





Abstract
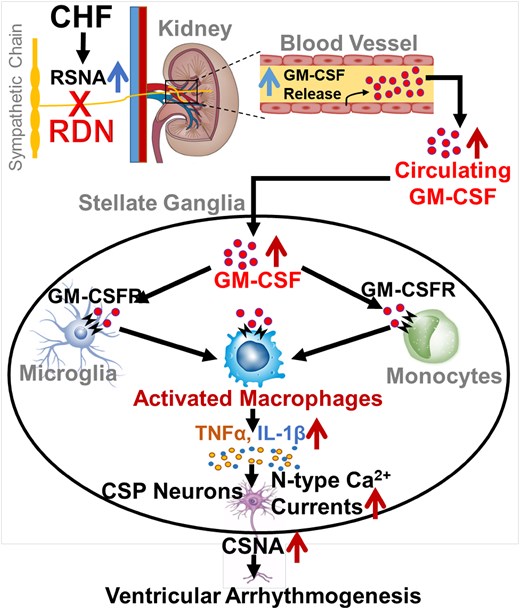
Renal denervation (RDN) is widely investigated in multiple studies of sympathetically driven cardiovascular diseases. While the therapeutic potential of RDN for ventricular arrhythmia has been reported, the mechanisms responsible for its antiarrhythmic effect are poorly understood. Our recent study showed that macrophage expansion-induced neuroinflammation in the stellate ganglion (SG) was a critical factor for cardiac sympathetic overactivation and ventricular arrhythmogenesis in chronic heart failure (CHF). This study investigates if and how RDN decreases ventricular arrhythmias by attenuating neuroinflammation in cardiac sympathetic post-ganglionic (CSP) neurons in CHF.
Rat CHF was induced by surgical ligation of the left anterior descending (LAD) coronary artery. At 12 weeks after LAD ligation, completed bilateral RDN was achieved by surgically cutting all the visible renal nerves around the renal artery and vein, followed by applying 70% ethanol around the vessels. Immunofluorescence staining and western blot data showed that expression of granulocyte-macrophage colony-stimulating factor (GM-CSF) and its receptor-α subunit in SGs was increased in CHF rats. RDN not only reduced CHF-elevated GM-CSF levels in kidney, serum, and SGs but also attenuated macrophage expansion and neuroinflammation in SGs from CHF rats. Using flow cytometry, we confirmed that RDN reduced the percentage of macrophages in SGs, which is pathologically increased in CHF. RDN also decreased CHF-enhanced N-type Ca2+ currents in CSP neurons and attenuated CHF-elevated cardiac sympathetic nerve activity. Electrocardiogram data from 24-h continuous telemetry recording in conscious rats revealed that RDN improved CHF-induced heterogeneity of ventricular electrical activities and reduced the duration of spontaneous ventricular tachyarrhythmias in CHF rats.
RDN alleviates cardiac sympathetic overactivation and ventricular arrhythmogenesis through attenuating GM-CSF-induced macrophage activation and neuroinflammation within SGs in CHF. This suggests that manipulation of the GM-CSF signalling pathway could be a novel strategy for achieving the antiarrhythmic effect of RDN in CHF.
Time of primary review: 53 days
1. Introduction
Chronic heart failure (CHF) is the most common type of heart disease, with a prevalence of over 26 million worldwide.1 As a common complication in CHF, ventricular arrhythmia accounts for nearly 50–60% of mortality in patients with CHF.2 Alongside structural and electrophysiological remodelling in the ventricle,3 neuronal remodelling (such as sympathetic hyperactivation) plays a crucial role in the development and maintenance of ventricular arrhythmias in CHF.2 Renal denervation (RDN) is widely investigated in multiple studies of sympathetically driven cardiovascular diseases,4 in which RDN is shown to reduce cardiac sympathetic activity in hypertension and acute myocardial infarction (MI) models.5 Although RDN is a safe and cost-effective strategy to achieve long-term and sustained blood pressure lowering in patients with resistant hypertension,6–9 RDN-induced rare complications, including renal dysfunction and renal artery stenosis,10–14 as well as reinnervation post-RDN,15–17 have also been highlighted in some studies. Despite the therapeutic potential of RDN for ventricular arrhythmia has been reported,18–22 the mechanisms responsible for the antiarrhythmic effect of RDN remain largely unclear. Therefore, it is necessary to explore the mechanisms responsible for the antiarrhythmic effect of RDN and to further identify novel therapeutic targets against ventricular arrhythmias in CHF.
RDN causes remodelling of stellate ganglion (SG) neurons in normal dogs23 and suppresses ventricular arrhythmias through modulating SG neuronal activity in the long QT canine model.24 Additionally, stimulation of renal nerves facilitates ischemia-induced ventricular arrhythmias through increasing nerve activity of the SG.25 These facts suggest a pathophysiological link between the SG and kidney, with the possibility that RDN attenuates ventricular arrhythmias via reducing the cardiac sympathetic outflow in the SG in CHF. Given the fact that neuroinflammation and macrophage activation in the SG are well characterized in patients with cardiomyopathy and arrhythmias,26 our recent study further revealed that macrophage infiltration and neuroinflammation in SGs contributed to CHF-elevated N-type Ca2+ currents and cell excitability of cardiac sympathetic post-ganglionic (CSP) neurons, which subsequently promoted cardiac sympathetic overactivation and ventricular arrhythmogenesis in CHF.27 However, it remains poorly understood if and how RDN attenuates the cardiac sympathetic overactivation and ventricular arrhythmogenesis through modulating macrophage activation and CSP neuronal function in SGs in the CHF state.
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a pleiotropic cytokine secreted by a wide variety of cells.28 GM-CSF serves as a growth factor to prompt the proliferation of bone marrow-derived macrophages and granulocytes.29 Alongside its effects on chronic inflammation, GM-CSF also plays an important role in neuroinflammation in the central and peripheral nervous systems.30 Although the main source of serum GM-CSF has not been fully determined, increased serum GM-CSF levels were reported to correlate with disease severity in patients with CHF.31 More importantly, RDN reduced local tissue and serum GM-CSF levels by reducing the production of GM-CSF in kidney endothelial cells.32 Because GM-CSF induces M1 macrophage (proinflammatory phenotype) activation with the production of proinflammatory cytokines,33 GM-CSF itself may be a key factor responsible for CHF-elevated macrophage expansion and neuroinflammation in SGs. Based on the above background, and by using multifaceted technical approaches from whole-animals to cellular–molecular levels, our current study validated our hypothesis that RDN achieves its antiarrhythmic effect by inhibiting GM-CSF-induced macrophage activation and neuroinflammation in SGs, which subsequently decreases cardiac sympathetic overactivation, thereby alleviating ventricular arrhythmogenesis in CHF.
2. Methods
All experimental procedures were approved by the University of Nebraska Medical Center (UNMC) Institutional Animal Care and Use Committee and were carried out in accordance with the National Institutes of Health (NIH Publication No. 85-23, revised 1996). Urethane and α-chloralose were only used in a non-recovery procedure in the present study. Detailed methods and procedures are available online in the Supplementary material.
