





Abstract
Abnormal Ca2+ release from the sarcoplasmic reticulum (SR), associated with Ca2+-calmodulin kinase II (CaMKII)-dependent phosphorylation of RyR2 at Ser2814, has consistently been linked to arrhythmogenesis and ischaemia/reperfusion (I/R)-induced cell death. In contrast, the role played by SR Ca2+ uptake under these stress conditions remains controversial. We tested the hypothesis that an increase in SR Ca2+ uptake is able to attenuate reperfusion arrhythmias and cardiac injury elicited by increased RyR2-Ser2814 phosphorylation.
We used WT mice, which have been previously shown to exhibit a transient increase in RyR2-Ser2814 phosphorylation at the onset of reperfusion; mice with constitutive pseudo-phosphorylation of RyR2 at Ser2814 (S2814D) to exacerbate CaMKII-dependent reperfusion arrhythmias and cardiac damage, and phospholamban (PLN)-deficient-S2814D knock-in (SDKO) mice resulting from crossbreeding S2814D with phospholamban knockout deficient (PLNKO) mice. At baseline, S2814D and SDKO mice had structurally normal hearts. Moreover none of the strains were arrhythmic before ischaemia. Upon cardiac I/R, WT, and S2814D hearts exhibited abundant arrhythmias that were prevented by PLN ablation. In contrast, PLN ablation increased infarct size compared with WT and S2814D hearts. Mechanistically, the enhanced SR Ca2+ sequestration evoked by PLN ablation in SDKO hearts prevented arrhythmogenic events upon reperfusion by fragmenting SR Ca2+ waves into non-propagated and non-arrhythmogenic events (mini-waves). Conversely, the increase in SR Ca2+ sequestration did not reduce but rather exacerbated I/R-induced SR Ca2+ leak, as well as mitochondrial alterations, which were greatly avoided by inhibition of RyR2. These results indicate that the increase in SR Ca2+ uptake is ineffective in preventing the enhanced SR Ca2+ leak of PLN ablated myocytes from either entering into nearby mitochondria and/or activating additional CaMKII pathways, contributing to cardiac damage.
Our results demonstrate that increasing SR Ca2+ uptake by PLN ablation can prevent the arrhythmic events triggered by CaMKII-dependent phosphorylation of RyR2-induced SR Ca2+ leak. These findings underscore the benefits of increasing SERCA2a activity in the face of SR Ca2+ triggered arrhythmias. However, enhanced SERCA2a cannot prevent but rather exacerbates I/R cardiac injury.
1. Introduction
Ischaemia/reperfusion (I/R) injury is one of the leading causes of heart failure, cardiovascular disease, and sudden death in the western world.1 The consequences of cardiac ischaemia include reduced force production, arrhythmias, and cell death. To limit these detrimental effects, reperfusion therapies are the standard treatment for patients suffering myocardial infarction. However, re-establishing blood flow is usually associated with additional cell damage, a phenomenon referred to as I/R injury, exacerbating the effect of the preceding ischaemia. Indeed, it was shown that reperfusion may trigger life-threatening arrhythmias and accounts for up to half of the total I/R-induced infarct size.2–5
Although the factors contributing to I/R injury are complex, experimental evidence reveals that loss of Ca2+ homeostasis is one of the major contributing mechanisms.3,6–8 It has been established that at least one of the signalling death pathways of this multifactorial injury involves Ca2+-calmodulin kinase II (CaMKII)—dependent phosphorylation RyR2-Ser2814, mitochondrial Ca2+ overload through the mitochondrial Ca2+ uniporter (MCU), cytochrome c release, and caspase-3 activation.3,9–11 Supporting and extending this signalling cascade, Joiner et al.12 showed that CaMKII-dependent phosphorylation of MCU increases Ca2+ entry into the mitochondria, favouring cell death.
In addition, alterations of RyR2 open probability have been associated with ventricular arrhythmias in different pathologies. For instance, RyR2 mutation is one of the causes of catecholaminergic polymorphic ventricular tachycardia (CPVT) and CaMKII-dependent phosphorylation of RyR2 at Ser2814 has been associated with heart failure and reperfusion-induced arrhythmias.4,13–15 Moreover, experimental evidence indicated that animals with constitutive pseudo-phosphorylation at the RyR2-Ser2814 site were more prone to stress-induced arrhythmias.16,17 The increase in sarcoplasmic reticulum (SR) Ca2+ leak produced by RyR2 alterations, if high enough, may evoke SR Ca2+ waves and enhance Ca2+ extrusion via the electrogenic Na+/Ca2+ exchanger (NCX). This electrogenic transport generates a depolarizing current, which—when sufficiently large—leads to delayed afterdepolarizations (DADs) and eventual triggering of ectopic action potentials (APs) and ventricular arrhythmias.18,19
In contrast to the clear participation of CaMKII-dependent phosphorylation of RyR2 on cardiac damage and ventricular arrhythmias during I/R, the role of increasing SR Ca2+ uptake remains uncertain due to controversial findings. Several studies support a beneficial effect of accelerating SR Ca2+ uptake in different pathologies, including experimental heart failure,20–23 while others suggest a detrimental role.24–27 The cause for these inconsistent findings may rest on the opposite effects that the augmented SR Ca2+ uptake has on cytosolic Ca2+. On one hand, increasing the rate of SR Ca2+ reuptake would increase SR Ca2+ content, favouring RyR2 Ca2+ sensitization, enhanced diastolic SR Ca2+ leak, and the consequent risk of Ca2+ waves and mitochondria damage. On the other hand, it would reduce cytosolic Ca2+, and therefore, the risk of Ca2+ diffusion throughout the cytosol and towards the mitochondria. Thus, a slight imbalance between SR Ca2+ uptake and leak may favour either a protective or a harmful outcome with opposite consequences on I/R cardiac damage and arrhythmias. Another possibility that can independently add variability to these complex outcomes, is the increase in energy consumption that occurs as a consequence of the enhanced SR Ca2+ uptake, which would conspire against post-ischaemic recovery as previously shown in PLNKO mice.24
In the present study, we tested the hypothesis that an increase in SR Ca2+ uptake produced by PLN ablation rescues the vulnerability to reperfusion arrhythmias and infarction, associated with RyR2 phosphorylation by CaMKII at Ser2814 and explored the underlying mechanisms. To this purpose, we used hearts from WT mice, whose transient phosphorylation of RyR2-Ser2814 at the onset of reperfusion is associated with reperfusion cardiac damage (infarction) and arrhythmias7,28; hearts from S2814D mice, with constitutive pseudo-phosphorylation of RyR2-Ser2814,16 in an attempt to exacerbate the effects of RyR2-Ser2814 phosphorylation at the onset of I/R,4,9,11 and hearts from PLNKO and SDKO mice, the latter resultant from crossbreeding PLNKO with S2814D mice. The results indicate that our hypothesis holds true for reperfusion arrhythmias. However, ablation of PLN exacerbates cardiac infarct associated with Ser2814-RyR2 phosphorylation. These distinct effects involved different sub-cellular mechanisms that were not related to increases in energy demand.
