results are representative data from 3–5 experiments performed in duplicate. The combined results of all experiments are summarized in Table 1.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/38/1/10.1016_S0008-6363%2897%2900324-6/1/m_38-1-198-fig2.gif?Expires=1750429472&Signature=or6r7AkHA6AYETkY73sDVusG3Pny6jJujSqT6y6Imarji~eZ1AIDO0ncWgo4w4V~JWHTDSb47bM4J5HXXMB0mO53h8bDZkMCSSR-BfliNQZRdyH~lKQdBZFIFHIlFkhDu3iAtvk4AbnTfMgKPEPjjh3QB6K-x~1eHWb81EIVlMshS1xsYENWcTbBR2z~afn4Bbdc9ZwzfZn7ws-vJC6s7Xrn~Y-O0~rLa4roxyk9oOERWGOBBCngE2O0RQ1qRhNbU1X-wj~NGC4-oTiiAVfxi4-dyBTxmEHMOEZxSBkb7tpmapmL1i1pnRnzN4iswe5h0UQRlTuvoao9gWIrJ3S8Dw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Displacement of the specific binding of [3H]AVP to cultured neonatal rat cardiomyocytes. Unlabeled compounds added to the binding assay are as follows: ▴, AVP; ○, YM087; □, OPC-21268; ♦, OPC-31260. Specific binding of [3H]AVP is expressed as the percentage of the control binding. Results are representative data from 3–5 experiments performed in duplicate. The combined results of all experiments are summarized in Table 1.
![Binding of [3H]AVP to neonatal rat cardiomyocytes. (A) Saturation equilibrium binding of [3H]AVP to cardiomyocytes. Specific binding (○) was taken as the difference between binding in the absence (■) and in the presence (▴) of 1 μM unlabeled AVP. (B) Scatchard linear transformation of the data. Saturation experiments of [3H]AVP binding and its Scatchard plots were performed as described in Section 2. Each point represents the mean of duplicate determinations and is representative of 4 independent experiments.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/38/1/10.1016_S0008-6363%2897%2900324-6/1/m_38-1-198-fig1.gif?Expires=1750429472&Signature=WvBUc4AQMM5zx7O6U2vZQGedIH6hsjrtGXoxoADFL6a9~ElLeYRQPu5y8zzwXD7sXLL50rZGe7uA~UFlOIsIYobkdV7OJm91e08Z8-E-PQTqghbUfaZsJJQHpHb6K6k-ftMMg5vWnOrAjrX7~vwNTGST-00AQNn9XYWNmqUjPveou4w61mVMDalyDRT8DVzhMcREfT0L3ME1-IIObcd9xIH5zA5iQLLsGKxQsUmIrCk~Hnlaaog2WxFhz4hataDYtzlhJj0WftSHxrDQu7XMk9~itNBsMvXvZGUwMZLN54kAnyddXzD3lKY-NTpyCp2l4sgM6C1hph0vK9L6W7c1fA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Binding of [3H]AVP to neonatal rat cardiomyocytes. (A) Saturation equilibrium binding of [3H]AVP to cardiomyocytes. Specific binding (○) was taken as the difference between binding in the absence (■) and in the presence (▴) of 1 μM unlabeled AVP. (B) Scatchard linear transformation of the data. Saturation experiments of [3H]AVP binding and its Scatchard plots were performed as described in Section 2. Each point represents the mean of duplicate determinations and is representative of 4 independent experiments.
Affinity of YM087 and reference compounds for AVP receptors in cultured neonatal rat cardiomyocytes
| Ki (nM) | |
|---|---|
| AVP | 2.03±0.86 |
| oxytocin | 97.5±59.1 |
| d(CH2)5Tyr(Me)AVP | 0.54±0.09 |
| YM087 | 0.63±0.27 |
| OPC-21268 | 13.0±4.18 |
| OPC-31260 | 1440±289 |
| Ki (nM) | |
|---|---|
| AVP | 2.03±0.86 |
| oxytocin | 97.5±59.1 |
| d(CH2)5Tyr(Me)AVP | 0.54±0.09 |
| YM087 | 0.63±0.27 |
| OPC-21268 | 13.0±4.18 |
| OPC-31260 | 1440±289 |
Binding assays were performed as described in Section 2.
Each value is mean±S.E.M. of 3–5 independent experiments.