2.1 Study design, timeline, and interventions
Male Sprague–Dawley rats (6–7 weeks of age) were randomly assigned to sham and CHF groups in this current study. CHF rats underwent surgical ligation of the left anterior descending coronary artery (LAD) to induce MI-related CHF, and sham rats underwent the same surgery without LAD ligation. Implantation of radiotelemetry and labelling of CSP neurons were performed at 11 weeks after MI. At 12 weeks post-MI or sham surgery, rats were further assigned to four subgroups for RDN, including sham, sham + RDN, CHF, and CHF + RDN. RDN was performed at 12 weeks after LAD ligation. Ventricular arrhythmogenesis-related electrocardiogram (ECG) makers, heart rate variability (HRV), and spontaneous ventricular tachycardia/fibrillation (VT/VF) from 24-h radiotelemetry ECG recording in conscious rats were evaluated weekly before and after RDN (1, 2, and 3 weeks post-RDN) in all groups. Terminal experiments, including cardiac sympathetic nerve activity (CSNA) recording, measurement of inducibility of ventricular arrhythmias, and haemodynamic recording in anesthetized rats, were performed 3 weeks after RDN (see Supplementary material online, Figure S1). After finishing in vivo experiments, all the rats were euthanized with 0.39 mL/kg of Fatal-Plus euthanasia solution (about 150 mg/kg pentobarbital, i.p.), and then whole blood was collected to extract serum for enzyme-linked immunosorbent assay (ELISA) measurements, and SGs and kidneys were removed to perform flow cytometry, western blot, immunofluorescence staining, and patch-clamp recording in isolated CSP neurons (see Supplementary material online, Figure S1).
2.2 CHF rat model
Rats in the CHF group were anesthetized with 2% isoflurane for surgical ligation of the LAD, and sham rats underwent the same surgery without LAD ligation. To confirm the CHF model, echocardiography was performed to measure left ventricular end-diastolic diameter (LVDd), left ventricular end-systolic diameter (LVDs), ejection fraction (EF), fractional shortening (FS), left ventricular end-diastolic volume (LVd Vol), and left ventricular end-systolic volume (LVs Vol). A colorimetric technique was used to measure the infarct size, and a Millar pressure transducer was used to determine left ventricular end-diastolic pressure (LVEDP) and systolic pressure (LVSP) in the terminal experiment.
2.3 Renal denervation
Bilateral RDN or RDN-control was performed at 12 weeks after LAD ligation. Briefly, bilateral RDN was achieved by surgically cutting all the visible renal nerves around the renal artery and vein, followed by the application of 70% ethanol around the vessels using a cotton swab. RDN-control followed the same procedure as for denervation without cutting and ethanol application. Tyrosine hydroxylase (TH) staining of renal vessels was used to confirm the completeness of RDN.
2.4 Implantation of the ECG telemeter, and measurement of HRV and spontaneous VT/VF in conscious rats
Under anesthetized conditions (2% isoflurane), a radiotelemetry ECG transmitter was placed into the abdominal cavity. One week after the ECG telemeter implantation, a 24-h continuous ECG recording was performed to quantify the cumulative duration of VT/VF in conscious rats. HRV parameters, including low-frequency power (LF, 0.2–0.75 Hz), high-frequency power (HF, 0.75–2.5 Hz), and LF/HF ratio; ventricular arrhythmogenesis-related ECG markers, including QT and corrected QT (QTc) intervals, QT and QTc dispersions, and T-peak to T-end (Tpe) interval were calculated from the ECG recording in conscious rats.
2.5 Measurement of inducibility of ventricular tachyarrhythmia and recording of CSNA in anesthetized rats
Under anesthetized conditions and mechanical ventilation, a left thoracotomy was performed to expose the heart. The inducibility of ventricular tachyarrhythmia was determined by programmed electrical stimulation (PES) in the heart. After the determination of ventricular arrhythmia, a left thoracotomy was performed to expose the left cardiac sympathetic nerve, and CSNA was recorded by a bipolar electrode. Please see detailed information in the Supplementary material.
2.6 Labelling and isolation of SG neurons and whole-cell patch-clamp recording of N-type Ca2+ currents in CSP neurons
Since SG neurons project not only to the heart, but also to other target organs, we used a transported fluorescent dye (red colour DiI) to retrograde-label SG neurons projecting to the cardiac myocardium. Under anesthetized conditions (2% isoflurane), 20 injections (2 μL DiI for each injection) were made sub-epicardially into left and right atria and ventricles. The patch-clamp recording was performed 4 weeks after surgery to allow the dye to diffuse to CSP neurons. SG neurons were isolated by a two-step enzymatic digestion protocol, after which voltage-gated Ca2+ currents were recorded only in DiI-labelled SG neurons (i.e. CSP neurons) by the whole-cell patch-clamp technique.
2.7 ELISA, western blot, immunofluorescence staining, and flow cytometry analysis of macrophage expansion in SGs
The serum GM-CSF levels were determined in all groups using an ELISA kit. For immunofluorescence staining, isolated SG was fixed and cut into sections that were 10 μm thick. The sections were then incubated with primary antibodies and appropriate fluorescence-conjugated secondary antibodies. Slides were observed under a laser-scanning confocal microscope. For western blot analysis, SGs and kidneys were rapidly removed, immediately frozen in liquid nitrogen, and stored at −80°C until analysed. Target protein levels were measured using primary antibodies against a target or housekeeping protein and appropriate secondary antibodies. For flow cytometry analysis, SG neurons were isolated by enzymatically digested in 0.1% collagenase. Isolated live cells from SGs were then incubated with a mix of fluorochrome-conjugated antibodies against CD45 and CD11b, and then analysed by an LSRII cell analyser. Macrophages were identified as CD45+/CD11b+ cells.
2.8 Statistical analysis
SigmaPlot 12 was used for data analysis, and all data are presented as means ± SEM. Statistical significance was determined by one-way or two-way ANOVA with post hoc Bonferroni test for multi-group comparison. For incidence of ventricular arrhythmias, statistical significance was determined by a Fisher exact test. A Student’s unpaired t-test was used to perform a two-group comparison. Normal distribution of data was confirmed with the Kolmogorov–Smirov test, while equal variance was confirmed with Levene’s test. Statistical significance was accepted when P < 0.05.
3. Results
3.1 GM-CSF levels in the kidney, serum, and SG, and GM-CSF receptor expression in the SG
Although macrophage expansion in SGs from CHF rats was well characterized by our recent study,27 it remains largely unclear how CHF induces macrophage activation in SGs. Given that GM-CSF is a key factor in prompting the proliferation of macrophages,29 we first determined the GM-CSF levels in SGs from MI-induced CHF rats. The data from the western blot showed that protein expression of GM-CSF was non-detectable in SGs from sham rats, whereas its expression in SGs was markedly increased in CHF rats (Figure 1A). Since serum GM-CSF levels were reported to correlate with disease severity in patients with CHF,31 we then used ELISA to test if serum GM-CSF levels were increased in MI-induced CHF rats. Our data demonstrated that serum GM-CSF levels were higher in CHF rats than in age-matched sham rats (21.64 ± 1.87 pg/mL in CHF rats vs. 3.42 ± 0.52 pg/mL in sham rats, P < 0.05; Supplementary material online, Figure S2A). Considering that endothelial cells in the kidney serve as the key source of increased levels of serum GM-CSF in transverse aortic constriction (TAC)-induced CHF mice,32 we further determined protein expression of GM-CSF in the kidneys of MI-induced CHF rats. The data from the western blot showed that protein expression of GM-CSF was non-detectable in the kidneys of sham rats; however, its expression was significantly increased in the kidneys of CHF rats (see Supplementary material online, Figure S2B). Following the discovery of CHF-elevated GM-CSF levels, we also tested the protein expression of GM-CSF receptor (GM-CSFR) within SGs in CHF rats. The data from the western blot demonstrated a lower level of GM-CSF receptor-α subunit (GM-CSFRα) expression in SGs in sham rats, whereas its expression in the SGs of CHF rats was significantly increased (Figure 1B). The data from immunofluorescence staining confirmed the western blot data. Furthermore, it showed that GM-CSFRα staining (red colour) did not merge with TH (green colour, an adrenergic neuronal marker) staining in SGs from CHF rats (Figure 1C), raising the possibility that there was no GM-CSFR expression in CSP neurons. Since GM-CSFR was reported to predominantly express in macrophages,30 double staining of Iba1 (a protein maker for activated macrophages) and GM-CSFRα was conducted by immunofluorescence staining to further explore the location of GM-CSFR expression in SGs. The image for co-localization of GM-CSFRα and Iba1 demonstrated that the GM-CSFRα image did merge with Iba1 staining (Figure 2D), confirming that GM-CSFR is mainly expressed in the macrophages but not CSP neurons in SGs.