2. Methods
An expanded Methods section is available in the Supplementary material online.
2.1 Animals
Experiments were performed in 3–4-month-old male mice: PLN deficient mice,29 RyR2-S2814D knock-in,16 and PLN deficient/RyR-S2814D, resulting from crossbreeding PLNKO and RyR2-S2814D mice (SDKO mice).17 C57BL/6 mice that constituted the background for PLNKO and S2814D mice, were used as controls (WT mice). All experiments were performed in accordance with the Guide for Care and Use of Laboratory Animals (NIH Publication No.85-23, revised 2011) and approved by the Ethics committee of the Faculty of Medicine, La Plata, Argentina (CICUAL # P03-01-15).
2.2 Langendorff perfusion and experimental protocol
Animals were anaesthetized with an intraperitoneal injection of ketamine/diazepam (100 mg/kg and 5 mg/kg, respectively). Central thoracotomy and heart excision were performed immediately after Phase III anaesthesia was reached, verified by the loss of pedal withdrawal reflex. Isolated hearts were perfused according to the Langendorff technique at constant temperature (37°C) and flow (2–3 mL/min), as previously described.28,30 Hearts were subjected to a protocol of either 15 or 45 min of no-flow ischaemia followed by reperfusion, to assess reperfusion arrhythmias,4 mechanical recovery, infarct size,9 lactate dehydrogenase (LDH) release,9 and mitochondrial integrity.31
2.3 Histology
Cardiac wall, myocyte dimensions and interstitial fibrosis were assessed in hearts from SDKO and WT mice. After paraffin embedding and sectioning, samples were stained with haematoxylin-eosin for cell morphology and with Picrosirius techniques for interstitial fibrosis, as previously described.32
2.4 Western blot analysis
Cardiac homogenates were prepared as previously described.32 Protein was measured by the Bradford method using BSA as standard. Lysates were subjected to SDS-PAGE.33 After western blotting, membranes were probed overnight with antibodies raised against total PLN (Thermo Scientific MA3-922, Waltham, MA, USA), phosphor-Ser2814 (Badrilla A010-31, Leeds, UK), and total CaMKII (Badrilla A010-56AP, Leeds, UK), total RyR2 (Thermo Scientific MA3-916, Waltham, MA, USA), SERCA2a (Thermo Scientific MA3-919, Waltham, MA, USA), and NCX (Millipore MAB1590, Burlington, MA, USA), and appropriate secondary antibodies. Glyceraldehyde phosphate dehydrogenase (GAPDH, Millipore, MA, USA) was used as a loading control.
2.5 Echocardiographic examination
Baseline cardiac geometry and function were evaluated by two-dimensional M-mode echocardiography, as previously described.17
2.6 Intracellular Ca2+ and APs
Rhod-2 and Di-8-ANEPPS (Invitrogen, USA) were used to assess intracellular Ca2+ transients at the cytosol (Rhod-2) and transmembrane APs, respectively. Measurements were performed at the epicardial layer of intact mouse hearts using a custom-made setup for pulse local-field fluorescence microscopy.34
2.7 Ca2+ sparks and waves in the intact heart
Spontaneous local SR Ca2+ release was evaluated at the epicardial layer of intact mouse hearts, as previously described, using Fluo-4 AM (Invitrogen, OR, USA) and laser scanning confocal microscopy.6 A modified upright confocal microscope (OlympusFV 1000) with long-working distance and high-numerical apertures (0.80/40x and 1.0/20x) objectives was used for the fluorescence recordings. Both line scan recordings and two-dimesional images were obtained from the left ventricular epilayer, as previously described.6
2.8 Infarct size
After reperfusion, infarct size was assessed by the triphenyltetrazolium chloride technique.9
2.9 LDH determination
Cardiac injury was also evaluated by LDH released in the perfusion effluent during the first 10 min of reperfusion.11
2.10 Cardiac energetics
The energetics of isolated mouse hearts were measured inside a flow-calorimeter at 37°C, as previously described,35 while perfusing them with Krebs-2.5 mM Ca2+ at 1.5–2 mL/min.
2.11 Isolation and function of mouse heart mitochondria
Two groups of mouse cardiac mitochondria were obtained. The first set was isolated from perfused hearts that were not submitted to I/R (control mitochondria); the second set of mitochondria was isolated from hearts submitted to I/R (15/10 min), after completion of the reperfusion period. The perfused hearts were immediately washed and mitochondria were obtained by differential centrifugation as previously described.31 Mitochondrial potential changes were evaluated by measuring rhodamine-123 (RH-123, Sigma-Aldrich, St Louis, MO, USA) fluorescence quenching and mitochondrial swelling by light scattering.31
2.12 Statistics
Continuous variables were expressed as mean ± SEM and were evaluated with either unpaired Student’s t-test or one or two-way ANOVA followed by Tukey's post hoc test, to compare differences among groups. The Kruskal–Wallis test was used to compare differences among groups with non-normal data distribution. Categorical data were expressed as percentages and compared with Fisher’s exact test. A P-value <0.05 was considered significant.
