





Abstract
The incidence of calcific aortic valve disease (CAVD) has risen over the last decade and is expected to continue rising; however, pharmacological approaches have proven ineffective. In this study, we evaluated the role and underlying mechanisms of human antigen R (HuR)–mediated post-transcriptional regulation in CAVD.
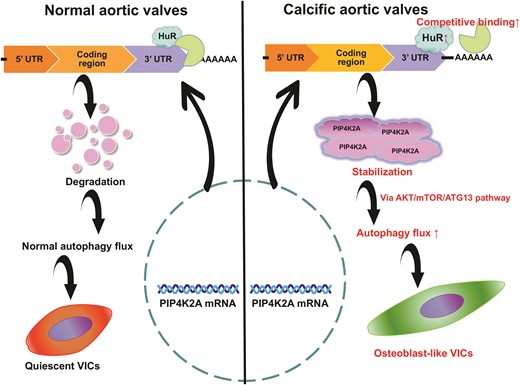
We found that HuR was significantly upregulated in human calcified aortic valves and primary aortic valvular interstitial cells (VICs) following osteogenic stimulation. Subsequent functional studies revealed that HuR silencing ameliorated calcification both in vitro and in vivo. For the first time, we demonstrated that HuR directly interacted with the transcript of phosphatidylinositol-5-phosphate 4-kinase, type II, alpha (PIP4K2A), which mediates phosphatidylinositol signalling, facilitates autophagy, and acts as an mRNA stabilizer. HuR positively modulated PIP4K2A expression at the post-transcriptional level and consequently influenced the AKT/mTOR/ATG13 pathway to regulate autophagy and CAVD progression.
Our study provides new insights into the post-transcriptional regulatory role of HuR in modulating autophagy-positive factors to regulate the pathogenesis of CAVD. Our findings highlight the potential of HuR as an innovative therapeutic target in CAVD treatment.
Time of primary review: 35 days
1. Introduction
Calcific aortic valve disease (CAVD) is a chronic disorder characterized by ectopic mineralization of the aortic valve,1 which may progress to aortic stenosis, left ventricular hypertrophy, and, eventually, cardiovascular death.2 The incidence of CAVD has been increasing over the last 10 years and is expected to continue increasing.3,4 Currently, surgical and transcatheter aortic valve replacements (TAVR) remain the most effective interventions for patients with late-stage CAVD; however, they are invasive, costly, and risky with several complications.5,6 Furthermore, pharmacological approaches are ineffective.7 Therefore, it is essential to explore the regulatory mechanisms that contribute to CAVD pathogenesis. A fundamental hallmark of CAVD progression is the activation of quiescent valvular interstitial cells (VICs), which then undergo phenotypic switching to osteoblast-like cells.8,9 Strategies to effectively inhibit this transformation may lead to novel pharmacological interventions.
RNA–binding proteins (RBPs) regulate gene expression at the post-transcriptional level, largely by modulating target mRNA localization, stability, and translation.10 Human antigen R (HuR), a member of the embryonic lethal abnormal visual (ELAV) protein family,11 is a well-known RBP that stabilizes the mRNAs of target genes by specifically binding to adenylate/uridylate-rich elements (AREs) in the 3′-untranslated region (3′-UTR), which mediate rapid degradation of mRNAs.12 In general, HuR functional activity is regulated by dynamic subcellular localization. HuR is primarily localized in the nucleus under physiological conditions, but upon exposure to intrinsic or extrinsic stress, HuR translocates to the cytoplasm, where it acts with its target mRNAs.13 Thus, HuR plays a strong regulatory role in many cellular processes, including proliferation, senescence, autophagy, apoptosis, and differentiation,14,15 as well as dysfunctional processes such as cancers,11 dyslipidemia,16 and cardiovascular diseases.17 Furthermore, it has been reported that the lncRNA MAARS can interact with HuR and regulate the HuR targets associated with apoptosis, such as p53, caspase-9, and BCL2, to influence atherosclerosis progression.18 In particular, it is well known that the pathogenesis of CAVD shares many similarities with that of atherosclerosis.9 However, the role and underlying mechanisms of HuR–mediated post-transcriptional regulation in CAVD require further study.
Autophagy is a housekeeping subcellular process that degrades and recycles damaged cellular components in eukaryotes.19,20 Most living cells perform a physiological range of basal autophagy to maintain normal cellular homeostasis and energy balance.21 However, autophagy is upregulated following stresses, including nutrient starvation, hypoxia, infection, and increased phosphate levels.22 Dysregulated autophagy is extensively implicated in various diseases including cancer,23 neurodegeneration,24 and cardiovascular diseases.7 Emerging evidence also implies that autophagy is activated during osteoblast differentiation.25 The autophagosomes found in osteoblasts are packed with calcified hydroxyapatite, comprising calcific nodules,26 suggesting that autophagy is crucial during the process of phenotypic switching in CAVD progression. However, the exact role of autophagy in regulating CAVD has not yet been fully elucidated. In the present study, we investigated the function and underlying mechanism of HuR in post-transcriptional regulation and autophagy in CAVD.
2. Method
2.1 Clinical samples
Calcified aortic valve leaflets (CAVs) were explanted from the patients who underwent surgical aortic valve replacement. Control samples (without thickening or calcium deposition in leaflets) were obtained from sex- and age-matched patients who underwent heart transplantation, Bentall surgery, or aortic regurgitation at the hospital between December 2020 and June 2022. The exclusion criteria included rheumatic aortic valve disease, congenital valve disease, bicuspid aortic valves, autoimmune diseases, and infective endocarditis. The clinical characteristics of the patients are presented in Supplementary material online, Table S1. All studies involving human subjects complied with the Declaration of Helsinki and were approved by the Ethics Committee of The Second Affiliated Hospital, School of Medicine, Zhejiang University (no. 2014-159). Written informed consent was obtained from all the patients before participating in the study.
2.2 Primary cell culture
To isolate human aortic valvular interstitial cells (hVICs), non-calcified aortic valves were obtained from patients undergoing surgical aortic valve replacement or heart transplant procedures. Primary hVICs were isolated using the collagenase II digestion method, as described previously.9,27 The clinical characteristics of the patients are summarized in Supplementary material online, Table S1. VICs between passages three and seven were used for further experiments. To induce osteogenic differentiation of hVICs, osteogenic medium (OM), consisting of complete medium (DMEM) supplemented with 10 mmol/L β-glycerophosphate (G9422, Sigma-Aldrich, St. Louis, USA), 100 nmol/L dexamethasone (D4902, Sigma-Aldrich), and 50 µg/mL ascorbic acid (A5960, Sigma-Aldrich), was used as previously described.28 The medium was changed every 2 days.
2.3 RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted using an RNA purification kit (EZBioscience, Shanghai, China) following the manufacturer’s instructions. RNA was reverse transcribed using the PrimeScript RT Reagent Kit (Takara Bio, Kusatsu, Japan). qRT-PCR was performed using the SYBR Premix Ex Taq II kit (Takara Bio). Relative gene expression data were analysed using the 2−ΔΔCT method. The primers used are listed in Supplementary material online, Table S3.