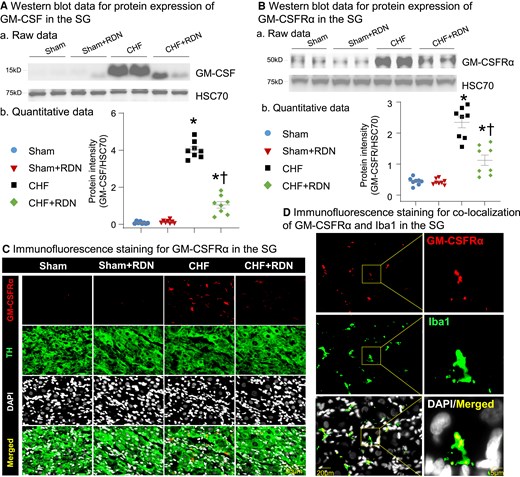
Effect of RDN on CHF-elevated GM-CSF levels and its receptor expression in SGs. (A) Representative images (a) and quantitative data (b) showing the expression of GM-CSF in SGs from all groups, measured by western blot. (B) Raw images (a) and quantitative data (b) of western blot analysis showing the expression of GM-CSF receptor-α subunit (GM-CSFRα) in SGs from all groups. (C) Raw images of immunofluorescence staining showing expression of GM-CSFRα in SGs from all groups. (D) Representative images of immunofluorescence staining showing co-localization of GM-CSFRα and Iba1 (a protein marker for activated macrophages) in SG from the CHF group. n = 8 rats per group. Statistical significance was determined by one-way ANOVA with post hoc Bonferroni test. Data are means ± SEM. *P < 0.05 vs. sham; †P < 0.05 vs. CHF.
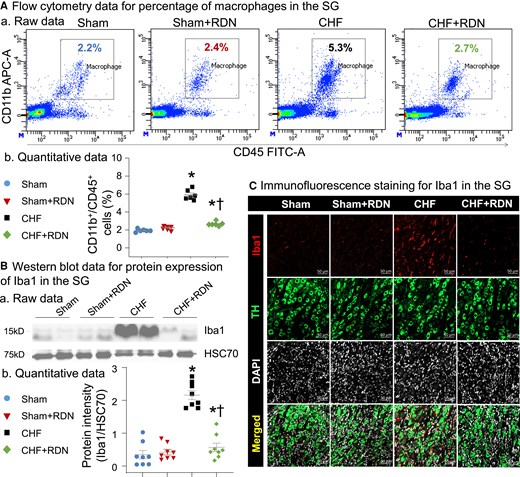
RDN attenuated CHF-increased macrophage activation in SGs. (A) Raw images (a) and quantitative data (b) for flow cytometry analysis showing macrophage expansion in SGs from all groups. (B) Representative images (a) and quantitative data (b) of western blot analysis showing that CHF-increased expression of Iba1 was reduced by RDN. (C) Raw images of immunofluorescence staining showing levels of macrophage activation in SGs from all groups. Iba1, ionized calcium-binding adaptor molecule 1 protein (a protein marker for macrophage activation); TH, tyrosine hydroxylase (an adrenergic neuronal marker); 4',6-diamidino-2-phenylindole (DAPI) nuclear marker. n = 6–8 rats per group. Statistical significance was determined by one-way ANOVA with post hoc Bonferroni test. Data are means ± SEM. *P < 0.05 vs. sham; †P < 0.05 vs. CHF.
3.2 Effect of RDN on CHF-elevated GM-CSF levels and GM-CSFR expression in SGs
First, immunofluorescence staining of renal arteries with TH was used to confirm the success of the RDN procedure. Our data demonstrated that TH (red colour) staining was widely detected around the renal artery in control rats without RDN, whereas the TH staining totally disappeared around the vessels at 3 weeks after RDN (see Supplementary material online, Figure S3). Given that RDN was reported to reduce local tissue and serum GM-CSF levels by reducing the production of GM-CSF in kidney endothelial cells,32 we then tested whether RDN-attenuated GM-CSF levels in SGs and serum by reducing GM-CSF production in the kidney. Our data demonstrated that RDN markedly reduced CHF-increased protein expression of GM-CSF in the kidney (see Supplementary material online, Figure S2B). Additionally, western blot and immunofluorescence staining data also revealed that RDN not only decreased CHF-elevated serum GM-CSF levels (see Supplementary material online, Figure S2A), but also reduced the CHF-elevated protein expression of GM-CSF (Figure 1A) and GM-CSFRα (Figure 1B and C) in SGs. RDN in sham rats had no effect on protein expression of GM-CSF (Figure 1A) and GM-CSFRα in SGs (Figure 1B and C), and GM-CSF levels in serum and kidney (see Supplementary material online, Figure S2A and B).
3.3 RDN reduced CHF-increased macrophage expansion and neuroinflammation in SGs
Our flow cytometry data showed that the percentage of macrophages (defined as CD45+/CD11b+ cells) in SGs was markedly higher in CHF rats than in sham rats (5.90 ± 0.24% in CHF rats vs. 1.93 ± 0.07% in sham rats, P < 0.05, Figure 2A), suggesting the CHF-increased macrophage expansion in SGs. The data from western blots (Figure 2B) and immunofluorescence staining (Figure 2C) also revealed that protein expression of Iba1 in SGs was higher in CHF rats than in age-matched sham rats, indicating that macrophages were highly activated within SGs in CHF. Data from immunofluorescence staining also illustrated that the expression of proinflammatory cytokines, including tumour necrosis factor alpha (TNFα) and IL-1β, in SGs were markedly increased in CHF rats (see Supplementary material online, Figure S4). To observe the effect of RDN on CHF-induced macrophage activation and neuroinflammation in SGs, we measured macrophage expansion at 3 weeks after RDN. Data from flow cytometry demonstrated that CHF-elevated macrophage expansion in SGs was reduced by RDN, as evidenced by the reduced percentage of macrophages in the CHF + RDN group (Figure 2A). Additionally, data from western blots and immunofluorescence staining further confirmed that RDN decreased the CHF-induced macrophage activation in SGs (Figure 2B and C), and attenuated CHF-elevated expression of TNFα and IL-1β in SGs (see Supplementary material online, Figure S4).
3.4 RDN attenuated CHF-increased N-type Ca2+ currents in CSP neurons
To test electrophysiological changes in CSP neurons, we recorded voltage-gated Ca2+ currents in DiI-labelled SG neurons using the whole-cell patch-clamp technique. In our experiment, N-type Ca2+ currents were separated from other types of Ca2+ currents by treatment of ω-conotoxin GVIA (a specific N-type Ca2+ channel blocker, Figure 3A). Our data showed that N-type Ca2+ currents in CSP neurons were markedly increased in CHF rats (Figure 3B), which was in line with the results from our previous study.34 RDN normalized CHF-increased N-type Ca2+ currents towards the levels seen in sham rats, as evidenced by the reduction in N-type Ca2+ currents (29.76 ± 1.44 pA/pF in the CHF + RDN group vs. 51.38 ± 1.95 pA/pF in the CHF group, P < 0.05, Figure 3B) in the CHF + RDN group.