3. Results
3.1 SDKO mice exhibit a combined functional phenotype of PLN ablation and RyR2-S2814 phosphorylation
Knock-in mice in which aspartic acid replaces serine at RyR2-2814 (S2814D) to mimic constitutive phosphorylation of RyR2 by CaMKII were crossbred with PLNKO. The resultant offspring (SDKO) did not exhibit any significant structural difference when compared with WT mice. On western blots, no significant alterations were observed in the main proteins related to Ca2+ handling, but RyR2 expression was decreased by approximately 33% in agreement with previous findings17,29 (Supplementary material online, Figures S1 and S2).
The basal mechanical and Ca2+ handling characteristics of SDKO hearts were also examined. Figure 1A–D shows the results of different basal mechanical parameters of Langendorff perfused S2814D, SDKO, PLNKO, and WT mouse hearts. In SDKO hearts, left ventricular developed pressure (LVDP) was similar to S2814D (Figure 1A). However, maximal rate of rise of LVDP (+dP/dt) and maximal velocity of relaxation (−dP/dt) (Figure 1B and C) were significantly higher than S2814D and WT hearts and lower than PLNKO. Half relaxation time (t½) (Figure 1D), was significantly shorter than in S2814D and WT but similar to PLNKO. Figure 1E shows that the Ca2+ transient amplitude, measured at the epicardial layer of the intact beating heart, was significantly higher in PLNKO hearts than in the other three groups. Half relaxation time of Ca2+ transient (Figure 1F) was significantly shorter in both the SDKO and PLNKO compared with WT and S2814D hearts. Taken together, these results indicate that SDKO hearts exhibit the combined functional phenotype of PLNKO and S2814D hearts.
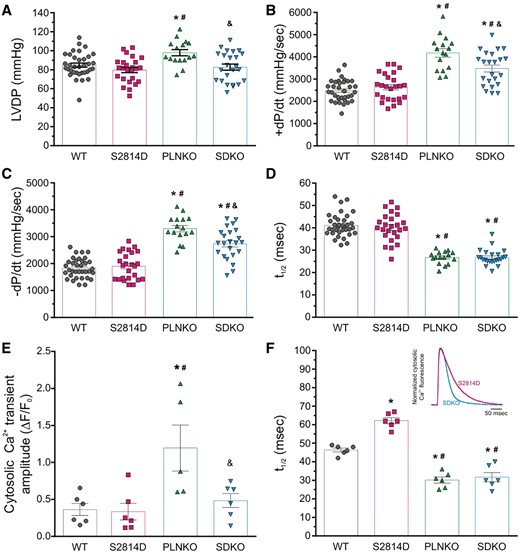
Mechanical and Ca2+ transients parameters under basal conditions in the intact heart of the different strains studied. (A) LVDP was significantly higher in PLNKO mice when compared with the other strains. (B and C) +dP/dt and relaxation rate (−dP/dt) were significantly increased in PLNKO and SDKO mice vs. S2814D and WT hearts. (D) PLNKO and SDKO mice also showed a shorter half relaxation time (t1/2). (E) Cytosolic Ca2+ transients at the epicardial layer of the intact heart were significantly higher in PLNKO mice when compared with the other strains. (F) Relaxation of Ca2+ transient was faster in PLNKO and SDKO, and slower in S2814D respect to WT hearts. Bars represent mean ± SEM. One-way ANOVA followed by Tukey’s post hoc test, was used to compare different groups *P < 0.05 vs. WT, #P < 0.05 vs. S2814D, and &P < 0.05 vs. SDKO. N = 16–33 hearts in the different groups for mechanical data and 5–6 hearts in the different groups for cytosolic Ca2+ data.
3.2 Reperfusion arrhythmias are not exacerbated in S2814D mice
Previous experiments in WT hearts showed that auto-phosphorylation of CaMKII and phosphorylation of RyR2-Ser2814 are significantly increased by approximately two-fold at the onset of reperfusion with respect to basal levels (pre-ischaemic).4,9 In those studies, CaMKII-dependent phosphorylation of RyR2 was tightly associated to reperfusion arrhythmias and cardiac damage.4,9 However, the observed phosphorylation of RyR2-Ser2814 was transient, as it peaked between 1 and 3 min of reperfusion and then returned to pre-ischaemic values.36 Thus, we used homozygous S2814D mice, which have constitutively phosphorylated Ser2814 in all four RyR2 subunits/channels,36 and are more prone to stress-induced arrhythmias,16,17 to determine whether this permanent pseudo-phosphorylation is associated with increased propensity to reperfusion-induced arrhythmias compared with the transient phosphorylation in WT hearts.4,9 To that purpose, we subjected WT and S2814D hearts to I/R (15/30 min) in the presence of blebbistatin to minimize movement artefacts. Hearts were paced at 5 Hz during pre-ischaemia. After 1 min of establishing no-flow ischaemia, stimulation was stopped and the hearts beat spontaneously for the rest of the protocol. Figure 2 presents a scheme of the experimental protocol (Figure 2A) and shows typical examples (Figure 2B and C) of WT and S2814D hearts submitted to the I/R protocol. During reperfusion, spontaneous beats presented a similar occurrence in both WT and S2814D hearts. Left bars of Figure 2E and F shows that both the incidence and frequency of spontaneous beats in S2814D hearts is not significantly different, when compared with WT hearts. These results indicated that, in contrast to stress-induced arrhythmias,17 S2814D hearts have a propensity to reperfusion arrhythmias similar to WT mice. They also suggest that permanent CaMKII-dependent phosphorylation of the S2814 in RyR2 does not enhance the proarrhyhtmic action associated with the transient increase in S2814 phosphorylation of WT hearts at the onset of reperfusion.4 Possible explanations for these somewhat unexpected results are that: (i) the transient but significant increase in CaMKII-dependent phosphorylation of RyR2 in WT hearts at the onset of reperfusion is sufficient to account for CaMKII-induced reperfusion arrhythmias; and (ii) SR Ca2+ load at the beginning of reperfusion is lower in S2814D vs. WT hearts. Indeed, this possibility is supported by previous experiments showing that basal SR Ca2+ load was lower in S2814D than in WT isolated myocytes16,17 (see Discussion below).
