





Commentary on ‘A network of macrophages supports mitochondrial homeostasis in the heart’ by J.A. Nicolás-Ávila et al., Cell 2020.
The heart is an exceptional machine that must never cease to pump. To maintain sufficient energy supply for such strenuous mechanical workload, cardiac myocytes are densely packed with mitochondria, which occupy at least 35% of their cellular volume. Being terminally differentiated, cardiomyocytes have a limited ability to proliferate and thus cannot dilute their accumulating waste material among daughter cells. As such, long-lived cardiomyocytes predominantly rely on cellular quality control mechanisms to eliminate dysfunctional and potentially harmful organelles. One major homeostatic mechanism is macroautophagy, herein referred to as autophagy, which is a cellular self-digestion process responsible for surveillance and clearance of dysfunctional organelles including mitochondria.1,2 In fact, a growing body of evidence implicates dysfunctional autophagy in a plethora of cardiovascular diseases, as well as in age-related cardiac decline.3,4 In this context, Nicolás-Ávila et al.5 have recently discovered a novel mechanism supporting cardiac homeostasis, whereby cardiac resident macrophages closely cooperate with cardiomyocytes to eliminate their dysfunctional mitochondria. During this process, cardiomyocytes eject part of their autophagic cargo, including mitochondrial debris, into the interstitium within subcellular vesicles that are then phagocytosed and degraded by the nearby cardiac-resident macrophages (cMacs), thus coupling the cell-autonomous process of autophagy to macrophage-dependent heterophagy (Figure 1).
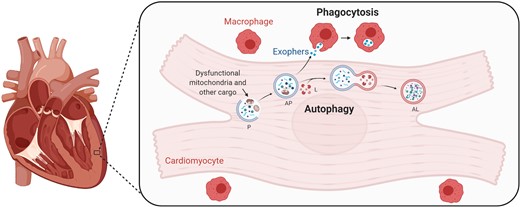
Cardiomyocytes partly outsource the degradation of dysfunctional mitochondria to resident macrophages. Schematic illustration of the process of autophagy in cardiomyocytes, whereby waste material, including dysfunctional mitochondria, is degraded. Shown also is how part of that cargo is exported in so-called exophers to the extracellular space, where it is phagocytosed by cardiac resident macrophages. AL, autolysosome; AP, autophagosome; L, lysosome; P, phagophore (created with BioRender.com).
In detail, Nicolás-Ávila et al. showed that cMacs are distributed throughout the heart but are particularly dense in and around the ventricular myocardium. Using high resolution imaging, they estimated that an average of 5 cMacs surround each cardiomyocyte. To determine the role of these cMacs under normal conditions (i.e. in the absence of infection or disease), the authors genetically depleted cMacs in mice. cMac-ablated adult mouse hearts exhibited higher numbers of mitochondria, which were structurally abnormal, as indicated by lower cristae density. Functionally, isolated mitochondria from cMac-ablated hearts showed reduced ATP generation, suggesting that these hearts might be incapable of meeting the high metabolic and energy demands necessary to sustain normal cardiac function. Indeed, 3 weeks after cMac ablation, mice exhibited compromised cardiac output and slower relaxation. Interestingly, following a subsequent recovery period of 3 weeks, cMac-depleted mice restored normal cardiac function, coinciding with cMac repopulating the heart. In a series of exhaustive experiments, employing genetic tools and state-of-the-art fluorescence microscopy, the authors elegantly demonstrated that cardiomyocytes extrude cellular material mainly composed of mitochondrial fragments in so-called cardiac exophers. These exophers were detectable nearby and inside neighbouring cMacs where they were targeted for degradation.
The authors went on to show that autophagy plays a central role in this process, as cardiac-specific deletion of the essential autophagy gene Atg7 significantly reduced the formation and release of exophers, while pharmacological activation of autophagy by rapamycin increased exopher production and mitochondrial transfer to the interstitium for subsequent uptake by cMacs. Furthermore, the authors demonstrated that cMacs modulate, albeit indirectly, autophagic activity in cardiac myocytes. Of note, extracellular accumulation of exophers in cMac-depleted hearts results in the activation of NLRP3 inflammasome, which in turn inhibits autophagy, leading to progressive decline in exopher export. In an effort to evaluate the role of cMacs under pathological conditions, the authors subjected cMac-depleted mice to myocardial infarction or β-adrenergic stress. The failure of cMac-depleted mice to up-regulate exopher formation in response to these cardiac insults worsened disease outcome. In a final twist of their fascinating study, Nicolás-Ávila et al. found that the tyrosine kinase receptor Mertk mediates the phagocytosis of exophers by cMacs. Mertk−/− mice exhibited increased extracellular accumulation of exophers, despite higher numbers of cMacs. Moreover, Mertk−/− mice closely recapitulated the functional and metabolic phenotype observed in cMac-depleted mice.
The authors are to be commended on their diligent efforts to elucidate a novel type of co-ordination between autophagy (cell-autonomous self-digestion) and heterophagy (phagocyte-mediated destruction). Macrophages, especially in the heart, exert multifaceted roles that go beyond immunity and infection control. For instance, macrophages actively contribute to cardiac electrical conduction, and their depletion causes a progressive atrioventricular block.6 Since the current study lacked detailed electrophysiological data,5 it is impossible to vaticinate whether, and to which extent, lower heart rates may contribute to the cardiac defects observed in cMac-depleted mice, especially reduced cardiac output and slower relaxation. Like any other pioneering study, this work raises multiple questions, which will need to be addressed by future research. Notably, it will be important to examine the physiological role of macrophages in maintaining mitochondrial homeostasis in other organs. For instance, unlike the liver for which Nicolás-Ávila et al. showed that macrophage depletion does not affect mitochondrial homeostasis,5 the central nervous system might turn out to involve similar mechanisms of autophagy–heterophagy coupling. Indeed, neurons, like cardiomyocytes, are long-lived, post-mitotic cells that are energy-demanding, rich in mitochondria and heavily rely on cell-autonomous homeostatic mechanisms like autophagy for their quality control.7 Moreover, at this point it is still unclear whether there is any direct dialogue between myocytes and macrophages that would regulate exopher ejection and accumulation in the extracellular space, as inhibiting the NLRP3 inflammasome only partially restored exopher formation in cMac-depleted hearts.
Finally, although the authors showed that extracellular exopher-like vesicles are present in the hearts from patients undergoing mitral valve replacement, further human testing will be necessary in order to translate these finding into actionable targets and effective therapeutics. Thus, it will be crucial to delineate the dynamics of this mechanism in healthy human hearts compared to common cardiomyopathies. For instance, in coronary artery disease, mitochondrial degradation may constitute a double-edged sword that might be beneficial during ischaemia but detrimental upon reperfusion,8 raising questions on the timing of therapeutic interventions on autophagy and heterophagy. Similarly, macrophages play an ambiguous role in the pathogenesis of atherosclerosis,9 meaning that targeting them for therapy will require fine-tuning their phagocytic vs. pro-inflammatory activities. To achieve this, novel molecules with high specificity will need to be developed. Until then, clinically-approved or naturally-occurring substances (e.g. metformin and spermidine, respectively) that are known to exert pro-autophagic and anti-inflammatory effects are worth testing.3,4 In doing so, future clinical trials will need to consider both the disease stage as well as the role of autophagy, being beneficial or detrimental, in disease pathogenesis.2,8
In conclusion, the findings reported by Nicolás-Ávila et al. substantiate an essential role for tissue-resident macrophages in cardiovascular health,10 that relies on the co-ordinated removal of dysfunctional mitochondria by autophagy and heterophagy.
Conflict of interest: G.K. is the scientific co-founder of Samsara Therapeutics, a company dedicated to the development of small molecule autophagy enhancer. He holds several patents and patent applications on autophagy inducers.
Funding
M.A. acknowledges support from the European Society of Cardiology, the Austrian Society of Cardiology, and the Medical University of Graz.
References
Authors