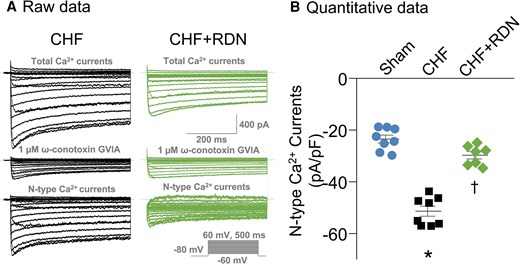
Effect of RDN on N-type Ca2+ currents in CSP neurons from CHF. (A) Original recording of total Ca2+ currents and N-type Ca2+ currents of CSP neurons from CHF and CHF + RDN rats. (B) Quantitative data for N-type Ca2+ currents recorded in CSP neurons from sham, CHF, and CHF + RDN. ω-Conotoxin GVIA: a specific N-type Ca2+ channel blocker. Data are means ± SEM; n = 7–8 neurons from 3 to 6 rats per group. Statistical significance was determined by one-way ANOVA with post hoc Bonferroni test. Data are means ± SEM. *P < 0.05 vs. sham; †P < 0.05 vs. CHF.
3.5 RDN improved CHF-induced cardiac sympathetic overactivation in both conscious and anesthetized rats
A direct relationship between the alteration of N-type Ca2+ channels in CSP neurons and cardiac sympathetic overactivation in CHF was documented by our recent study,34 in which inhibition of N-type Ca2+ channels in CSP neurons by transfection of Cav2.2-α short hairpin RNA (shRNA) into SGs markedly reduced cardiac sympathetic overactivation in CHF rats. Here, we wanted to determine whether RDN also attenuates cardiac sympathetic overactivation in CHF. Since HRV measurement is a widely used approach for the determination of autonomic function in the conscious state in the clinic, it was employed to compare sympathetic activation in conscious rats between groups in the current study. The power spectral analysis of HRV from a 24-h ECG recording demonstrated that LF power (Figure 4Aa and B) and LF/HF ratio (index of cardiac sympathetic activation, Figure 4Ad) were increased, whereas HF power (a marker of cardiac parasympathetic activation, Figure 4Aac) was decreased in conscious CHF rats, compared to sham rats.
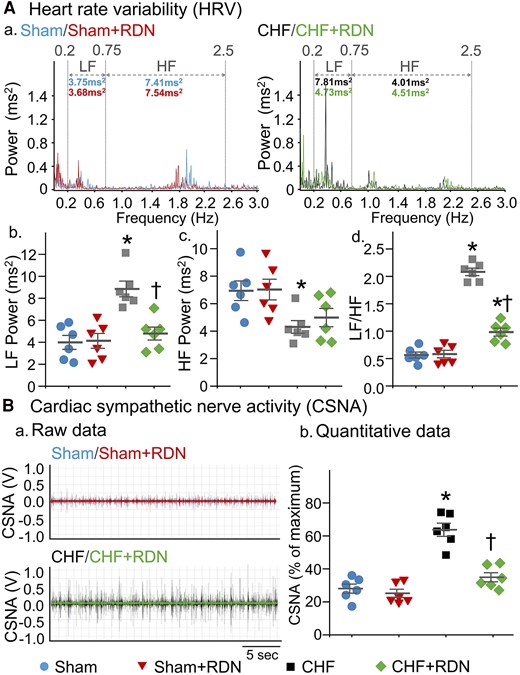
RDN reduced CHF-induced cardiac sympathetic overactivation in both conscious and anesthetized CHF rats. (A) Representative (a) and quantitative (b–d) data of HRV analysed from 24-h ECG recordings in all groups of conscious rats. Spectral power was quantified for LF from 0.2 to 0.75 Hz and HF from 0.75 to 2.5 Hz. (B) Representative tracings (a) and quantitative data (b) for CSNA recording in all groups of anesthetized rats. n = 6 rats per group. Statistical significance was determined by one-way ANOVA with post hoc Bonferroni test. Data are means ± SEM. *P < 0.05 vs. sham; †P < 0.05 vs. CHF.
More importantly, RDN in CHF rats markedly lowered LF power (4.78 ± 0.59 ms2 in the CHF + RDN group vs. 8.84 ± 0.72 ms2 in the CHF group, P < 0.05, Figure 4Aa and B) and LF/HF ratio (0.98 ± 0.07 in the CHF + RDN group vs. 2.08 ± 0.06 in the CHF group, P < 0.05, Figure 4Ad), but only induced a slight increase in HF power in CHF rats (4.98 ± 0.66 ms2 in the CHF + RDN group vs. 4.31 ± 0.46 ms2 in the CHF group, P > 0.05, Figure 4Ac). When HRV parameters before and after RDN were compared (1, 2, and 3 weeks post-RDN), a gradual decrease in LF power (see Supplementary material online, Figure S5A, P < 0.05) and LF/HF ratio (see Supplementary material online, Figure S5C, P < 0.05) and a slight increase in HF power (see Supplementary material online, Figure S5B, P > 0.05) were found from 1 to 3 weeks after RDN. A significant reduction in LF power and LF/HF ratio started around 2 weeks after RDN, reaching a maximum reduction at 3 weeks after RDN (see Supplementary material online, Figure S5A and C). Additionally, when HRV parameters before and after RDN was compared in sham, sham + RDN, and CHF groups, there was no significant alteration in LF power, HF power, and LF/HF ratio (see Supplementary material online, Figure S5).
Alongside the measurement of HRV in conscious rats, we also directly recorded CSNA in anesthetized rats to further confirm the RDN-achieved cardiac sympathetic reduction. RDN markedly reduced the CHF-elevated CSNA towards the levels seen in sham rats (34.92 ± 2.76% in the CHF + RDN group vs. 63.72 ± 3.99% in the CHF group, P < 0.05, Figure 4B). Contrary to the effect of RDN in CHF rats, RDN in sham rats had no effect on LF power, HF power, and LF/HF ratio in a conscious state (Figure 4A), nor was any effect seen on CSNA in anesthetized conditions (Figure 4B).
3.6 RDN restored CHF-increased heterogeneity of ventricular electrical activities
Since the heterogeneity of ventricular electrical activity is a critical marker of ventricular arrhythmogenesis, we calculated ventricular arrhythmogenesis-related ECG markers, including QT and QTc intervals, QT and QTc dispersions, and Tpe interval from 24-h continuous ECG recording. Our data demonstrated that CHF prolonged QT and QTc intervals (Figure 5A–C), QT and QTc dispersions (Figure 5D and E), and Tpe interval (Figure 5A and F), compared to sham rats. These data suggested that CHF increased the spatial and transmural dispersion of ventricular repolarization. Additionally, RDN significantly alleviated CHF-increased heterogeneity of ventricular electrical activity, as evidenced by significant shorting in QT and QTc intervals (67.52 ± 4.44 and 164.27 ± 5.64 ms in the CHF + RDN group vs. 87.73 ± 3.09 and 202.75 ± 5.82 ms in the CHF group, P < 0.05, Figure 5B and C), QT and QTc dispersions (30.97 ± 3.16 and 68.40 ± 5.89 ms in the CHF + RDN group vs. 49.45 ± 3.63 and 97.57 ± 5.08 ms in the CHF group, P < 0.05, Figure 5D and E), and Tpe interval (36.07 ± 2.19 ms in the CHF + RDN group vs. 55.23 ± 2.66 ms in the CHF group, P < 0.05, Figure 5F) in the CHF + RDN group towards the levels in the sham rats. When comparing the ventricular arrhythmogenesis-related ECG markers before and after RDN (1, 2, and 3 weeks post-RDN), we found that RDN markedly improved CHF-elevated heterogeneity of ventricular electrical activity in time-dependent manner, as demonstrated by a gradual shortening in QT and QTc intervals (see Supplementary material online, Figure S6A and B), QT and QTc dispersions (see Supplementary material online, Figure S6C and D), and Tpe interval (see Supplementary material online, Figure S6E) from 1 to 3 weeks after RDN in the CHF + RDN group. A significant reduction in these ECG markers started around 2 weeks after RDN and reached the maximum improvement at 3 weeks after RDN in the CHF + RDN group (see Supplementary material online, Figure S6). When ventricular arrhythmogenesis-related ECG markers before and after RDN were compared in sham, sham + RDN, and CHF groups, we did not find any significant alterations in these ECG parameters (see Supplementary material online, Figure S6).