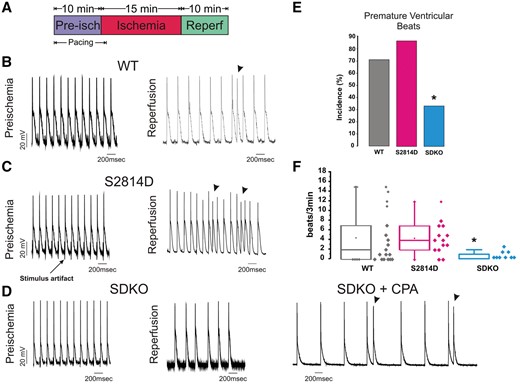
PLN ablation rescues the reperfusion ventricular arrhythmias produced by CaMKII-dependent phosphorylation of RyR2 at the S2814 site in the intact heart. (A) Schematic representation of I/R protocol. (B–D) Representative action potential tracings during pre-ischaemia (left) and at the onset of reperfusion (right), in WT (B), S2814D (C), SDKO, and SDKO hearts in the absence and presence of CPA (D). Arrow in C indicates electrical stimulus artefact on AP recording with microelectrodes. Arrowheads in C and D indicate premature beats. (E and F) Premature ventricular beat incidence and frequency (box plot), respectively. Categorical data (E), expressed as percentages, were compared with Fisher’s exact test. Non-parametric data (F) were compared with the Kruskal–Wallis test. A P-value <0.05 was considered significant. *P < 0.05 vs. WT and S2814D mice. N = 9–17 hearts in the different groups.
3.3 PLN ablation rescues reperfusion arrhythmias typical of WT and S2814D mouse hearts
We then tested the hypothesis that an increase in SR Ca2+ uptake, produced by PLN ablation rescues the vulnerability to reperfusion arrhythmias associated with CaMKII phosphorylation of Ser2814-RyR2. To this purpose, we subjected SDKO hearts to a protocol of 15/30 min I/R in the presence of blebbistatin, as described above. The results of these experiments are depicted in Figure 2D and the last bars of Figure 2E and F. Ablation of PLN diminishes reperfusion arrhythmias observed in both WT and S2814D hearts. As a proof of concept, we performed additional experiments in SDKO hearts treated with 1 µM of the SERCA2a inhibitor cyclopiazonic acid (CPA), in an attempt to diminish SR Ca2+ uptake and mimic S2814D (and WT) arrhythmic behaviour. Figure 2D shows the results of one of these experiments (SDKO + CPA). As expected, the decrease in SR Ca2+ uptake by CPA, recapitulates the phenotype of S2814D, i.e. reappearance of reperfusion arrhythmias. Overall results of these experiments are shown in Figure 2E. Taken together, our findings demonstrate that PLN ablation is capable of significantly diminishing reperfusion-induced arrhythmias.
3.4 PLN ablation increased reperfusion SR Ca2+ leak but rescued I/R-induced Ca2+ waves
Next, we sought to elucidate the underlying mechanisms by which increasing SR Ca2+ uptake by PLN ablation prevents reperfusion arrhythmias. To that purpose, we use confocal microscopy to study spontaneous Ca2+ release events in WT, S2814D, and SDKO hearts. Hearts were stabilized and loaded with the calcium-sensitive fluorescent dye under spontaneous beating and then submitted to 15/30 min I/R protocol in the presence of a combination of tetrodotoxin/carbamylcholine and blebbistatin to minimize the intrinsic electrical activity of the heart and cardiac movements, respectively (see Methods section). Figure 3A displays typical examples of line scan recordings, showing epicardial Ca2+ sparks obtained during reperfusion. Ca2+ sparks were more frequent in WT and SDKO than in S2814D hearts. In several cases, as in the example shown in Figure 3A for SDKO hearts, spontaneous Ca2+ sparks can be visualized as clusters of high frequency events, reminiscent of the repetitive releases at individual sites, described in these hearts by Hüser et al.37 These in turn could trigger additional sparks in immediate vicinity.37 Our findings are consistent with the enhanced SR Ca2+ uptake and the reduced refractoriness of PLNKO hearts, similar to that of SDKO, that we previously described.17,Figure 3B represents the binned histogram distribution of the kinetic characteristics of Ca2+ sparks in SDKO hearts: full width at half maximum (FWHM) and full duration at half maximum (FDHM). Superimposed for comparison are the curves (in fuchsia) of the corresponding histograms of S2814D hearts, showing a similar FWHM in SDKO and S2814D hearts, but a shorter FDHM in SDKO vs. S2814D, consistent with a higher SR Ca2+ uptake and/or an increased cytosolic Ca2+ buffering in the hearts of SDKO mice. Figure 3C shows that Ca2+ spark frequency is significantly higher in SDKO than in S2814D hearts. Figure 3D and E displays averaged Ca2+ spark mass (calculated as spark amplitude * 1.206 * FWHM3)38 and the estimated spark-mediated SR Ca2+ leak [defined as Ca2+ spark frequency (CaSpF) * spark mass], respectively, which are higher in SDKO than in WT and S2814D hearts. Figure 3 of supplementary information shows a scatter plot indicating the variation in spark frequency, spark mass, and spark-mediated Ca2+ leak in WT, S2814D, and SDKO hearts. The significantly higher spark mass of SDKO vs. S2814D and WT hearts, would strongly suggest a higher SR Ca2+ load in SDKO hearts, upon reperfusion.39 Moreover, the significantly lower Ca2+ spark frequency and spark mass in S2814D mice vs. WT would support the idea that the non-stimulated I/R protocol, used to visualize spontaneous events,6 further decreases SR Ca2+ load with respect to WT hearts. Interestingly, although smaller, SR Ca2+ leak in S2814D did not differ significantly from that of WT mice (Figure 3C).