2.4 RNA sequencing (RNA-Seq)
Total RNA was isolated and purified using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. The quantity and purity of each RNA sample were determined using NanoDrop ND-1000 (NanoDrop, Wilmington, DE, USA). RNA integrity was assessed using Agilent 2100 with an RIN number > 7.0. The poly(A) RNA was purified from the total RNA (5 µg) using poly-T oligo–attached magnetic beads and two rounds of purification. Next, the poly(A) RNA was fragmented into small pieces using divalent cations under a high temperature. Subsequently, the cleaved RNA fragments were reverse transcribed to cDNA, which was used to synthesize U-labelled second-stranded DNAs with Escherichia coli DNA polymerase I, RNase H, and dUTP. Then, an A-base was added to the blunt ends of each strand to facilitate their ligation to the indexed adapters. Each adapter contained a T-base overhang for ligating the adapter to the A-tailed fragmented DNA. The single- or dual-index adapters were ligated to the fragments, and size selection was performed with the AMPureXP beads. After the heat-labile UDG enzyme treatment of the U-labelled second-stranded DNAs, the ligated products were amplified via PCR under the following conditions: initial denaturation at 95°C for 3 min; 8 cycles of denaturation at 98°C for 15 s, annealing at 60°C for 15 s, and extension at 72°C for 30 s; final extension at 72°C for 5 min. The average insert size for the final cDNA library was 300 bp (±50 bp). Finally, we performed 150 bp paired-end sequencing on Illumina X Ten (LC Bio, Hangzhou, China) following the vendor’s recommended protocol.
2.5 Western blot
Human aortic valve tissues and hVICs were lysed on ice in RIPA buffer for 30 min in the presence of protease and phosphatase inhibitors (Roche, Basel, Switzerland). The protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Equal amounts of protein were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to PVDF membranes (MilliporeSigma, Burlington, USA), and then incubated with the relevant primary antibodies. After incubation with the corresponding horseradish peroxidase–conjugated secondary antibodies, the bands were visualized and quantified using ImageJ 2.1.0 (NIH, Bethesda, USA). All antibodies and dilutions used are listed in Supplementary material online, Table S4. Protein quantification of samples was normalized to actin or GAPDH levels. At least six samples per group were analysed.
2.6 Immunofluorescence staining
Immunofluorescence staining was used to detect protein expression in the aortic valve tissues and hVICs. Briefly, primary human VICs or aortic valve tissue sections were washed twice in phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for another 15 min, and blocked with 5% bovine serum albumin (BSA) for 30 min. Next, the cells were incubated with primary antibodies overnight at 4°C, followed by incubation with fluorescence-conjugated secondary antibodies and counterstaining with DAPI (Vector Laboratories, Burlingame, USA). All antibodies and dilutions used are listed in Supplementary material online, Table S4. Images were acquired by a Leica fluorescence microscope (Leica, Wetzlar, Germany) and analysed using the ImageJ software.
2.7 Cell transfection
For transient transfections, hVICs at 70% confluency were treated with targeting small interfering RNA (siRNA) or control siRNA (GenePharma, Shanghai, China) at a concentration of 50 nM. Lipofectamine (Invitrogen, Waltham, USA) was used as the transfection reagent. Seventy-two hours after siRNA transfection, hVICs were collected and assayed to test knockdown efficiency. For stable transfections, lentiviruses expressing full-length human HuR (oe-HuR), PIP4K2A (oe-PIP4K2A), HuR short hairpin RNA (shRNA) (sh-HuR), or PIP4K2A shRNA (sh-PIP4K2A) were purchased from GeneChem (Shanghai, China). hVICs were seeded in six-well plates at a density of 1 × 105 cells per well and infected with lentiviruses at a multiplicity of infection (MOI) of 50 in complete media containing 8 µg/mL polybrene (GenePharma). The media was replaced with fresh media after incubating for 6–8 h. Transfection efficiency was monitored based on GFP expression 48–72 h after infection, and the expression was confirmed using western blotting.
2.8 Measurements of alkaline phosphatase (ALP) activity
ALP activity was detected using an ALP detection assay kit (Beyotime), following the manufacturer’s instructions. Briefly, hVICs were harvested 7 days after osteogenic differentiation and lysed in RIPA lysis buffer. p-Nitrophenyl phosphate (pNPP) was added as the substrate and incubated for 15 min at 37°C. ALP activity was measured as the change in absorbance at 405 nm by catalyzing pNPP to p-nitrophenol (pNP). The results were presented as the ALP activity normalized to the protein level.
2.9 Electron microscopy
Treated human calcific aortic valve tissues and hVICs were prefixed in 2% glutaraldehyde in PBS at 4°C overnight, treated with 1% OsO4 at 4°C for 3 h, dehydrated in a graded series of ethanol, and flat embedded in Epon. Ultrathin sections were double stained with uranyl acetate and examined using a transmission electron microscope (Thermo Fisher Scientific, Waltham, USA).
2.10 Alizarin red staining
Alizarin red staining was performed as described previously.29 After treatment with OM for 21 days, hVICs or aortic valve tissue sections were washed twice with PBS, fixed with 4% formaldehyde for 10 min, and stained with alizarin red solution for 15 min. Red staining indicated calcified nodule formation. Images were acquired by a Leica microscope (Leica, Wetzlar, Germany) and analysed using the ImageJ software.
2.11 Actinomycin D assay
hVICs were seeded in 12-well plates at equal densities once they reached 70% confluency and then treated with sh-HuR or control shRNA (GeneChem) at a concentration of 50 nM. Forty-eight hours later, the cells were exposed to 2 µg/mL actinomycin D (Abcam, Cambridge, UK) for 0, 2, and 4 h, respectively. The cells were subsequently harvested, and relative mRNA levels of PIP4K2A and GAPDH were analysed using qRT-PCR.
2.12 RNA pull-down assay
Pull-down assay with biotinylated PIP4K2A mRNA was performed as previously described.30 Briefly, the biotin-labelled RNA probes for PIP4K2A were transcribed in vitro using the Pierce RNA 3′ End Desthiobiotinylation kit (Thermo Fisher Scientific) and T7 RNA polymerase (Vazyme Biotech, Nanjing, China).To generate biotin-labelled RNA–coated magnetic beads, the biotin-labelled RNA was resuspended in wash/binding buffer (0.5 mol/L NaCl, 20 mmol/L Tris-HCl, pH 7.5, and 1 mmol/L EDTA), followed by incubation with Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific, USA) at 4°C for 4 h. Subsequently, hVICs were lysed and incubated with biotin-labelled RNA or negative control RNA at 4°C overnight. After treatment with wash/binding buffer, the RNA complexes bound to the beads were isolated for western blot analysis.