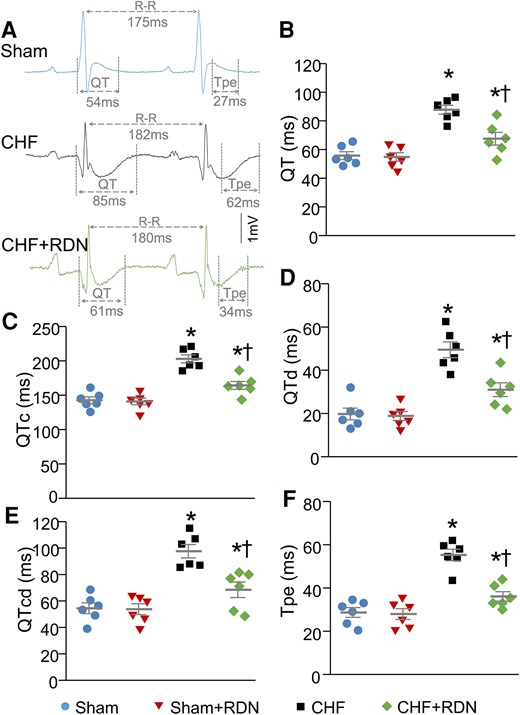
RDN improved CHF-increased heterogeneity of ventricular electrical activities in conscious rats. (A) Representative tracings for QT and Tpe intervals in sham, CHF, and CHF + RDN rats. Quantitative data for QT interval (B), QTc interval (C), QT dispersion (D), QTc dispersion (E), and Tpe interval (F) in all groups. n = 6 rats per group. Statistical significance was determined by one-way ANOVA with post hoc Bonferroni test. Data are means ± SEM. *P < 0.05 vs. sham; †P < 0.05 vs. CHF.
3.7 RDN ameliorated CHF-increased ventricular arrhythmogenesis in both conscious and anesthetized rats
Data from 24-h radiotelemetry ECG recordings in conscious rats showed that no premature ventricular contractions (PVCs) and VT/VF were detected in sham rats (Figure 6Aa), whereas 83% (5 out of 6) of the CHF rats had VT/VF (Figure 6Ab). Compared with sham rats, the cumulative duration of VT/VF was significantly increased in CHF rats (Figure 6Aac). Although RDN did not significantly reduce the incidence of VT/VF [33% (2 out of 6) in the CHF + RDN group vs. 83% (5 out of 6) in the CHF group, P > 0.05, Figure 6Ab], it markedly ameliorated the cumulative duration of VT/VF in CHF rats (2.24 ± 1.62 s/h in the CHF + RDN group vs. 13.22 ± 3.19 s/h in the CHF group, P < 0.05, Figure 6Ac). A comparison of spontaneous VT/VF in the conscious state (before and after RDN) demonstrated that RDN gradually reduced the cumulative duration of VT/VF, which reached the maximum reduction at 3 weeks after RDN in the CHF + RDN group (see Supplementary material online, Figure S7). There were no alterations in the cumulative duration of VT/VF before and after RDN in sham, sham + RDN, and CHF groups (see Supplementary material online, Figure S7).
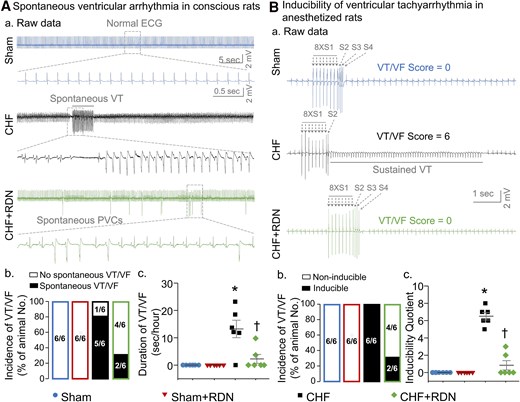
RDN ameliorated CHF-increased ventricular arrhythmogenesis in both conscious and anesthetized rats. (A) Raw ECG recordings (a) of VT/VF in conscious sham, CHF, and CHF + RDN rats, and mean data for incidence (b) and cumulative duration of VT/VF (c) in all groups of conscious rats. (B) Raw data (a) for PES-evoked VT/VF in anesthetized sham, CHF, and CHF + RDN rats, and quantitative data for incidence (b) and inducibility quotient (b) of PES-evoked VT/VF in all groups of anesthetized rats. n = 6 rats per group. Statistical significance was determined by Fisher's exact test for the incidence of VT/VF (Ab and Bb) and one-way ANOVA with post hoc Bonferroni test for cumulative duration (Ac) and inducibility quotient (Bc) of VT/VF. Data are means ± SEM. *P < 0.05 vs. sham; †P < 0.05 vs. CHF.
In addition to determining the spontaneous ventricular arrhythmias in conscious rats, we also evaluated the PES-triggered inducibility of ventricular arrhythmias in anesthetized rats to further clarify the antiarrhythmic effect of RDN in CHF. PES did not trigger the occurrence of VT/VF, and the inducibility quotient was zero in sham rats (Figure 6B). However, PES induced VT/VF with a high incidence and inducibility quotient in CHF rats, compared with sham rats (Figure 6Bbc). RDN in CHF rats reduced the inducibility quotient (0.83 ± 0.54 in the CHF + RDN group vs. 6.50 ± 0.43 in the CHF group, P < 0.05, Figure 6Bc), but failed to decrease the incidence of VT/VF in anesthetized rats [33% (2 out of 6) in the CHF + RDN group vs. 100% (6 out of 6) in the CHF group, P > 0.05, Figure 6Bb]. RDN in sham rats did not induce any cardiac arrhythmia in both conscious (Figure 6A) and anesthetized conditions (Figure 6B).
3.8 RDN failed to improve the performance of the failing heart
To confirm the success of the MI-induced CHF model, we used echocardiography to quantify cardiac function from sham and CHF rats (see Supplementary material online, Figure S8 and Supplementary material online, Table S1). Compared with sham rats, cardiac performance was markedly reduced in CHF rats. This was demonstrated by the significant reductions in EF and FS, and marked increases in LVDd, LVDs, LVd Vol, and LVs Vol in CHF rats (see Supplementary material online, Table S1). Besides the echocardiography measurements, the cardiac performance was also evaluated by quantifying haemodynamic and morphological parameters of the heart from all groups in our current study (Figure 7). A huge infarct size was presented in the left ventricles of both CHF and CHF + RDN rats (Figure 7A and B). However, RDN failed to improve LAD ligation-induced MI in late-stage CHF because there was no significant difference in infarct size between CHF and CHF + RDN rats (Figure 7B). Additionally, since no significant improvements in heart weight (Figure 7C), lung weight (Figure 7E), heart weight-to-body weight ratio (Figure 7F), lung weight-to-body weight ratio (Figure 7G), LVSP (Figure 7H), and LVEDP (Figure 7I) were detected in CHF + RDN rats, we suggested that RDN in CHF rats failed to improve the CHF-induced left ventricular contractile dysfunction, cardiac hypertrophy, and substantial pulmonary congestion. Moreover, we did not find any significant differences in body weight (Figure 7D), mean arterial pressure (Figure 7J), and heart rate (HR) (Figure 7K) between groups. RDN in sham rats did not affect cardiac performance (Figure 7A–K).