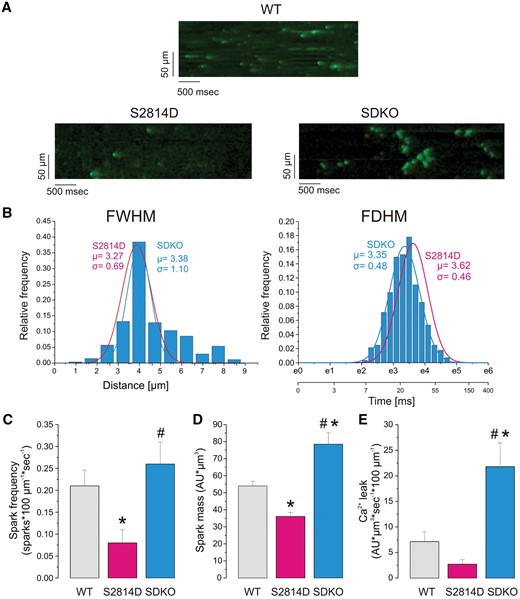
The frequency of epicardial Ca2+ sparks and SR Ca2+ leak at the onset of reperfusion is higher in SDKO than in WT and S2814D hearts. (A) Typical examples of line scan recordings of epicardial Ca2+ sparks obtained in the intact hearts of WT, S2814D, and SDKO at the beginning (3 min) of reperfusion after 15 min of ischaemia. (B) Linear binned distribution of FWHM and log-binned distribution of FDHM of Ca2+ sparks of the intact SDKO hearts. The corresponding curves of S2814D hearts are superimposed for comparison. FDHM was shorter in SDKO vs. S2814D. (C) Frequency of reperfusion Ca2+ sparks in WT, S2814D, and SDKO intact hearts. (D and E) Mass of Ca2+ sparks and estimated spark-mediated SR Ca2+ leak in WT, S2814D, and SDKO at the onset of reperfusion, respectively. Spark frequency at the onset of reperfusion was similar in SDKO than WT hearts and in both cases, higher than in S2814D. Spark mass and spark-dependent RS Ca2+ leak were significantly higher in SDKO than in S2814D and WT hearts and lower in S2814D than in WT hearts. Temperature: 32°C. (C–E) Bars represent mean ± SEM. One-way ANOVA followed by Tukey’s post hoc test, was used to compare different groups. A P-value <0.05 was considered significant. *P < 0.05 vs. WT mice, #P < 0.05 vs. S2814D. Data were obtained from 354, 318, and 281 events from five different WT, S2814D, and SDKO hearts, respectively.
Figure 4A and B shows typical confocal images of Ca2+ waves obtained during reperfusion in WT and SDKO. Whereas WT hearts presented full Ca2+ waves upon reperfusion, SDKO showed only a few detectable full propagating Ca2+ waves, despite severe spark-mediated SR Ca2+ leak (Figure 3). In contrast, they presented uncompleted Ca2+ waves (mini-waves). In some experiments, the spontaneous events appear as a series of mini-events, like necklace beads (red arrows in the expanded view of Figure 4B), conserving the shape of a wave, that finally fades out. Figure 4C shows the overall results of the experiments. In reperfusion, SR Ca2+ wave frequency was lower in SDKO vs. WT hearts. In contrast, either fragmented waves or mini-waves prevailed over Ca2+ waves in SDKO hearts. As a proof of concept, in four of the five experiments performed in SDKO hearts, SERCA2a inhibitor CPA was added after 10 min of reperfusion to diminish SR Ca2+ uptake. Notably, and similar to the behaviour observed when measuring ventricular arrhythmias, Figure 4D shows that SDKO hearts changed their typical mini-wave reperfusion pattern after the addition of CPA (red arrows), showing full instead of broken Ca2+ waves (right panel of Figure 4D). Similar results were obtained in three additional experiments of this type. Supplementary material online, Figure S4 shows additional examples of the interrupted Ca2+ waves observed during reperfusion in both SDKO and PLNKO hearts, which were transformed in non-fragmented Ca2+ waves in the presence of CPA. In addition, Supplementary material online, Videos S1 and S2 show the different Ca2+ wave pattern elicited by reperfusion in WT and SDKO hearts under confocal microscopy in the intact heart (see also the time series confocal images). Taken together, the results suggest that the enhancement of SR Ca2+ uptake, although able to persistently produce spontaneous Ca2+ events, averts Ca2+ wave propagation and transforms Ca2+ waves into fragmented, non-entire cell propagated SR Ca2+ release events.