Affinity of YM087 and reference compounds for AVP receptors in cultured neonatal rat cardiomyocytes
| Ki (nM) | |
|---|---|
| AVP | 2.03±0.86 |
| oxytocin | 97.5±59.1 |
| d(CH2)5Tyr(Me)AVP | 0.54±0.09 |
| YM087 | 0.63±0.27 |
| OPC-21268 | 13.0±4.18 |
| OPC-31260 | 1440±289 |
| Ki (nM) | |
|---|---|
| AVP | 2.03±0.86 |
| oxytocin | 97.5±59.1 |
| d(CH2)5Tyr(Me)AVP | 0.54±0.09 |
| YM087 | 0.63±0.27 |
| OPC-21268 | 13.0±4.18 |
| OPC-31260 | 1440±289 |
Binding assays were performed as described in Section 2.
Each value is mean±S.E.M. of 3–5 independent experiments.
3.2 Effect of YM087 on AVP-induced increase in [Ca2+]i
The basal [Ca2+]i in cardiomyocytes was 108±3.83 nM. AVP induced an increase in [Ca2+]i in a concentration-dependent manner with an EC50 value of 1.64 (0.99–2.71) nM (Fig. 3A). When the effect of various antagonists was studied in preparations stimulated with 100 nM AVP, which induced intensive but still submaximal stimulation, pretreatment of the cardiomyocytes with YM087 caused a potent inhibition of the AVP-induced increase of [Ca2+]i with an IC50 value of 7.58 (4.50–12.8) nM (Fig. 3B). When tested under the same experimental conditions, d(CH2)5Tyr(Me)AVP also potently inhibited the AVP-induced [Ca2+]i increase with an IC50 value of 14.0 (11.4–17.2) nM. The inhibitory potency of OPC-21268 and OPC-31260 were 110 and 1800 times lower than that of YM087, and this potency profile was comparable to its binding affinity. Furthermore, all antagonists including YM087 did not change the basal value of [Ca2+]i when tested alone (up to 1 μM) (data not shown).
![Effect of YM087 on AVP-induced [Ca2+]i increase in neonatal rat cardiomyocytes. (A) AVP-stimulated [Ca2+]i increase in cardiomyocytes. (B) Inhibitory effect of YM087 and reference compounds on the response to a submaximal concentration of AVP (100 nM). Cells were preincubated with the indicated concentrations of compounds for 1 min. Values are mean±S.E.M. of 3–9 independent experiments. *Significant difference from cells treated with vehicle or 100 nM AVP (p<0.05).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/38/1/10.1016_S0008-6363%2897%2900324-6/1/m_38-1-198-fig3.gif?Expires=1750429472&Signature=nJxOo4gXNQVBJHpbZrFdo49t1EY46GZu9pCApBZwVWRwJBACorPqhtkcmgiTXNwOlVfxziR6UHzfoAULwD~kbi4kh4h-M-vEhhc94IH3k8xHUC663s7DgB3sCLDRg6dx2WkGtgOnPx5ZI7hdg95yHZLdWeCM~2muQ0tux56jmMTA1PpIjOqLcwp35LZ46bgwn~JeXmIBX8GwDTRfV9Y3e067u2o6wHbJPWiU35I7IHpC28B8JzQMMT5QpzI~gqcXPLDAzv4sLfXG88IBngCphlU8O8hl8qyV1fcV0bFJIBJz8NU-RWQ~usJod7wQHBaKcLxhDF-XKuUpMOx2KwvpOg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of YM087 on AVP-induced [Ca2+]i increase in neonatal rat cardiomyocytes. (A) AVP-stimulated [Ca2+]i increase in cardiomyocytes. (B) Inhibitory effect of YM087 and reference compounds on the response to a submaximal concentration of AVP (100 nM). Cells were preincubated with the indicated concentrations of compounds for 1 min. Values are mean±S.E.M. of 3–9 independent experiments. *Significant difference from cells treated with vehicle or 100 nM AVP (p<0.05).
3.3 Effect of YM087 on AVP-induced MAP kinase activation
In response to AVP, MAP kinase activity reached peak level 3–5 min after the addition of AVP (100 nM). This was followed by a gradual decrease in MAP kinase activity. However, during the 30 min observation period, MAP kinase activity level remained above the basal level (Fig. 4). The following studies were therefore performed after incubation for 5 min. AVP induced activation of MAP kinase with maximal increase to 214% of the control value and an EC50 value of 1.13 (0.35–3.59) nM (Fig. 5A). YM087 concentration-dependently inhibited 100 nM AVP-induced activation of MAP kinase with an IC50 value of 4.12 (0.65–26.2) nM (Fig. 5B), but had no agonistic activity up to 1 μM (data not shown). When tested under the same experimental conditions, d(CH2)5Tyr(Me)AVP also potently inhibited AVP-induced activation of MAP kinase with an IC50 value of 0.76 (0.42–1.40) nM. The inhibitory potency of OPC-21268 and OPC-31260 were 96 and 330 times lower than that of YM087.