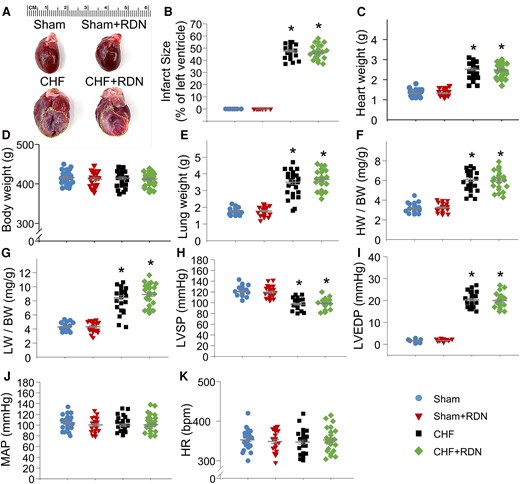
Effect of RDN on haemodynamic and morphological characteristics in all groups. (A) Representative images illustrating the infarct size of the left ventricle, measured at 15 weeks after MI in all groups. Quantitative data showing infarct size (B), heart weight (HW) (C), body weight (BW) (D), lung weight (LW) (E), HW to BW ratio (F), LW to BW ratio (G), LVSP (H), LVEDP (I), mean arterial pressure (MAP, J), and HR (K) in all groups. n = 21–27 rats per group. Statistical significance was determined by one-way ANOVA with post hoc Bonferroni test. Data are means ± SEM. *P < 0.05 vs. sham; †P < 0.05 vs. CHF.
4. Discussion
4.1 Major findings
This study not only provides direct evidence showing the contribution of GM-CSF (and its consequence of macrophage activation and neuroinflammation in SGs) to cardiac sympathetic overactivation and ventricular arrhythmogenesis in CHF but also reveals the involvement of GM-CSF in the antiarrhythmic effect of RDN. We demonstrate for the first time that RDN significantly reduces the CHF-elevated GM-CSF levels in the kidney, serum, and SG, which further decreases GM-CSF receptor expression within macrophages in SGs, thereby alleviating macrophage activation and neuroinflammation in the SG of CHF rats. Additionally, alongside its inhibition of N-type Ca2+ currents in CSP neurons, RDN markedly improves CHF-induced cardiac sympathetic overactivation and heterogeneity of ventricular electrical activities. More importantly, RDN not only decreases susceptibility to ventricular arrhythmias in anesthetized CHF rats but also significantly reduces the duration of VT/VF in conscious rats. However, RDN failed to improve CHF-induced cardiac contractile dysfunction. This direct evidence clarifies the contribution of the GM-CSF signalling pathway to the RDN-reduced macrophage expansion and neuroinflammation in SGs, as well as the consequent attenuation of cardiac sympathetic overactivation and ventricular arrhythmogenesis in the CHF state.
4.2 Renal sympathetic innervation and RDN-reduced GM-CSF levels
Given that increased renal sympathetic nerve activity (RSNA) plays an important role in the pathogenesis of sympathetically driven cardiovascular diseases,4 the modulation of RSNA has drawn increasing scientific and clinical interest over the last decade. Since the kidney is innervated with renal sympathetic nerve fibres that are directly in contact with the renal vasculatures, the juxtaglomerular granular cells, and the renal tubules,35 increased RSNA has been thought to contribute to the deterioration of the diseases through three main mechanisms which include: (i) elevating norepinephrine release from nerve terminals innervating the renal vasculatures, which primarily leads to contraction of smooth muscle cells of the renal arterial resistance vessels, thereby reducing renal blood flow and glomerular filtration rate;36 (ii) increasing sodium and water reabsorption at renal tubular epithelial cells;35 and (iii) elevating release of renin from the juxtaglomerular cells, which subsequently activates the renin–angiotensin–aldosterone cascade.37 In consistent with the data from other studies,38 our present study confirmed that RSNA was significantly increased in MI-induced CHF rats, compared with age-matched sham rats (see Supplementary material online, Figure S9). Given that RDN represents the most traditional means for neuronal inhibition of the RSNA to achieve its therapeutic roles in pathologies, our current study revealed its antiarrhythmic role in MI-induced CHF rats. Although RDN suppresses ventricular arrhythmias by modulating SG neural activity in the long QT canine model,24 the pathophysiological link between the kidney and SG is not fully explored.
In line with the data from other studies,32,39 we demonstrated that RDN significantly reduced renal GM-CSF levels (see Supplementary material online, Figure S2B). Corresponding with a decrease in GM-CSF levels within the denervated kidneys, we also found a significant decrease in GM-CSF levels in serum (see Supplementary material online, Figure S2A) and SGs (Figure 1A) in CHF + RDN rats. While the main source of CHF-elevated GM-CSF levels in SGs was not clarified fully, and how RDN reduces GM-CSF levels remains unclear, several possible sources contributing to CHF-elevated and RDN-reduced GM-CSF levels are considered below. First, kidney endothelial cells are thought to be the major source of GM-CSF because RDN decreases GM-CSF levels in kidney tissue and serum by reducing its production and secretion in kidney endothelial cells isolated from TAC-operated mice,32 raising the possibility that RDN may reduce SG GM-CSF levels in the same manner. Secondly, renal tubular cells were considered to be another source of CHF-elevated GM-CSF levels because elevated GM-CSF was also detected in renal tubular epithelial cells isolated from diseased kidneys.40 Although renal tubules are innervated by renal sympathetic nerves,35 there is no evidence that RDN can modulate renal tubular epithelial cell-derived GM-CSF. Thirdly, renal vascular smooth muscle cells are also potent producers of GM-CSF. While the renal efferent nerves densely branch the cortex and outer medulla, renal sympathetic innervation is also detected around the vascular smooth muscle cells in the afferent and efferent arterioles and along the inner medulla.36 Although no study has detected GM-CSF expression in renal smooth muscle cells, GM-CSF was reported to be expressed in vascular smooth muscle cells.41 Nevertheless, whether RDN can modulate GM-CSF production in renal smooth muscle cells and tubular epithelial cells should be addressed in future studies. Fourth, besides the renal parenchymal cells, infiltrating inflammatory cells in the kidney can produce GM-CSF.42 Since RDN mitigates renal inflammation and immune cell activation in a hypertensive state,43 it is possible that RDN reduces renal GM-CSF production by deactivating renal immune cells. However, because RDN has been shown to achieve its therapeutic roles through reducing renal blood flow, increasing sodium excretion, and attenuating plasma renin activity in experimental CHF,44 it is unclear whether RDN-reduced GM-CSF levels are because of the direct effect of RDN on inflammatory signalling or secondary to its beneficial effects on other pathways. Future studies are required to address these issues.