2.13 RBP immunoprecipitation (RIP) assay
RNA immunoprecipitation was conducted using a human anti-HuR antibody (Cell Signaling Technology, Danvers, USA) and the corresponding negative control IgG (Cell Signaling Technology). hVICs were then lysed in a solution of 150 mmol/L KCl, 25 mmol/L Tris-HCl, pH 7.4, 5 mol/L MEDTA, 0.5% Triton X-100, and 5 mmol/L dithiothreitol (DTT) supplemented with RiboLock RNase Inhibitor (Thermo Fisher Scientific) and proteinase inhibitor cocktail (Roche). The lysates were then mixed with HuR antibody- or IgG-coupled Sepharose beads and rotated at 4°C for 4 h. Following six washes in lysis buffer, the immunoprecipitated RNAs were extracted, and the enrichment levels of PIP4K2A or GAPDH mRNA were determined by qPCR analysis.
2.14 Luciferase reporter assay
The luciferase reporter assay was performed in HEK-293T cells as previously described.31 For the PIP4K2A mRNA and HuR interaction, either wild-type (WT) or mutant PIP4K2A mRNA 3′-UTR fragments containing putative HuR binding sites, and the PIP4K2A promoter reporter vector, were inserted into the pGL3-firefly luciferase vector (GeneChem). Luciferase activity was detected using the Dual-Luciferase Reporter Assay Kit (Vazyme Biotech, Nanjing, China), following the manufacturer’s instructions. The ratio of firefly to Renilla luciferase activity was determined to eliminate variation in transfection efficiency.
2.15 RNA-fluorescence in situ hybridization (RNA-FISH) assay
A Cy3-labelled oligonucleotide probe for PIP4K2A was designed and synthesized by GenePharma. RNA-FISH assays of aortic valve tissue sections were performed using a fluorescence in situ hybridization kit (GenePharma) according to the manufacturer’s instructions. Images were acquired using a fluorescence microscope (Leica) and analysed using the ImageJ software.
2.16 Animal experiments
Adult WT and low-density lipoprotein receptor (LDLR)−/− mice (C57BL/6 background) aged 6–8 weeks were housed in a pathogen-free, temperature-controlled environment under a 12:12 h light-dark cycle and fed a 0.2% high-cholesterol diet (HCD) for 24 weeks to develop aortic valve calcification or normal diet (ND) for control, as described previously.8 The HuR inhibitor MS-444 or the negative control solvent (20 mg/kg) was administered twice a week via intraperitoneal injection. At the end of the protocol, mice were anesthetized using 2.5% isoflurane and echocardiographic parameters were determined by transthoracic echocardiography using a Vevo 2100 imaging system (VisualSonics, Toronto, Canada). Finally, mice were euthanized by intravenous injection of a lethal dose of pentobarbital sodium (100 mg/kg), with blood and aortic valve samples collected for further analysis, including metabolic parameter detection, haematoxylin and eosin (H&E) staining, alizarin red staining, von Kossa staining, and RNA-FISH, as previously described.28 All procedures involving animals were approved by the Animal Care and Utilization Committee of the Zhejiang University (No. 2019–177), and in accordance with the European Communities Council Directive 2010/63/EU for the protection of animals used for experimental purposes.
2.17 Von Kossa staining in aortic valve tissue
Aortic valve tissue sections were stained using the von Kossa staining kit (Abcam). Deparaffinized sections were incubated in silver nitrate solution (5%) for 30 min with exposure to ultraviolet light. After three washes in distilled water, the sections were treated with sodium thiosulfate solution (5%) for 2–3 min. After two rinses with distilled running water, the sections were incubated with a nuclear fast red solution for 5 min. The calcium deposits were stained brown to black. Images were acquired by a Leica microscope (Leica, Wetzlar, Germany) and analysed using the ImageJ software.
2.18 Statistical analysis
Data were analysed using GraphPad Prism version 9.0 and were presented as mean ± standard deviation (SD). Statistical differences were determined using two-tailed unpaired Student’s t-test for two groups and one-way analysis of variance (ANOVA) for multigroup analyses. The association between two variables was evaluated using two-tailed Pearson correlation analysis. Statistical significance was set at P < 0.05.
3. Results
3.1 HuR is upregulated in human calcific aortic valves and associated with osteogenic differentiation of VICs
To evaluate whether HuR expression was altered in human calcific aortic valves, we compared the expression of HuR in aortic valves from 10 patients with CAVD and 10 sex- and age-matched non-CAVD controls (see Supplementary material online, Table S1, for detailed clinical characteristics). HuR protein levels were significantly elevated in the subjects with CAVD compared to those in the non-CAVD controls (Figure 1A), and the mRNA expression of HuR was strongly positively correlated with that of the osteoblastic differentiation markers ALP and RUNX2 (see Supplementary material online, Figure S1A). As the phenotypic transition of hVICs into osteoblast-like cells is an indispensable step in CAVD pathogenesis,32 we explored whether HuR played a critical role in hVIC osteogenic differentiation. We isolated hVICs from non-calcified human aortic valves and exposed them to OM to stimulate osteogenic differentiation.8 We found that both HuR protein and mRNA levels were significantly upregulated in hVICs after osteogenic induction compared with those in controls (Figure 1B and C, respectively), and the mRNA expression of HuR was strongly positively correlated with that of the osteoblastic differentiation markers ALP and RUNX2 (Figure 1D).
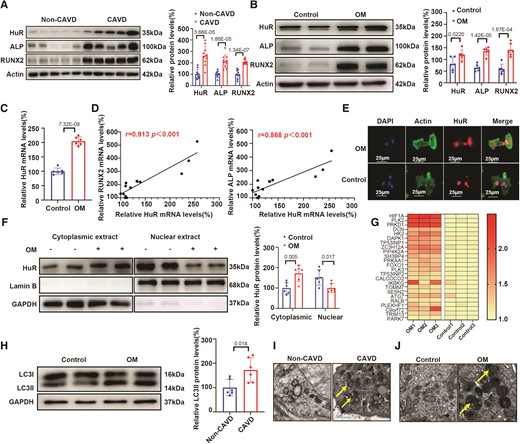
HuR is upregulated in human calcific aortic valves and associated with osteogenic differentiation of VICs. (A) HuR protein expression in valves of patients with calcific CAVD and non-CAVD controls (n = 10). (B) Protein expression of HuR, ALP, and RUNX2 in hVICs after osteogenic induction using OM (n = 6). (C) HuR mRNA expression in hVICs after osteogenic induction (n = 6). (D) Pearson’s correlation analysis between mRNA levels of HuR and osteogenic differentiation markers ALP and RUNX2 in VICs after osteogenic induction (n = 12). (E) Fluorescence-labelled HuR protein shuttling from the nucleus to the cytosol in hVICs after osteogenic induction. Nuclei are stained using DAPI (), scale bar = 25 μm. (F) HuR protein expression in cytosolic and nuclear fractions from hVICs after osteogenic induction by OM (n = 6). (G) Heatmap showing upregulation of positive regulators of autophagy upon OM stimulation. (H) Autophagy marker LC3 protein expression in hVICs after osteogenic induction compared with expression levels in controls (n = 6). (I, J) TEM analysis was performed on human calcified and non-calcified aortic valves as well as hVICs stimulated with OM to identify autophagic vesicles (arrows). Scale bar = 1 μm. Actin (A–B) or GAPDH (F, H) protein levels were used for normalization. Values are mean ± SD. Statistical differences were determined using two-tailed unpaired Student’s t-test for two groups (A–C, F, and H). The association between two variables was evaluated using two-tailed Pearson correlation analysis (D). Statistical significance was set at P < 0.05.