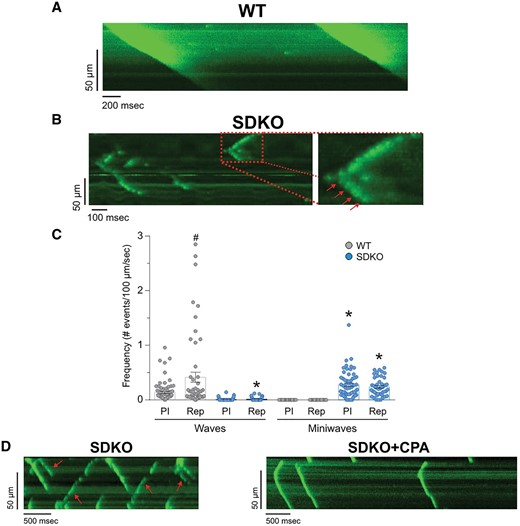
Epicardial Ca2+ waves at the onset of reperfusion are fragmented into non-propagated events in SDKO hearts. (A and B) Protocol and typical examples of line scan recordings of epicardial Ca2+ waves obtained at the beginning of reperfusion in intact WT and SDKO hearts. (C) Frequency of Ca2+ waves and mini-waves during pre-ischaemia and at the beginning of reperfusion in WT and SDKO hearts. (D) Protocol and typical recordings of line scan before and after the addition of the SERCA2a inhibitor CPA in SDKO hearts. Red arrows in B and D indicate fragmented SR Ca2 waves. One-way ANOVA followed by Tukey’s post hoc test, was used to compare different groups. A P-value <0.05 was considered significant. *P < 0.05 vs. WT mice, #P < 0.05 vs. pre-ischaemia. Data are obtained from five WT and five SDKO hearts.
3.5 PLN ablation exacerbates the damage produced by I/R in S2814D hearts
Previous experiments showed that when Ser2814D hearts were subjected to a protocol of I/R (45/120 min), there was a significant increase in infarct size and HDL release with respect to WT mice. Moreover, prevention of S2814 phosphorylation diminished I/R injury, i.e. necrotic and apoptotic death, and improved functional recovery in the intact heart.9 Thus, the following series of experiments was designed to test whether ablation of PLN could prevent I/R cardiac damage, induced by RyR2-Ser2814 phosphorylation at the onset of reperfusion. To this purpose, SDKO, PLNKO, S2814D, and WT hearts were subjected to two different protocols of I/R: 45/120 and 15/30 min. In both protocols, hearts were paced at 5 Hz during pre-ischaemia and reperfusion. Stimulation was stopped during the ischaemic period, after 1 min of cessation of coronary flow.7,28,30,Supplementary material online, Figure S5A and B shows that whereas left ventricular end diastolic pressure was similarly affected in the four groups of hearts, the prolonged ischaemic period (45 min) did not affect LVDP in S2814D relative to WT hearts, in agreement with previous findings.9 However, LVDP was greatly diminished in PLNKO and SDKO hearts vs. WT, in consonance with previous findings in PLNKO hearts.24 Similarly, infarct size and LDH release during reperfusion were significantly greater in SDKO and PLNKO hearts compared with WT and S2814D (Supplementary material online, Figure S5C and D).
Since the long I/R protocol produced an almost null mechanical recovery and an infarct of nearly 70% of the total risk area in SDKO and PLNKO hearts, we explored the effect of a shorter I/R period (15/30 min) in an attempt to uncover any potential differences between PLNKO and SDKO hearts. The ischaemic period of 15 min was chosen because it is known to produce a reversible functional injury, without structural cardiac damage in WT (control) hearts.40,41,Figure 5 shows the results of this group of experiments. Whereas the mechanical recovery was similar between WT and S2814D hearts, it was significantly lower in SDKO hearts (Figure 5A and B). The infarct size was not significant in WT and S2814D hearts; the 5–10% infarct of these hearts (arrows) mostly corresponds to the damage produced by stimulation electrodes. In contrast, infarct size significantly increased in PLNKO and SDKO hearts. Notably, the infarct size and LDH release were significantly higher in SDKO than in PLNKO hearts (Figure 5C and D). It is important to note that S2814D hearts suffered a significantly higher infarct size than WT hearts only when submitted to the prolonged I/R protocol (Supplementary material online, Figure S5). These findings suggest that the permanent phosphorylation of Ser2814, which should be associated with a constant increased SR Ca2+ leak during the whole I/R period, increases cardiac injury over that produced by the transient phosphorylation of S2814 in WT at the onset of reperfusion. In addition, our results indicate that the increase in SR Ca2+ reuptake produced by PLN ablation is unable to rescue the damage evoked by I/R in WT and S2814D hearts. On the contrary, cardiac injury was even higher in SDKO hearts than in all other groups, including PLNKO. An additional finding of these experiments is that the mechanical recovery tended to be dissociated from the structural damage in S2814D and SDKO hearts, when compared with WT and PLNKO hearts, at least within the first 30 min of reperfusion. Although we did not explore the reason for this uncoupling, a possible explanation may lie in the opposite effects of CaMKII-dependent phosphorylation at RyR2-Ser2814 site: an increase in fractional Ca2+ release, which would contribute to support LVDP42 and an enhancement of SR Ca2+ leak, which would possibly exacerbate cardiac damage.16,17
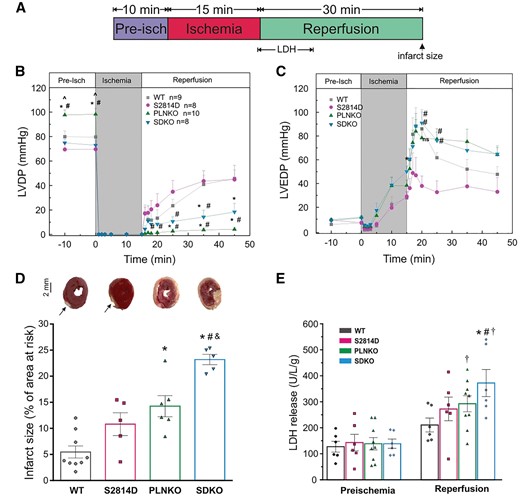
PLN ablation produces mechanical dysfunction and cardiac damage in S2814D hearts submitted to short-term I/R. (A) Experimental protocol. LDH indicates the time period of samples recollection for LDH measurements. Head arrow indicates the time point of infarct determination. (B and C) Mice with dual mutation (SDKO) had a slightly better mechanical recovery compared to PLNKO but both groups had a significantly greater deterioration with respect to WT and S2814D mice. (D and E) After short-term ischaemia, SDKO hearts have the larger infarct area and the highest LDH release in reperfusion, compared with the other groups. Of note, in S2814D infarct size and LDH release were not different from values in WT hearts. Arrows indicate the damaged area, mostly corresponding to the insertion of stimulation electrodes. One-way ANOVA followed by Tukey’s post hoc test, was used to compare different groups. A P-value <0.05 was considered significant. *P < 0.05 vs. WT mice, #P < 0.05 vs. S2814D, ^P < 0.05 vs. SDKO, &P < 0.05 vs. PLNKO, and †P < 0.05 vs. the corresponding pre-ischaemic values (Paired t-test). N = 5–9 hearts in the different groups.