4.3 GM-CSF signalling axis and its contribution to macrophage activation and neuroinflammation in SGs
GM-CSF is a pleiotropic cytokine that has been reported to prompt the proliferation of bone marrow-derived macrophages and granulocytes.29 Increased serum GM-CSF levels were reported to correlate with disease severity in patients with CHF.31 Although the main source of GM-CSF is less clearly identified under physiological conditions, multiple cellular sources of GM-CSF have been described in pathological conditions, which include endothelial cells, monocytes, macrophages, T cells, microglia, epithelial cells, fibroblasts, and smooth muscle cells, etc.41 In addition to its effects as a growth factor, GM-CSF also plays an essential role in neuroinflammation in the central nervous system,28 and serves as a crucial mediator in the development of chronic inflammation.29 Reports have identified monocytes as the primary target of GM-CSF.29 Since GM-CSF can initiate the monocyte-to-macrophage transition, it is considered the prime regulator of monocyte differentiation.45 It has been reported that GM-CSF signalling in monocytes is critical for macrophage activation in affected tissues.46 Multiple reports have shown that GM-CSF induces M1 macrophage activation with the production of proinflammatory cytokines, including TNFα and IL-1β.33 In vitro treatment of macrophages with GM-CSF also triggered overproduction of TNFα and IL-1β.47
Considering that GM-CSF levels were markedly elevated in SGs from CHF (Figure 1A), we assumed that CHF-elevated GM-CSF levels in the SGs might be a prime mediator of macrophage activation and neuroinflammation in SGs. As a result, a significant increase in macrophage infiltration (Figure 2) and levels of TNFα and IL-1β (see Supplementary material online, Figure S4) were observed in the SG of CHF rats, when compared with sham rats. In support of these results, increased inflammatory infiltration and macrophage activation in SGs have been identified in patients with cardiomyopathy and arrhythmias.26 Proinflammatory cytokines such as TNFα and IL-1β are also elevated in SGs from patients with various cardiomyopathies and are correlated with the prognosis and severity of CHF.48 Although the main source of TNFα and IL-1β in SGs was not identified, activated macrophages in SGs (including resident macrophages, converted from microglia, and recruited macrophages, derived from circulating monocytes) are considered to be the primary source of cytokine release in local tissues.49 Although macrophage activation in the ischaemic heart after MI has been reported extensively, there is limited literature available for macrophage activation in the SGs during CHF. Therefore, it is hard to speculate the origin of macrophages in SGs in CHF. However, it is well known that resident macrophages are seeded in many organs during embryonic development and self-renew independently from blood monocytes, and these tissue macrophages are joined and replaced by recruited monocyte-derived macrophages under inflammation.50 Therefore, we propose that both resident and monocyte-derived macrophages are considered as the origin of macrophages identified in SGs in CHF. However, there are some controversies regarding the source of macrophage expansion in tissues. Some studies have shown that tissue-resident macrophages are predominantly differentiated from monocytes in the steady state,51 whereas monocytes can be rapidly recruited from the systemic circulation into the inflamed sites where they differentiate into macrophages during tissue inflammation.52 In contrast, other studies have suggested that most tissue macrophage populations are derived from embryonic precursors, which are seeded before birth and are maintained by self-renewal in adults.53 Future studies are needed to identify the source of macrophage expansion in SGs in the CHF state.
Following the discovery of increased GM-CSF levels in SGs, we also detected GM-CSFR receptor expression in the SG of CHF rats. The GM-CSF heterodimeric receptor comprises two transmembrane glycoprotidic subunits, including a GM-CSF-specific α subunit and a signal-transducing β chain.46 Our data demonstrated that the expression of GM-CSFRα is highly increased in macrophages infiltrating SGs in CHF (Figure 1B). The α-subunit specifically binds the ligand (i.e. GM-CSF) with low affinity.54 Once binding with its ligand, it dimerizes with the β-subunit.55 Although GM-CSFR lacks intrinsic catalytic activity, it can govern the proliferation, differentiation, and inflammatory signature by triggering the activation in different signalling pathways.56 After detecting the expression of GM-CSFR in SGs, we further identified the precise location of GM-CSFR expression in SGs. Our immunofluorescence study illustrating the co-localization of GM-CSFRα and Iba1 demonstrated that the GM-CSFRα image did merge with Iba1 staining (a maker for activated macrophages, Figure 2D), suggesting that GM-CSFR is mainly expressed in macrophages within SGs. These data are supported by the study from Donatien et al.,30 who demonstrated that GM-CSFR is predominantly expressed in microglia/macrophages in the central and peripheral nervous systems. Based on the evidence presented above, we believe that CHF-elevated GM-CSF directly stimulates its receptor, which is predominantly expressed in the macrophages of SGs. This, in turn, activates a GM-CSF cascade within the macrophages of SGs, thereby inducing macrophage activation and overproduction of proinflammatory cytokines within SGs in CHF. Given that RDN decreases GM-CSF levels in kidney tissue and serum through reducing its production and secretion in kidney endothelial cells isolated from TAC-operated mice,32 we propose that RDN reduces neuroinflammation in SGs through decreasing the overproduction of the GM-CSF in the kidney endothelial cells, which subsequently decreases GM-CSF release from the kidney to the circulation and further to SGs, thereby attenuating GM-CSF-induced macrophage activation and neuroinflammation in SGs.
4.4 GM-CSF signalling pathway could be a potential therapeutic target for achieving the antiarrhythmic effect of RDN
Given that RDN has been widely shown to achieve its therapeutic effect by reducing sympathetic overactivation in multiple sympathetically driven cardiovascular diseases,5 targeting the renal sympathetic nerves could be a logical therapeutic strategy for treating ventricular arrhythmias. Compared with its potential treatment in refractory ventricular arrhythmias,18–22 RDN has been widely reported to offer an innovative treatment for patients with hypertension. While RDN serves as a safe and effective procedure to achieve long-term and sustained blood pressure lowering in patients with resistant hypertension,6–9 RDN-related complications were highlighted in some studies.10–17 Several issues raised by RDN remain to be solved in a few patients post-RDN. First, insufficient RDN would result in less efficacy,36 while completed RDN would induce adverse complications such as renal dysfunction and renal artery stenosis, partially limiting its use in the clinic.12,14 Secondly, analyses of the anatomical distribution of peri-arterial renal sympathetic nerves are suggested to optimize the RDN procedure and achieve more effective and consistent denervation.36 However, due to the lack of procedural metrics on successful renal sympathetic nerve ablation, individual variability in treatment responses was remarkable.57 Thirdly, whether or not the regeneration of renal nerves has an effect on the long-term therapeutic responses to RDN is controversial. Although reinnervation of the renal nerves post-RDN in CHF remains to be established, some studies have demonstrated that reinnervation of renal nerves can begin as early as 28 days in humans.15
From the evidence provided above, we believe that there is an urgent need to develop an effective strategy that can achieve the therapeutic effects of RDN while avoiding its limitations. Although the important role of macrophage activation and neuroinflammation in SGs on cardiac sympathetic overactivation and ventricular arrhythmogenesis in CHF was well documented in our previous study,20 the mechanisms responsible for CHF-elevated macrophage activation and neuroinflammation in SGs were not known. Our current study provides direct evidence showing that CHF-elevated GM-CSF is a critical factor responsible for the macrophage activation and neuroinflammation observed in SGs. In addition to measurements of GM-CSF levels, we also observed high expression levels of GM-CSFR in activated macrophages within SGs in the CHF state. More importantly, our present study clearly shows that RDN achieves its antiarrhythmic effect by attenuating GM-CSF-induced macrophage activation and neuroinflammation within SGs in CHF. These data suggest that targeting the GM-CSF signalling pathway could be a potential therapeutic strategy to achieve the antiarrhythmic effect of RDN.
4.5 Possible contribution of other pathways to the beneficial effect of RDN
Although our study demonstrated that RDN reduced cardiac sympathetic overactivation-related ventricular arrhythmia by attenuating neuroinflammation in SGs, we believe that RDN-reduced neuroinflammation is not restricted to SGs but potentially involves other peripheral ganglia, afferent neurons (dorsal root ganglia), and central nerve systems. Since systemic inflammation after MI indeed serves as the primer for the subsequent occurrence of neuroinflammation in CHF,58 we propose that RDN-reduced GM-CSF also mitigates the neuroinflammation in those regulatory centres. Additionally, based on the data from Xiao et al. who reported that RDN also affected the function of dendritic cells in the spleen,43 we believe that RDN-reduced neuroinflammation in SGs could be partially attributed to its inhibitory effects on inflammatory cells in the spleen. However, the mechanisms responsible for the RDN-inhibited inflammatory cells in the spleen remain unclear. Moreover, as RDN plays a beneficial role by attenuating the renin–angiotensin–aldosterone cascade in CHF,59 we believe that the renin–angiotensin–aldosterone system (RAAS) might be partially involved in the beneficial effects of RDN on reduction in cardiac sympathetic overactivation and ventricular arrhythmogenesis seen in RDN-treated CHF rats. The effect of RAAS is viewed as a potential explanation and will be tested in our future studies.