To study the effects of osteogenic differentiation on HuR subcellular localization, we measured HuR protein levels in the nucleus and cytoplasm following induction with OM. We observed that in hVICs stimulated to undergo osteogenic differentiation, HuR shuttled from the nucleus to the cytoplasm, as shown by immunofluorescence (IF) (Figure 1E). Additionally, fractionation studies confirmed higher HuR expression in the cytosolic fraction and lower expression in the nuclear fraction of hVICs treated with OM than that in the controls (Figure 1F). This suggests that osteogenic differentiation regulates cytosolic shuttling of HuR to an active state.
To determine the presence of autophagy alteration in hVICs during the progression of CAVD, RNA was derived from three hVICs from non-calcified human aortic valves and three counterparts exposed to osteogenic differentiation using RNA-Seq profiling to capture the differentially expressed mRNAs. Several positive regulators of autophagy including HIF1A, PLK2, PRKD1, and PIP4K2A were upregulated upon stimulation with OM compared with expression levels in controls (Figure 1G). We also found increased expression of the autophagy marker LC3II in the CAVD valve tissue samples (see Supplementary material online, Figure S1B) and hVICs exposed to OM compared with that in controls (Figure 1H). Consistent with the western blot analysis, transmission electron microscopy (TEM) analysis was performed on calcified and non-calcified human aortic valves and hVICs stimulated with OM to investigate the presence of autophagic vesicles. Normal levels of autophagy were observed in the non-calcified aortic valves and control hVICs, whereas a significant build-up of autophagic vesicles was observed in the calcified aortic valves and hVICs stimulated to undergo osteogenic differentiation (Figure 1I and J). These data indicate that HuR may participate in CAVD progression through its association with osteoblast differentiation and autophagy in hVICs.
3.2 HuR silencing ameliorates osteogenic differentiation of hVICs in vitro
To investigate the role of HuR in osteogenic differentiation of hVICs, we designed shRNA to silence HuR in hVICs (Figure 2A). VICs transfected with sh-HuR or the negative control (sh-NC) were exposed to OM to induce osteogenic differentiation for 7–21 days. The results showed that shRNA–mediated silencing of HuR in hVICs ameliorated the OM–induced increase in the expression of the osteogenic markers ALP and RUNX2 (Figure 2B), ALP activity (Figure 2C), and calcium deposition (Figure 2D) compared with those in sh-NC–treated cells. Overexpression of HuR (Figure 2E) increased the expression of osteogenic markers (Figure 2F), ALP activity (Figure 2G), and calcium deposition (Figure 2H) compared with that in controls. These results indicated that HuR played a positive role in the osteogenic differentiation of VICs. Furthermore, we applied the HuR small molecule inhibitor MS-444 to hVICs treated with osteogenic differentiation medium and observed reduced expression of the osteogenic markers ALP and RUNX2 (Figure 2I), ALP activity (Figure 2J), and calcium deposition (Figure 2K) compared with those in OM-treated hVICs without MS-444. Lastly, we found a decrease in the expression of the autophagy marker LC3II upon HuR inhibition (Figure 2L) and an increase upon HuR overexpression (Figure S1C) compared with those in the controls, suggesting a link between HuR and autophagy.
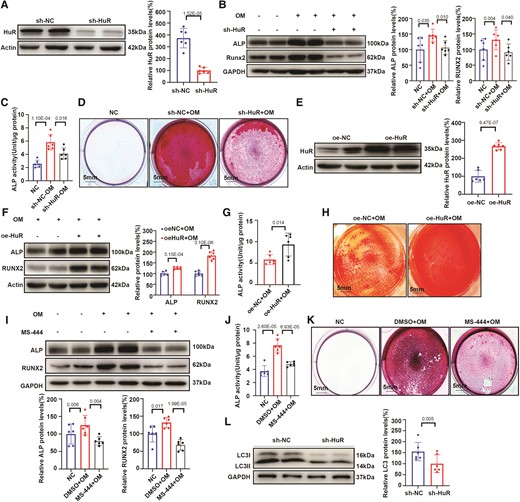
HuR knockdown inhibits osteogenic differentiation of hVICs in vitro. (A) Silencing efficiency of HuR with shRNA in human valvular interstitial cells (hVICs). (B–D) hVICs were transfected with sh-HuR or sh-NC and stimulated with osteogenic differentiation medium (OM) (n = 6). (B) Protein expression of the osteogenic differentiation markers ALP and RUNX2 was evaluated by western blot (n = 6). (C) ALP activity was evaluated by spectrophotometry (n = 6). (D) Mineralized bone matrix formation was evaluated by alizarin red staining, scale bar = 5 mm. (E) Overexpression efficiency of HuR with lentivirus transfection in VICs (n = 6). (F, G) Expression of osteogenic differentiation markers (F) and ALP activity (G) in hVICs transfected with NC or HuR overexpression lentivirus and stimulated with OM (n = 6). (H) Mineralized bone matrix formation was evaluated by alizarin red staining, scale bar = 5 mm. (I–K) Expression of osteogenic differentiation markers (n = 6) (I), ALP activity (n = 6) (J), and alizarin red staining (K) in hVICs treated with the HuR small molecule inhibitor MS-444 and stimulated with OM, scale bar = 5 mm. (L) Protein expression of the autophagy marker LC3 in hVICs transfected with sh-HuR or sh-NC (n = 6). Actin (A, E–F) or GAPDH (B, I, and L) protein levels were used for normalization. Values are mean ± SD. Statistical differences were determined using two-tailed unpaired Student’s t-test for two groups (A–C, E–G, I, J, and L). Statistical significance was set at P < 0.05.
3.3 HuR directly interacts with PIP4K2A mRNA and regulates its expression
Among the most differentially expressed genes associated with autophagy identified using RNA-Seq, phosphatidylinositol-5-phosphate 4-kinase, type II, alpha (PIP4K2A) was predicted to have several binding sites for HuR in different databases (https://starbase.sysu.edu.cn and https://www.uniprot.org/uniprot/Q15717; Figure 3A). We explored how HuR regulates PIP4K2A expression in CAVD. PIP4K2A protein levels were significantly elevated in the CAVD valve tissue samples compared to those in the non-CAVD controls (see supplementary material online, Figure S1E), and the mRNA expression of HuR was strongly positively correlated with PIP4K2A in the CAVD valve tissue samples (see supplementary material online, Figure S1F). Western blot and qPCR analyses showed that both protein and mRNA levels of PIP4K2A were significantly reduced by downregulation of HuR compared with that in controls (Figure 3B and C, respectively). This is consistent with decreased protein levels of the autophagy marker LC3 (Figure 2L). Moreover, HuR expression was not affected by PIP4K2A knockdown (Figure 3D). Lastly, HuR silencing strongly reduced the stability of PIP4K2A mRNA, as indicated by the shortened half-life after treatment with the transcriptional inhibitor actinomycin D compared with that in the control (Figure 3E).