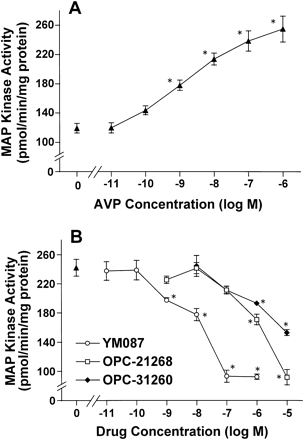
Effect of YM087 on AVP-induced MAP kinase activations in neonatal rat cardiomyocytes. Cells were incubated with increasing concentrations of AVP (A) or antagonists and AVP (100 nM) (B) for 5 min. MAP kinase activity was determined as described in the legend to Fig. 4. Values are mean±S.E.M. of 4 independent experiments. *Significant difference from cells treated with vehicle or 100 nM of AVP (p<0.05).
![The time course of stimulatory effects of AVP on MAP kinase activity of neonatal rat cardiomyocytes. Cells were serum starved for 24 h and stimulated by the addition of vehicle (△) or 100 nM AVP (▴) in medium with 0.1% BSA. The reaction was stopped by washing cells with ice-cold phosphate-buffered saline at the indicated time, and cells were lysed for the MAP kinase assay. The activity of MAP kinase was assessed by the incorporation of into MBP from [γ-32P]ATP. Values are mean±S.E.M. of 4 independent experiments. *Significant difference from cells treated with vehicle (p<0.05).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/38/1/10.1016_S0008-6363%2897%2900324-6/1/m_38-1-198-fig4.gif?Expires=1750429472&Signature=puIX3ho2jOEUULA43W3gPaOsD2QgGgS1cWUsBSuaGcG67ERjgsrDggcoRZTUOIoNwQtYNzbh6rIv6q7BZJz1Hhwyf08YGrsBp5fYSgja9zPMSkVB7k2Ff7m7geYXk9UT1OTnJuT6qWM4R4XOyaZakQvp5iZHDEUcRmP~K5UnDfNY5yhRXqDvUrJVivF05yCM5Z0EOepe6g2ezuC5thgNMwTdhR~kel0rBdggFhp49sx~CRRvLX2U9EhGXWCmrYnG~RAMq96To6jZTCGagnRDxEcPExP-FboU0vxcAxxaib1gAufcFhmIe2uclIkjxGAsuZNNh7dvwMWLE3WVmzwcvA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The time course of stimulatory effects of AVP on MAP kinase activity of neonatal rat cardiomyocytes. Cells were serum starved for 24 h and stimulated by the addition of vehicle (△) or 100 nM AVP (▴) in medium with 0.1% BSA. The reaction was stopped by washing cells with ice-cold phosphate-buffered saline at the indicated time, and cells were lysed for the MAP kinase assay. The activity of MAP kinase was assessed by the incorporation of  into MBP from [γ-32P]ATP. Values are mean±S.E.M. of 4 independent experiments. *Significant difference from cells treated with vehicle (p<0.05).
into MBP from [γ-32P]ATP. Values are mean±S.E.M. of 4 independent experiments. *Significant difference from cells treated with vehicle (p<0.05).
3.4 AVP causes protein synthesis of cardiomyocytes
We examined the effects of AVP on protein synthesis and the rate of DNA synthesis in cardiomyocytes. AVP produced concentration-dependent (10−11–10−6 M) increases in protein synthesis, as reflected by [3H]leucine incorporation, with an EC50 value of 0.93 (0.23–3.75) nM (Fig. 6A). In contrast, AVP (10−11–10−6 M) did not increase DNA synthesis as measured by [3H]thymidine incorporation (Fig. 6B).