3.6 The enhanced cardiac damage of SDKO vs. S2814D hearts is associated with mitochondrial dysfunction but not with augmented energy consumption
Several possible explanations can account for the higher damage produced by I/R in SDKO hearts vs. S2814D hearts. Previous experiments indicated that PLNKO hearts are more vulnerable to I/R injury due to the high energy cost inherent to the enhanced Ca2+ turnover and increased contractility typical of PLN ablation.24 Thus, the higher energy consumption in SDKO mice, associated with PLN ablation, could explain the observed difference.
To assess whether energy metabolism is altered, we measured the energetic demand in all four groups of hearts submitted to a protocol of 15/30 min I/R, as the one used to measure infarct size and cardiac mechanics. Hearts were paced at 5 Hz during pre-ischaemia and reperfusion and stimulation was stopped after one minute of flow cessation during the ischaemic period. As shown in Supplementary material online, Figure S6, the pre-ischaemic total heat production (Ht), was similar in SDKO, PLNKO, and S2814D and in all cases greater than in WT hearts. These results suggest that the higher reperfusion damage, observed in SDKO vs. S2814D hearts, cannot be attributed to a higher energetic demand in SDKO hearts vs. S2814D. Moreover, total Ht in SDKO hearts was lower than in PLNKO during the protocol of I/R, consistent with the higher infarct size and mitochondria dysfunction of double mutant hearts (see Figures 5D and 6B, C ).
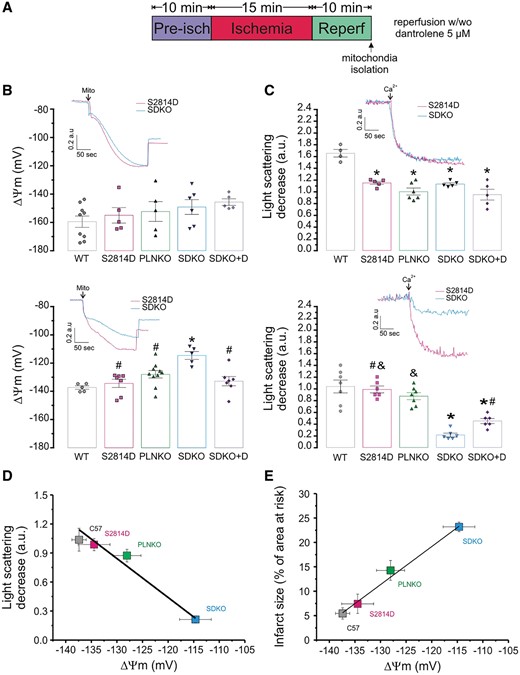
The enhanced cardiac damage of SDKO hearts is associated with mitochondrial dysfunction. (A) Experimental protocol. (B) Typical rhodamine fluorescence changes, expressed in arbitrary units (a.u.), in mitochondrial isolated from perfused (upper panel) and ischaemic-reperfused (lower panel) hearts of S2814D and SDKO hearts, and mean values of mitochondrial membrane potential (ΔΨm, mV) in all groups (WT, S2814D, PLNKO, and SDKO). (C) Typical traces of light scattering after the addition of 50 μM Ca2+ to mitochondrial samples isolated from perfused hearts (upper panel) and ischaemic-reperfused hearts (lower panel) of S2814D and SDKO hearts, and mean values of LSD in mitochondrial samples from all groups. (C) Correlation between LSD and ΔΨm. (D) Correlation between infarct size and ΔΨm. One-way ANOVA followed by Tukey’s post hoc test, was used to compare different groups. A P-value <0.05 was considered significant. *P < 0.05 vs. WT, #P < 0.05 vs. SDKO, and &P < 0.05 vs. SDKO + dantrolene. N = 5–9 hearts in the different groups.
A second possible explanation that may account for the higher damage, observed during I/R in SDKO hearts, is the increased in SR Ca2+ leak exhibited in SDKO described above (Figure 3). If the increase in SR Ca2+ leak is involved in the bigger infarct size of SDKO mice, it is reasonable to expect a more prominent mitochondrial dysfunction produced by mitochondrial Ca2+ overload in SDKO than in S2814D and WT hearts, which in turn might be circumvented by inhibiting SR Ca2+ leak. To test this hypothesis, we evaluated mitochondrial state in isolated mitochondria, obtained from the four different groups of hearts that were submitted to a protocol of I/R (15/10 min) and paced during pre-ischaemia and reperfusion (stimulation was halted after 1 min of ischaemia). Figure 6B shows the overall results of mitochondrial membrane potential (ΔΨm; expressed in mV) of S2814D and SDKO mitochondria under basal conditions (i.e. pre-ischaemia, upper panel) and after the protocol of I/R (i.e. reperfusion, lower panel). Overall results obtained from WT and PLNKO are shown for comparison. The insets in the Figure display typical traces of rhodamine fluorescence changes (ΔF) expressed in arbitrary units, used for ΔΨm determinations in mitochondria isolated from S2814D and SDKO hearts previously submitted to I/R. Under basal conditions, there is no significant difference in ΔΨm among the different groups. In contrast, the short protocol of I/R significantly increased mitochondrial depolarization in SDKO vs. the other groups, including PLNKO mitochondria. Figure 6C shows typical examples and overall results of mitochondria swelling, measured as light scattering decrease (LSD), in samples derived from the four groups of hearts after addition of 50 µM Ca2+. In mitochondria, which were not submitted to I/R (basal conditions), the LSD produced by the addition of Ca2+ was significantly lower in S2814D, SDKO, and PLNKO than in WT, suggesting that mitochondria from these groups are initially more loaded with Ca2+ than WT mitochondria. In mitochondria submitted to the modest I/R challenge, LSD produced by addition of Ca2+ was not significantly different in S2814D and PLNKO, when compared with WT mitochondria. However, LSD was significantly lower in SDKO mitochondria vs. all groups. Figure 6D shows a tight negative correlation between mitochondrial swelling and ΔΨm. Of note, comparison of the LSD and ΔΨm alterations of the four groups, shows that they followed a similar pattern to that observed when infarct size and LDH release were explored (Figure 6D and E), i.e. infarct size increases as mitochondrial damage increases (Figure 6E). Taken together, the results indicate that mitochondrial damage is significantly more important in SDKO than in S2814D hearts. Moreover, in the presence of 5 µM dantrolene, used to inhibit RyR2, mitochondrial damage was significantly prevented (last bars of bottom panel of Figure 6A and B). These results support the idea that the greater SR Ca2+ leak of SDKO hearts is significantly involved in the enhanced cardiac damage produced by I/R in these hearts.