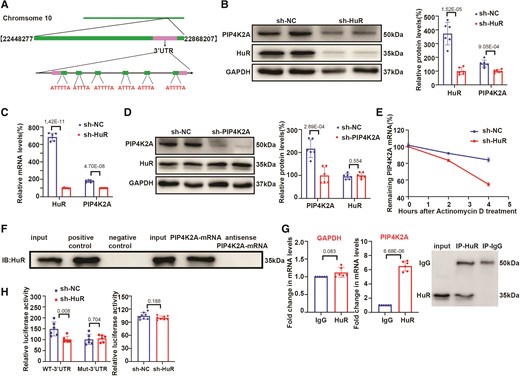
HuR directly interacts with PIP4K2A mRNA and regulates its post-transcriptional expression. (A) Putative binding sites of HuR with PIP4K2A mRNA in the 3′-UTR. (B–C) PIP4K2A protein (B) and mRNA (C) levels in human valvular interstitial cells (hVICs) transfected with sh-HuR or sh-NC (n = 6). (D) HuR protein expression in hVICs transfected with sh-PIP4K2A or sh-NC (n = 6). (E) Expression of PIP4K2A mRNA in hVICs transfected with sh-HuR or sh-NC and treated with actinomycin D as detected by qRT-PCR (n = 6). (F) Detection of HuR protein using western blotting with the eluate pulled down by the biotinylated PIP4K2A mRNA and negative control (antisense PIP4K2A mRNA) probe. (G) RBP immunoprecipitation (RIP) assays were performed using normal mouse IgG or anti-HuR. Relative expression levels of PIP4K2A and GAPDH mRNAs were detected by qRT-PCR (n = 6). (H) Relative luciferase activities of PIP4K2A WT or mutated 3′-UTR reporter (left) (n = 6), and PIP4K2A mRNA promoter reporter in VICs transfected with sh-HuR or sh-NC (right) (n = 8) detected using the dual-luciferase reporter assay. GAPDH (B, D) protein levels were used for normalization. Values are mean ± SD. Statistical differences were determined using two-tailed unpaired Student’s t-test for two groups (B–D, G, and H). Statistical significance was set at P < 0.05.
To further verify whether HuR directly interacted with PIP4K2A, we performed a pull-down assay using biotin-labelled T7 in vitro–transcribed PIP4K2A mRNA or antisense mRNA incubated with whole cell protein lysates from hVICs. HuR protein was only detectable in the eluate of biotin-labelled PIP4K2A mRNA and whole cell lysates (input) compared with that in the negative control (Figure 3F). In reverse pull-down experiments, PIP4K2A mRNA expression was increased by more than six-fold in RNA isolated after HuR immunoprecipitation compared with that isolated with the IgG control (Figure 3G). This was specific to PIP4K2A mRNA, compared with the GAPDH transcript, a known non-targeting control. Additionally, among the differentially expressed autophagy-associated genes, PIP4K2A mRNA expression showed the maximum increase when compared to the others (Figure S1D) in the RBP immunoprecipitation experiment, which indicated that PIP4K2A is the best candidate. These results indicated that HuR directly interacted with PIP4K2A mRNA.
To confirm the effect of HuR on PIP4K2A mRNA, human PIP4K2A mRNA 3′-UTR fragments containing six putative binding sites, as predicted in the database (https://www.uniprot.org/uniprot/Q15717), for the HuR reporter vector and the PIP4K2A promoter reporter vector were constructed. Dual-luciferase reporter assays showed that HuR silencing significantly decreased the luciferase activity of the WT PIP4K2A 3′-UTR reporter compared with that in the control but had no effect on the luciferase activity of the mutated 3′-UTR reporter (Figure 3H). Next, we constructed six plasmids by mutating the six predicted AREs (see Supplementary material online, Table S5). The PIP4K2A mRNA-HuR interaction remarkably decreased upon ARE mutation in vitro, as shown by the reduced luciferase activity in the dual-luciferase reporter assays (see supplementary material online, Figure S1G), indicating that all six sites could bind to HuR. In contrast, there was no significant change in the PIP4K2A promoter activity after HuR inhibition in VICs (Figure 3H). These findings indicate that HuR may play a critical role in osteogenic differentiation through autophagy by targeting PIP4K2A mRNA to enhance its stability at the post-transcriptional level in hVICs.
3.4 Knockdown of PIP4K2A inhibits osteogenic differentiation by activating the PI3K/AKT/mTOR/ATG13 pathway in vitro
We further validated the role of PIP4K2A in regulating calcification. VICs transfected with sh-PIP4K2A or the negative control (sh-NC) were cultured in OM to induce osteoblast differentiation. shRNA-mediated knockdown of PIP4K2A in hVICs (Figure 4A) ameliorated the OM-induced increase in the expression of the osteogenic markers ALP and RUNX2 (Figure 4B), ALP activity (Figure 4C), and calcium deposition (Figure 4D) compared with that in controls. Moreover, protein levels of the autophagy marker LC3 were markedly reduced after VICs were transfected with sh-PIP4K2A compared with that in the control, as shown by western blot analysis (Figure 4E) and mRFP-GFP-LC3 adenovirus (Figure 4F).
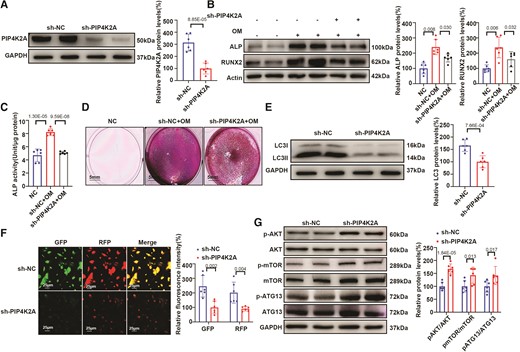
PIP4K2A knockdown inhibits OM-induced osteogenic differentiation by activating the PI3K/AKT/mTOR/ATG13 pathway in vitro. (A) Silencing efficiency of shRNA targeting PIP4K2A in VICs (n = 6). (B–D) Expression of osteogenic differentiation markers (n = 6) (B), ALP activity (n = 6) (C), and alizarin red staining (D) in hVICs transfected with sh-PIP4K2A or sh-NC and stimulated with osteogenic differentiation medium (OM), scale bar = 5 mm. (E) Protein expression of the autophagy marker LC3 in hVICs transfected with sh-PIP4K2A or sh-NC (n = 6). (F) RFP–positive and GFP–positive LC3–labelled autophagosomes in hVICs transfected with sh-PIP4K2A or sh-NC (n = 6), scale bar = 25 μm. (G) Western blot analysis of proteins involved in the AKT/mTOR/ATG13 signalling pathway in hVICs transfected with sh-PIP4K2A or sh-NC (n = 6). Actin (B) or GAPDH (A, E, and G) protein levels were used for normalization. Values are mean ± SD. Statistical differences were determined using two-tailed unpaired Student’s t-test for two groups (A–C and E–G). Statistical significance was set at P < 0.05.