![Effect of AVP on [3H]leucine (A) and [3H]thymidine (B) incorporation in cardiomyocytes. (C) Effect of YM087 on AVP-stimulated incorporation of [3H]leucine into cardiomyocytes. Cells were incubated with the increasing concentrations of AVP or antagonists and AVP (100 nM) in medium with 0.5% FCS and 0.1% BSA for 48 h. [3H]leucine and [3H]thymidine incorporation was determined as described in Section 2. Values are mean±S.E.M. of 4–28 independent experiments. *Significant difference from cells treated with vehicle or 100 nM of AVP (p<0.05).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/38/1/10.1016_S0008-6363%2897%2900324-6/1/m_38-1-198-fig6.gif?Expires=1750429472&Signature=2y5l0YQEHoghjiU0VnRFLxGmzdmyD-M0uAGtVJcTo49GVynIiNgr2pvSqCYy~-3TI1Ugvz5GpgaKt4xns0G6wsE92K2iOCnPpd24yKMNekOZlrrULdOIsgc~ArQdBLSeJp0~CEzB3RQeX~Wcl2UnA-F7ImV2n9OHc7T4CJ9PQGstQKjvWciJqYo9zRdHOFcIjYNngXykVfmeQjUp4gJmd5YJ3dF~-~gc97j9Kfl9T2nAb4tt6IuPKisladKDJkUBo8eTmK1LYJXbjlC~JAFkQnkNIRP2PoLSfyrwiML938qZSgDlI~zDvfiWN55y8Q2RVHkWWln~9OiEN~FgeqYkXg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of AVP on [3H]leucine (A) and [3H]thymidine (B) incorporation in cardiomyocytes. (C) Effect of YM087 on AVP-stimulated incorporation of [3H]leucine into cardiomyocytes. Cells were incubated with the increasing concentrations of AVP or antagonists and AVP (100 nM) in medium with 0.5% FCS and 0.1% BSA for 48 h. [3H]leucine and [3H]thymidine incorporation was determined as described in Section 2. Values are mean±S.E.M. of 4–28 independent experiments. *Significant difference from cells treated with vehicle or 100 nM of AVP (p<0.05).
3.5 Effect of YM087 on AVP-induced protein synthesis
YM087 strongly and concentration-dependently inhibited 100 nM AVP-induced protein synthesis with an IC50 value of 25.4 (7.76–83.1) nM (Fig. 6C). This inhibitor potency of YM087 was 10 times and 116 times higher than that of OPC-21268 (IC50=246.3 nM) and OPC-31260 (IC50=2950 nM), respectively. In contrast, d(CH2)5Tyr(Me)AVP did not exhibit potent and complete inhibition of AVP-induced protein synthesis. This inhibition of AVP-induced protein synthesis required even higher concentrations of d(CH2)5Tyr(Me)AVP than inhibition of [3H]AVP binding, [Ca2+]i increase and MAP kinase activation, with an IC50 value of 810 nM. Each antagonist itself had no effect on protein synthesis up to 1 μM (data not shown).
4 Discussion
The present study has demonstrated that AVP has a direct protein synthetic effect on cultured rat cardiomyocytes with concomitant increase in [Ca2+]i and activation of MAP kinase. It also shows that YM087, a nonpeptide AVP receptor antagonist, has a potent inhibitory effect on the AVP-induced protein synthesis in vitro. [3H]AVP binding to rat cardiomyocytes was saturable and identified a single class of specific, high affinity binding sites with a Kd of 1.60 nM and a Bmax of 47.0 fmol/mg protein, respectively. The high affinity was in agreement with Kd value already observed for rat cardiomyocytes, hepatocytes and vascular V1A receptors [5, 29, 30]. YM087 concentration-dependently inhibited the specific binding of [3H]AVP with a Ki value of 0.63 nM and a Hill coefficient value close to unity. The affinity rank of antagonists indicated that the homogenous population of sites exhibited the expected V1A receptor profile comparable to that identified on rat vascular and non-vascular tissues: d(CH2)5Tyr(Me)AVP ⩾ YM087 > OPC-21268 ≫ OPC-31260 [23, 30]. Furthermore, we observed a good general correlation between affinity of antagonists for cardiomyocytes V1A receptors and inhibition potency of antagonists on AVP-induced physiological responses elicited on [Ca2+]i increase, MAP kinase activation and protein synthesis. Nevertheless, for d(CH2)5Tyr(Me)AVP in particular, a 1500-fold difference was obtained between affinity and inhibition of protein synthesis. It has been reported that d(CH2)5Tyr(Me)AVP needs further preincubation to fully antagonize AVP-induced Ca2+ entry whereas OPC-21268 added simultaneously with AVP was completely efficient on rat cultured mesangial cells [31]. Other arguments involving the short biological half-life of this peptide could also be raised. In protein synthesis experiments, the antagonist was incubated with cells for a longer period of time (48 h) than in binding experiments (only 30 min). Such a difference remains unclear and has to be studied thoroughly. Many of the physiological responses to AVP, including contraction, can be induced by increasing concentration of intracellular Ca2+ and by stimulation of protein kinase C [32]. In the present experiments, AVP increased [Ca2+]i in a concentration-dependent manner and YM087 potently antagonized this increase in [Ca2+]i induced by AVP, with the IC50 value of 7.58 nM. In the absence of AVP, furthermore, YM087 did not increase [Ca2+]i in rat cardiomyocytes, indicating a lack of agonistic activity for V1A receptors. These results suggest that YM087 possesses potent V1A receptor antagonistic activity without agonistic activity.