Besides the efferent innervation of the kidney, most of the brainstem regions receive inputs from renal afferent nerves to modulate cardiovascular function.60 Given that renal afferent nerves have been reported to increase sympathetic nerve activity and blood pressure in the hypertensive status,61 it is possible that RDN reduces cardiac sympathetic overactivation partially via reducing renal afferent input to the central nervous system, thereby inhibiting preganglionic cardiac sympathetic neuron activity. Therefore, the beneficial effect of RDN might be achieved partially by ablating afferent renal nerves. Additionally, it has been reported that renal afferent nerves regulate the hypertensive response to deoxycorticosterone acetate (DOCA) salt, while renal inflammation might be mediated primarily by renal efferent sympathetic nerves because renal inflammation was reduced by total RDN but not affected by afferent-selective RDN.62 Data from Banek et al.63 further demonstrated the important contribution of renal nerves but not perfusion pressure in macrophage activation in the kidney from the rat model of DOCA salt hypertension. Moreover, Xiao et al.43 have also reported that the kidney is a major site of immune activation in angiotensin II-induced hypertension, which is mediated by efferent sympathetic nerves and their release of norepinephrine. These data suggested that renal efferent nerve per se, rather than pressure elevation and renal afferent nerve, participated in the renal inflammatory response in hypertension. Thus, it is also hard to propose that the chronically increased renal afferent activity61 alone could lead to neuroinflammation in renal afferent neurons in the dorsal root ganglia. Whether neuroinflammation occurs in dorsal root ganglia neurons and if it is caused by the increased renal afferent activity in CHF will be addressed in future studies, although it is beyond our current aim. Based on the data from the above studies, we would propose that renal efferent sympathetic nerves play a predominant role in regulating GM-CSF release from the kidney and its consequence of neuroinflammation in SGs in CHF. For the sake of clarifying and focusing on pathophysiological link between the kidney and SGs, we did not evaluate the contribution of the above potential pathways to the beneficial effect of RDN in current studies.
4.6 Study limitations
In the present study, bilateral RDN was achieved by surgically cutting all visible renal nerves around both the renal artery and vein and followed by the application of 70% ethanol around vessels. This method is known to ablate both efferent and afferent renal nerves. A limitation of the current study is that, based on the data presented here, we could not answer whether RDN-attenuated GM-CSF signalling and cardiac sympathetic overactivation are achieved through efferent or afferent renal nerves. As we discussed above, it is likely that the beneficial effect of RDN might be achieved by ablating both efferent and afferent renal nerves. Additionally, while both hypertension and heart failure are sympathetically driven cardiovascular diseases, their mechanisms responsible for renal sympathetic overactivation and renal inflammation might be different. Even in the same disease, different stages of heart failure might demonstrate various levels of pathological changes in sympathetic activation, inflammation, and arrhythmias because our previous study showed that malignant ventricular arrhythmias mainly occur in the early and late stages but not the middle stage of CHF.64 We propose that variability of ventricular arrhythmogenesis during the progression of CHF might be attributed to MI-induced time-dependent changes in the neuroendocrine and expression of various genes.65 Whether RDN in the different stages of CHF could play the same protective roles on renal inflammation and ventricular arrhythmias should be tested in future studies. Considering the clinical relevance of catheter-based RDN (denervating both efferent and afferent renal nerves), we did not design an experiment to test the effect of renal afferent denervation on renal inflammation in our current study. However, we believe that both efferent and afferent renal nerves are involved in mediating renal inflammation. Moreover, due to the limited study period, we only describe the short-term effects of RDN (3 weeks) on GM-CSF-mediated neuroinflammation and sympathetically driven ventricular arrhythmias in CHF. Whether prolonged RDN (months or years) can elicit the same beneficial effects is still unknown, and should be studied further.
4.7 Perspective
In addition to the RDN, techniques in pharmacological treatment are also widely used to achieve neuronal inhibition of sympathetic overactivation in pathologies. For example, adrenergic inhibition using pharmacological approaches such as β-blockers serves as the first-line treatment for ventricular arrhythmias by preventing sympathetic activation.66 However, such pharmacological strategies may not be ideal because β-blockers do not provide adequate protection against sudden cardiac death, and some patients are either intolerant or refractory to β-blockers therapy.67 Pharmacological treatment with β-blockers also lacks organ specificity, resulting in off-target side effects and even toxicologic effects.68 Therefore, concerns about safety and tolerance are emerging as a limitation to their use in the clinic.
To avoid the limitations of RDN and pharmacological approaches, our current study provides evidence to propose that the GM-CSF signalling pathway could be a potential therapeutic target for achieving the antiarrhythmic effect of RDN. Interestingly, the first successful application of Lenzilumab, a first-in-class recombinant monoclonal antibody targeting human GM-CSF, was recently shown to reduce immune hyper-response in patients with COVID-19 pneumonia.69 Additionally, GM-CSF neutralization with Lenzilumab successfully reduced cytokine release and neuroinflammation.70 These exciting results encourage us to conduct future studies to determine whether blocking the GM-CSF cascade by neutralizing GM-CSF with monoclonal GM-CSF antibodies could achieve the antiarrhythmic effect of RDN by attenuating neuroinflammation-induced cardiac sympathetic overactivation in CHF. Moreover, specifically targeting GM-CSF receptors of macrophages in the SG might be another strategy to achieve the antiarrhythmic role of RDN in CHF. We aim to further validate these possibilities in our ongoing studies.
RDN is emerging as an effective treatment for refractory hypertension and ventricular arrhythmias. Although RDN serves as a safe and cost-effective procedure to achieve sustained blood pressure lowering in patients with resistant hypertension, rare complications induced by RDN have been highlighted in some studies, partially limiting its use in the clinic. Our study reveals that RDN achieves its antiarrhythmic effect by attenuating GM-CSF-induced neuroinflammation within SGs in CHF. Therefore, we suggest that targeting the GM-CSF signalling axis could be a potential therapeutic strategy to achieve the antiarrhythmic effect of RDN in CHF. The translational perspective of this study is to open a new avenue in therapeutics working against refractory ventricular arrhythmias by neutralizing GM-CSF or blocking GM-CSF receptors in the SG of patients with CHF.
5. Conclusions
In summary, our present study demonstrates that RDN attenuates CHF-elevated macrophage activation and neuroinflammation by reducing GM-CSF levels in SGs. This subsequently alleviates cardiac sympathetic overactivation, thereby reducing ventricular arrhythmogenesis in CHF. These findings support the hypothesis that RDN achieves its antiarrhythmic effect by attenuating GM-CSF-induced macrophage activation and neuroinflammation within SGs in CHF. Therefore, targeting the GM-CSF signalling axis in SGs could be a potential therapeutic strategy for treating excessive cardiac sympathetic activity and ventricular arrhythmia in the CHF state.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Author contributions
D.Z. and Y.-L.L. conceived and designed the experiments, and wrote the paper. W.H., H.T., Y.-L.L., and D.Z. performed the experiments. W.H., H.T., M.C.W., Y.-L.L., and D.Z. analysed the data. D.Z., Y.-L.L., and M.C.W. contributed reagents/materials/analysis tools.
Acknowledgements
We are grateful to the Small Animal Ultrasound Core at the University of Nebraska Medical Center and Bryan T. Hackfort’s assistant on echocardiography measurement.
Funding
This work was supported by the Career Development Award [851929 to D.Z.] from American Heart Association, Great Plains IDeA-CTR Pilot Grant [to D.Z.] from the University of Nebraska Medical Center, and the National Institute of Health’s National Heart, Lung, and Blood Institute [1R21HL163509-01A1 to D.Z., R01HL137832 to Y.L.L., and R01HL144146 to Y.L.L.].
Data availability
Data supporting the findings of this study are available upon reasonable request to the corresponding author.
References
Author notes
Wenfeng Hu and Huiyin Tu contributed equally to the study.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}