To further investigate the possible targets of PIP4K2A, we analysed changes in the phosphorylation of key activators of the AKT/mTOR/ATG13 signalling pathway (including AKT, mTOR, ATG13, and their phosphorylated forms) by western blot analysis. Compared with that in the control group, PIP4K2A knockdown increased the expression levels of p-AKT/AKT, p-mTOR/mTOR, and p-ATG13/ATG13 in hVICs (Figure 4G), indicating that sh-PIP4K2A activated the AKT/mTOR/ATG13 signalling pathway in VICs to hinder the initiation of autophagy as it is proven that ATG13 participates in the formation of autophagosomes.
3.5 Overexpression of PIP4K2A and rapamycin treatment reversed autophagy and osteogenic differentiation induced by HuR shRNA in vitro
We performed a rescue experiment using an adenovirus expressing PIP4K2A to confirm whether HuR functions through PIP4K2A-mediated autophagy. We found that overexpression of PIP4K2A when HuR was knocked down (Figure 5A) partially abolished the inhibition of the levels of osteogenic differentiation (Figure 5B), autophagy flux (Figure 5C) markers, and calcium deposition (Figure S2A) in hVICs compared to that in the sh-HuR-treated cells. Furthermore, we used the mTOR-dependent inducer rapamycin (50 nM) and the mTOR-independent inducer carbamazepine (10 μM) to induce autophagy and then evaluated the osteogenic phenotype transition after HuR knockdown in hVICs exposed to OM. As expected, rapamycin and carbamazepine treatment also partially abrogated the effects of HuR shRNA on the osteogenic differentiation (Figure 5D and see supplementary material online, Figure S3A) and calcium deposition (Figure S2B and Figure S3B) of hVICs. Lastly, we applied rapamycin and carbamazepine to hVICs treated with sh-PIP4K2A and observed a rescue effect on the expression of the autophagy marker LC3II (Figure 5E and see supplementary material online, Figure S3C) and phosphorylation of mTOR (Figure 5F). Collectively, these results indicate that HuR targets PIP4K2A to promote osteoblast differentiation in hVICs by inducing autophagy.33
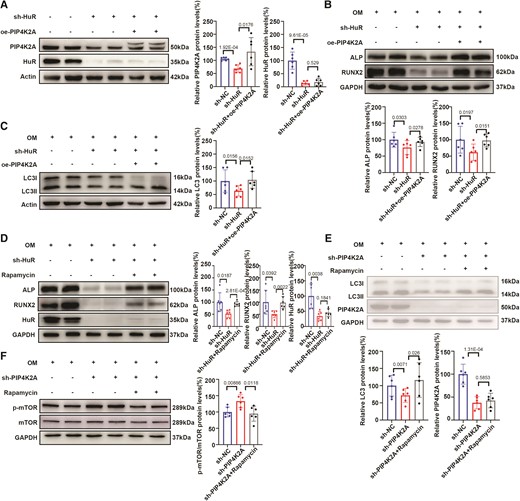
PIP4K2A overexpression and rapamycin treatment rescue autophagy levels and osteogenic differentiation induced by HuR knockdown in vitro. (A) Efficiency of HuR silencing and PIP4K2A overexpression in VICs (n = 6). (B, C) Overexpression of PIP4K2A reversed the decrease in osteogenic differentiation (B) and autophagy flux (C) marker levels in hVICs transfected with sh-HuR upon OM stimulation compared with that in controls (n = 6). (D) Rapamycin treatment reversed the decrease in osteogenic differentiation marker expression in hVICs transfected with sh-HuR upon OM stimulation compared with that in controls (n = 6). (E, F) Rapamycin treatment reversed the decrease in autophagy marker (E) and increase in p-mTOR/mTOR (F) expression in hVICs transfected with sh-PIP4K2A upon OM stimulation (n = 6). Actin (A, C) or GAPDH (B, D–F) protein levels were used for normalization. Values are mean ± SD. Statistical differences were determined using two-tailed unpaired Student’s t-test for two groups (A–F). Statistical significance was set at P < 0.05.
3.6 HuR inhibition in vivo ameliorates aortic valve calcification in LDLR−/− mice by reducing autophagy
To investigate the role of HuR inhibition in aortic valve calcification and autophagy in vivo, mice were divided into three groups and observed for 24 weeks: (i) the ND group (WT mice fed a ND), (ii) the HCD group (LDLR−/− mice fed a HCD to induce development of aortic valve calcification), and (iii) the MS-444 group (LDLR−/− mice treated with biweekly intraperitoneal administrations of 20 mg/kg MS-444 and fed an HCD). The clinical and echocardiographic measurements of the groups are shown in Supplementary material online, Table S2.
After 24 weeks, echocardiographic assessment of mice in the HCD group showed a significant increase in the transvalvular peak jet velocity and mean transvalvular pressure gradient compared with that in the ND group, whereas the administration of MS-444 significantly reversed these changes (Figure 6A). However, there were no significant differences in left ventricular diameter and systolic or diastolic function among the groups at the time of sacrifice (see Supplementary material online, Table S2). We then collected aortic valve samples from the three groups and evaluated valve leaflet morphology and calcification. Haematoxylin and eosin (H&E) staining showed that aortic valve leaflet thickness was higher in the HCD group than in the ND group (Figure 6B). This is consistent with the increased calcium deposits in the aortic valve leaflets of the animals in the HCD group compared with those in the ND group, as shown by alizarin red and von Kossa staining (Figure 6C and D, respectively). Strikingly, MS-444 treatment reduced the thickness (Figure 6B) and calcium deposits (Figure 6C and D) in the aortic valve leaflets compared with those in the HCD group. Moreover, protein levels of the osteogenic differentiation marker RUNX2 was significantly increased in aortic valves from the HCD group compared with that of valves from the ND group. MS-444 treatment reduced the levels of RUNX2 compared with that in the HCD group (Figure 6E).