The cardiomyocytes cultured from neonatal rats are a valuable model system which, following exposure to appropriate agonists including angiotensin II and endothelin-1, exhibits many of the features of the hypertrophic response in the adult heart in vivo [33]. It has been shown that growth of cardiomyocytes is primarily due to hypertrophy rather than to hyperplasia [25]and during the development of cellular hypertrophy, rapid activation of PKC is induced [34]. These findings suggest an important role of PKC in the signal transduction mechanism of cardiac hypertrophy, a hypothesis supported by PKC transfection experiments [35]. However, details of the signaling events downstream from PKC remain poorly defined. Recently, the MAP kinases have been identified as members of a family of serine–threonine kinases [36]that become tyrosine phosphorylated and activated in response to mitogenic growth factors [37]and which probably play a central role in diverse growth-related responses but are also likely to have additional physiological roles [38]. It is well established that AVP induces a rapid activation of PKC and MAP kinases in platelets and in cultured vascular smooth muscle cells [36, 38]. In the present experiments, AVP also induced rapid activation of MAP kinase and YM087 potently inhibited AVP-induced activation of MAP kinase in cardiomyocytes. The ability of V1A antagonists to block AVP-induced MAP kinase activation indicates that AVP is acting through this receptor. Vasoactive peptide hormones are involved in contraction and in the regulation of cellular growth. Among them, AVP is a potent cellular growth factor implicated in a variety of normal and pathophysiological responses in tissue proliferation or regeneration and cellular hypertrophy [39, 40]. In vascular smooth muscle cells, AVP induces hyperplasia and hypertrophy and stimulates the expression of early growth-response genes [32, 38]. The present results showing enhanced protein synthesis by AVP without DNA synthesis also lend strong support to the role of AVP as a hypertrophic factor for cardiomyocytes. On the other hand, YM087 did not affect basal MAP kinase activity and [3H]leucine incorporation, but potently inhibited AVP-stimulated MAP kinase activity and protein synthetic effects. These results indicate that YM087 can prevent AVP-induced cellular hypertrophy via V1A receptors.
Elevated levels of AVP have been reported in experimental models of heart failure and in decompensated heart failure patients [41–43]. Intravenous administration of d(CH2)5DAVP or d(CH2)5Tyr(Me)AVP, the peptide V1A receptor antagonist, has been known to produce hemodynamic improvement with a transient decrease in systemic vascular resistance and an increase in cardiac output in experimental models of heart failure and congestive heart failure patients [44, 45]. From these observations, it can be inferred that AVP could play an important role in the development of cardiac hypertrophy in congestive heart failure. The present experiments provide no information on the hypertrophic effects of AVP or the anti-hypertrophic effects of YM087 on myocardium in vivo; to determine this, additional studies are required to clarify the roles of AVP. From the present results, however, it is clear that AVP directly causes protein synthesis and YM087 is a potent inhibitor of AVP-induced protein synthesis of cardiomyocytes.
In conclusion, AVP caused an increase in [Ca2+]i, activation of MAP kinase and protein synthesis of rat cardiomyocytes through the V1A receptors. YM087 possesses high affinity for V1A receptors on cardiomyocytes and potently inhibited AVP-induced physiological responses. YM087 may have beneficial effects possible by preventing the development and by inducing regression of cardiomyocyte hypertrophy.
Acknowledgements
The authors acknowledge Drs. Toichi Takenaka, Noboru Satoh, Isao Yanagisawa and Gensei Kon (Yamanouchi Pharmaceutical Co., Ltd.) for their valuable comments and continuing encouragement.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}