Biography: Dr Mahmoud Abdellatif is a post-doctoral research fellow at the Department of Cardiology, Medical University of Graz (Austria), and the Institute Gustave Roussy (Paris, France). In his research, he studies the molecular mechanisms of cardiovascular ageing to exploit them in the development of effective interventions that extend lifespan and delay late-life chronic diseases, including heart failure. During his PhD, he contributed to the discovery of the cardioprotective and longevity-promoting effects of spermidine, a natural autophagy inducer and caloric restriction mimetic. He has received various prestigious awards for his work including, the Sanofi Aventis Prize (2017) and the Guido Tarone award of the European Society of Cardiology Heart Failure Association (2020). As a nucleus member of the European Society of Cardiology Scientists of Tomorrow (SoT), he is actively involved in the dissemination of basic science amongst young clinicians and scientists.

Biography: Prof. Guido Kroemer is currently Professor at the Faculty of Medicine of the University of Paris, Director of the research team ‘Metabolism, Cancer and Immunity’ of the French Medical Research Council (INSERM), Director of the Metabolomics and Cell Biology platforms of the Gustave Roussy Comprehensive Cancer Center, and Hospital Practitioner at the Hôpital Européen George Pompidou, Paris, France. He is also a Foreign Adjunct Professor at the Karolinska Institute, Stockholm, Sweden, as well as an Honorary Professor at the Systems Medicine Institute of the Chinese Academy of Sciences in Souzhou. Dr Kroemer’s work focuses on the pathophysiological implications of cell stress and death in the context of ageing, cancer, and inflammation. Kroemer is the founding Editor-in-Chief of five journals: Cell Death & Disease, Cell Stress, OncoImmunology, Microbial Cell, and Molecular & Cellular Oncology. He is member of the European Molecular Biology Organization (EMBO), German Academy of Sciences (Leopoldina), Austrian Academy of Sciences, Academia Europaea, European Academy of Sciences (EAS), European Academy of Sciences and Arts (EASA), European Academy of Cancer Sciences (EACS), and Institut Universitaire de France (IUF). He is the Founding Director of the European Research Institute for Integrated Cellular Pathology (ERI-ICP) and the Founding President of the European Academy of Tumor Immunology (EATI).

{kind=link}
{kind=link}
{kind=link}