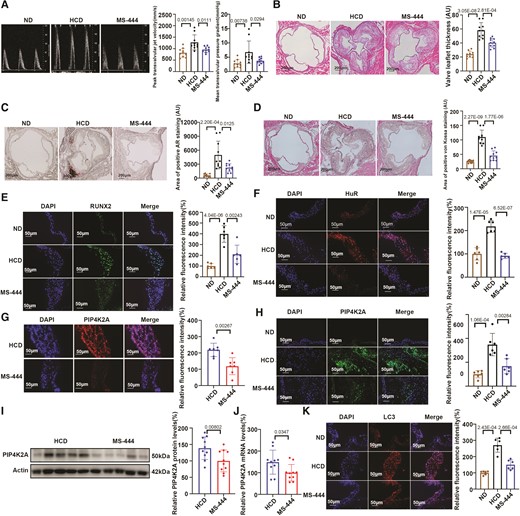
Targeted inhibition of HuR ameliorates aortic valve calcification in LDLR−/− mice by reducing autophagy. (A) Echocardiographic parameters, transvalvular peak jet velocity, and mean transvalvular pressure gradient, in the ND, HCD, and MS-444 groups (n = 10/group). (B) Representative haematoxylin and eosin (H&E) staining images and quantification of leaflet thickness in HCD and MS-444 groups (n = 10/group), scale bar = 200 μm. (C, D) Representative alizarin red (C) and von Kossa staining (D) images and quantification of calcium deposits in valve leaflets (n = 10/group), scale bar = 200 μm. (E) Fluorescence-labelled RUNX2 protein in valve leaflets of mice in the ND, HCD, and MS-444 groups was visualized by fluorescence microscopy (n = 6/group), scale bar = 50 μm. (F) Fluorescence-labelled HuR protein in valve leaflets of mice in the ND, HCD, and MS-444 groups (n = 6/group), scale bar = 50 μm. (G) RNA-FISH assay identifying Cy3-labelled PIP4K2A mRNAin valve leaflets of mice in the HCD and MS-444 groups (n = 6/group), scale bar = 50 μm. (H) Fluorescence-labelled PIP4K2A protein in valve leaflets of mice in the ND, HCD, and MS-444 groups (n = 6/group), scale bar = 50 μm. (I) PIP4K2A protein expression in heart tissues of mice in the HCD and MS-444 groups (n = 10/group). (J) Circulating PIP4K2A mRNA expression in HCD and MS-444 groups (n = 10/group). (K) Fluorescence-labelled LC3 protein in valve leaflets of mice in the ND and HCD groups (n = 6/group), scale bar = 50 μm. Nuclei are stained using DAPI. Actin (I) protein levels were used for normalization. Values are mean ± SD. Statistical differences were determined using two-tailed unpaired Student’s t-test for two groups (A–K). Statistical significance was set at P < 0.05.
As expected, HuR was higher in the aortic valve leaflets in the HCD group than in the ND group (Figure 6F) and was reduced upon MS-444 treatment (Figure 6F). Moreover, PIP4K2A protein levels were higher in the HCD group than in the ND group (Figure 6H), while HuR inhibition by MS-444 treatment significantly decreased PIP4K2A mRNA and protein expression in the mouse aortic valve (Figure 6G and H, respectively), protein levels in the whole heart (Figure 6I), and circulating PIP4K2A mRNA levels (Figure 6J) compared with that in mice receiving the vehicle treatment, as confirmed by RNA-FISH, IF, western blotting, and qPCR. Furthermore, LC3 expression was upregulated in the HCD group compared with that in the ND group, whereas it decreased upon MS-444 treatment (Figure 6K). Accordingly, p-AKT, p-mTOR, and p-ATG13 expression was reduced in the HCD group compared to that in the ND group, whereas it increased upon MS-444 treatment (see supplementary material online, Figure S2C, E, and G). However, the total AKT, mTOR, and ATG13 expression showed no significant changes (see supplementary material online, Figure S2D, F, and H), indicating that sh-PIP4K2A activated the AKT/mTOR/ATG13 signalling pathway in vivo to hinder autophagy. Lastly, the twice-weekly MS-444 dosing regimen did not cause significant changes in metabolic parameters, such as glucose, total cholesterol (TC), low-density lipoprotein (LDL), and triglyceride (TG) levels (see Supplementary material online, Table S2), suggesting that MS-444 treatment ameliorates aortic valve calcification independently of metabolic regulation. These results further verified the role of HuR in osteoblast differentiation and provided evidence for a therapeutic strategy targeting HuR in CAVD treatment.
4. Discussion
A rapid increase in the technology of surgical or percutaneous aortic valve replacement has occurred recently, prolonging the lifespan of end-stage CAVD patients.34 However, there remains an unmet medical need to investigate effective therapeutic targets for early treatment of aortic valve calcification to halt progression. CAVD was previously considered a passive process due to aging;21 however, emerging advances suggest that CAVD is an active process involving multiple molecular and cellular events, including endothelial, valve interstitial, and circulating immune cells.2 Osteogenic differentiation of valve interstitial cells plays a crucial role in the development and progression of CAVD,8 which we partly validated in our study. HuR silencing significantly attenuated the VIC phenotype following osteogenic stimulation in vitro, as shown by the decreased protein expression of the osteogenic differentiation markers RUNX2 and ALP. This is consistent with the reduced valve calcification in vivo, evidenced by alizarin red and von Kossa staining in aortic valve tissues, and echocardiographic valve parameters. Therefore, our findings provide direct evidence that the VIC phenotypic transition plays a crucial role in CAVD progression and suggests a novel therapeutic target.
Autophagy is a major event in the initiation and development of CAVD, but it is still controversial whether autophagy plays a protective or harmful role in CAVD. Several hypotheses have been proposed to support the view that excessive activation of autophagy promotes calcification. First, the role of autophagy may be mediated by the degradation of key regulatory proteins associated with modulation of the VIC phenotype.21 Second, autophagy regulates Ca2+ homeostasis, and dysfunctional autophagy flux causes an imbalance between Ca2+ uptake and release, which is an essential constituent of hydroxyapatite deposition.21,26 Furthermore, a new field of secretory autophagy has emerged, which is opposed to traditional degradative autophagy, whereby autophagosomes release microvesicles from osteogenic cells to facilitate hydroxyapatite formation in the extracellular matrix, accelerating valve calcification progression.35 In our study, both human and animal calcification valves demonstrated activation of autophagy, as shown by increased LC3 protein levels and autophagosomes in western blotting, IF, and TEM analyses. Phosphatidylinositol signalling affects a large and diverse number of cellular processes, including proliferation, survival, metabolism, and autophagy. A form of PIP4K2A exists in intracellular locations, including endosomes, lysosomes, and the nucleus, and plays a critical role in autophagy.36 Functional experiments revealed that both HuR knockdown and PIP4K2A inhibition led to decreased autophagy flux along with amelioration of calcification in primary hVICs and mouse valve tissues, suggesting that autophagy promotes calcification pathogenesis. Overall, these findings demonstrate the fundamental importance of autophagy in CAVD. Thus, pharmacological intervention in the autophagy process is a promising CAVD treatment.
The RBP HuR is a crucial modulator of various cardiovascular diseases. HuR is an established regulator of post-transcriptional gene expression and is overexpressed in various human cardiovascular diseases.37,38 Recently, Hu et al.17 confirmed that HuR regulates phospholamban expression by stabilizing or destabilizing phospholamban mRNA to affect cardiac remodelling. In addition, Cai et al.39 revealed that ADAR1 regulates abdominal aortic aneurysm (AAA) development by interacting with HuR to increase the stability of matrix metalloproteinase (MMP)-2 and MMP9 mRNA. We wondered whether the ADAR1-mediated RNA editing relevant for the HuR-mediated control of PIP4K2A in our study. We designed an siRNA to silence ADAR in the hVICs and found that ADAR knockdown did not affect the RNA expression of pre-PIP4K2A and m-PIP4K2A (see supplementary material online, Figure S4A). In addition, we used the ADAR RNA editing inhibitor 8-azaadenosine, which also did not affect the RNA expression of pre-PIP4K2A and m-PIP4K2A (see supplementary material online, Figure S4B). Moreover, ADAR knockdown did not affect PIP4K2A stability (see supplementary material online, Figure S4C) and its interaction with HuR (see supplementary material online, Figure S4D), as shown by the RNA stability and RIP experiments. Therefore, we concluded that ADAR1-mediated RNA editing was not relevant for the HuR-mediated control of the HuR target described in this study. Moreover, Simion et al.18 reported that HuR is a critical regulator of apoptosis and efferocytosis in atherosclerosis by targeting a series of mRNAs, including p53, caspase-9, and BCL2. Wang et al.31 revealed that MALAT1 could positively regulate HuR expression through sponging miR-191-3p, and HuR reversely had a role in stabilization of MALAT1, thus forming a positive feedback loop between HuR and MALAT1 to regulate the CAVD progression. We also explored the MALAT1-mediated role in PIP4K2A expression. We designed an siRNA to knock down the expression of lncMALAT1 in the hVICs and found that lncMALAT1 knockdown did not affect PIP4K2A expression (see Supplementary material online, Figure S5A) or the autophagy level (see supplementary material online, Figure S5B). In addition, we used the CRISPR/Cas9 SAM virus to overexpress lncMALAT1, and it still did not affect the expression of PIP4K2A (see supplementary material online, Figure S5C). Furthermore, knockdown of PIP4K2A did not affect the expression of MALAT1 (see supplementary material online, Figure S4D). Therefore, we inferred that LncMALAT1 does not affect the pathological process of valve calcification by affecting PIP4K2A and the downstream autophagy pathway. Additionally, the RIP test supported that PIP4K2A expression increased more than that of LncMALAT1 (see supplementary material online, Figure S4E) in the RNA isolated after HuR immunoprecipitation than in that isolated from the IgG control, revealing a binding preference of PIP4K2A to HuR. Therefore, the mechanism of our experimental results is independent of that of the previous study. We showed that HuR expression was induced in both calcified human aortic valves and primary aortic VICs following osteogenic stimulation. HuR silencing significantly attenuated valve calcification and phenotypic alterations, as shown by the significantly decreased expression of osteogenic differentiation markers and ALP activity in hVICs in vitro, and decreased calcium deposition and improved echocardiographic valve parameters in vivo. Moreover, HuR silencing decreased the expression of PIP4K2A and autophagy flux, indicating a novel role for HuR in targeting PIP4K2A to promote osteogenic differentiation in hVICs and accelerate valve calcification in vivo by upregulating autophagy. The mechanism elucidated in our study involves the HuR subcellular localization effect on the target and osteogenic differentiation, which provides a deeper and more detailed understanding of this phenomenon than that in the previous study. Furthermore, our experiments involved animal models and we further explored the HuR target downstream autophagy pathway, which is more conducive to the clinically specific therapeutic target search and is a powerful expansion, supplement, and progression in the exploration of the valve calcification mechanism.
This study has several limitations. First, we only identified the mRNA stabilizer function of HuR in the cytoplasm. In addition to being expressed in the VIC cytoplasm, HuR is also expressed in the nucleus where it regulates transcription and splicing of target genes.40,41 Further work is necessary to explore more functions of HuR. Second, we only found that HuR positively regulated PIP4K2A expression during post-transcriptional processing. As a single RBP can modulate multiple coding and noncoding RNAs, whether HuR targets other key regulatory genes involved in aortic valve calcification requires further exploration. Third, there are limitations of the HCD-fed LDLR−/− mouse model that may not faithfully represent the full progression of human CAVD. Further research is needed to verify whether HuR inhibition can ameliorate aortic valve calcification in animal models applicable to humans. Besides, for HuR’s extensive regulatory roles in various cellular processes and diseases, we didn’t overexpress HuR in vivo to observe the aortic valve calcification, which may increase in uncontrollable factors or fatal diseases that affect the lifespan of mice and the observation of end-point events. We hope to construct specific in vivo disease models in the future to better study the specific functions. Finally, we did not conduct an in vivo investigation using genetically modified mice as HuR knockout is lethal because of its essential function in mouse embryonic development.42 Although small molecule inhibition of HuR silencing is effective for validating loss-of-function studies, a conditional VIC-specific HuR knockout murine model would be more persuasive.
Taken together, our study identified a novel role for HuR in CAVD. HuR increases the expression of PIP4K2A, thereby activating autophagy and promoting osteogenic differentiation of hVICs. Mechanistically, HuR interacts with PIP4K2A mRNA to promote its stability in hVICs, leading to the activation of autophagy via the AKT/mTOR/ATG13 pathway, accelerating CAVD progression. Therefore, HuR may be a novel target for hindering CAVD development.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
X.L. conceived the project. J.F. performed most experiments and wrote the manuscript. Y.Q., J.C., D.X., N.C., and G.Z. provided the materials and collected the samples. W.H., H.H., N.Q., and S.Y. participated in data analysis. J.W. supervised the research and revised the paper.
Acknowledgements
We thank all patients with the donation of samples for kindly providing aortic valve leaflets.
Funding
This work was supported by the National Natural Science Foundation of China (81770252 to X.L. and 82030014 and 81870292 to J.W.), National Key R&D Program of China (2019YFA0110400 and 2016YFC1301204 to J.W. and 2017YFA0103700 to X.H.), Zhejiang Province Science and Technology Department Key R&D Program (2022C03063 to X.L.), and Key R&D Program of Zhejiang Province Science and Technology Department (2021C03097 to J.W.).
Data availability
The data related to this article will be available upon request to the corresponding author.
References
As the incidence of calcific aortic valve disease (CAVD) has been rising over the past decade with only invasive, expensive, and risky interventions available, it is imperative to explore novel approaches to halt aortic valve calcification. Currently, regulation of CAVD pathogenesis is not fully understood. Human antigen R (HuR), an RNA–binding protein, and autophagy are thought to play key roles in CAVD; however, the precise underlying mechanisms have not been elucidated. Therefore, we believe that the findings of this study are relevant to the field because they provide direct evidence of a critical role for HuR, human aortic valvular interstitial cell (hVIC) phenotypic transition, and autophagy in CAVD and suggest a novel target for hindering CAVD progression.
Author notes
Conflict of interest: None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}