Taken together, the present findings indicate that increased SR Ca2+ uptake is able to interfere and impair cytosolic Ca2+ wave propagation, produced by the enhanced SR Ca2+ leak, but ineffective in preventing the increased Ca2+ flux from the SR to the cytosol, particularly to the neighbouring mitochondria.
4. Discussion
The key finding of the present study is that PLN ablation reduces the propensity to reperfusion arrhythmias evoked by CaMKII-dependent phosphorylation of RyR2-Ser2814 in the intact heart. The clinical relevance of these findings is obviously significant with respect to reperfusion arrhythmias as well as to arrhythmias in failing hearts, since CaMKII expression, activity, and phosphorylation of RyR2 at S2814 are all enhanced in patients and animals with HF,14,43 as well as during reperfusion after cardiac ischaemia.9,11,30 However, PLN ablation fails to rescue the increased propensity to infarct size produced by this phosphorylation. Quite the opposite, it exacerbates cardiac reperfusion damage, i.e. infarct size and LDH release. This occurred not only when hearts were submitted to a long ischaemic period, but also after short ischaemia, which does not produce significant cardiac damage upon reperfusion in WT and S2814D mutant hearts.
The inconsistency between the reduced propensity to reperfusion arrhythmias and the worsening of reperfusion cardiac damage when increasing SR Ca2+ uptake, suggests different mechanisms involved in these two processes.
4.1 Constitutive pseudo-phosphorylation of S2814D did not exacerbate reperfusion arrhythmias
It was previously shown that the arrhythmic pattern, observed during the early reperfusion period, can be traced to ectopic sources, i.e. early reperfusion arrhythmias were triggered by both DADs and EADs.4,44,45 It was further shown that a main cause of the reperfusion arrhythmogenic behaviour is an increase in the phosphorylation of the RyR2-Ser2814 site at the onset of reperfusion. Mutation of this site to the non-phosphorylatable form (S2814A) critically diminished reperfusion arrhythmias.4 Thus, at first sight, the failure of S2814D to significantly increase reperfusion arrhythmias over those observed in WT hearts might appear unexpected. Indeed, previous results demonstrated that S2814D mice are more prone to stress-induced arrhythmias than WT mice.16,17 One can argue that the transient phosphorylation of WT mice at the onset of reperfusion, may account by itself for reperfusion arrhythmias, i.e. the ‘permanent RyR2 phosphorylation’ of S2814D would not add any significant influence on reperfusion arrhythmias. However, previous experiments indicated that SR Ca2+ load is significantly lower in S2814D than in WT myocytes under basal conditions. Indeed S2814D was shown to be more prone to arrhythmias only under stress situations, like β-adrenergic stimulation, increasing extracellular Ca2+ or stimulation frequency, all conditions known to enhance SR Ca2+ load.16,17 The lower SR Ca2+ content in association with the ‘permanent CaMKII-dependent phosphorylation’ of the Ser2814 site, may account for the similarity in reperfusion arrhythmic patterns presented by S2814D and WT hearts. The SR Ca2+ load of S2814D should be still lower, when compared with WT, when non-stimulated protocols were used to detect spontaneous events. Under these conditions, assessment of SR Ca2+ spark mass and frequency strongly indicate that SR Ca2+ content was significantly lower in S2814D than in WT mice at the beginning of reperfusion (Figure 3).
4.2 SR Ca2+ waves are the substrate of reperfusion arrhythmias
Reperfusion significantly increases SR Ca2+ wave frequency relative to ischaemia and pre-ischaemia6 and these reperfusion Ca2+ waves might constitute the substrate of reperfusion arrhythmias. The fact that in the present experiments SR Ca2+ waves were associated with reperfusion arrhythmias and that abortion of SR Ca2+ waves by PLN ablation concurrently decreased reperfusion arrhythmias, further confirmed this concept. Moreover, decreasing SR Ca2+ uptake by inhibiting SERCA2a in PLN ablated hearts, recapitulated the arrhythmogenic phenotype of WT and S2814D hearts. SR Ca2+ waves would lead to enhanced Ca2+ extrusion via the electrogenic NCX, generating a depolarizing current (transient inward current), which, when sufficiently large, leads to DADs and potentially triggers ectopic APs and ventricular arrhythmias.18,19
4.3 PLN ablation prevents reperfusion arrhythmias by aborting fully propagated SR Ca2+ waves
PLN ablation severely enhanced reperfusion SR Ca2+ leak in SDKO hearts. Paradoxically, SR Ca2+ waves were significantly reduced. An increase in SR Ca2+ leak associated with a decrease in Ca2+ waves indicates a limitation in Ca2+ wave propagation throughout the cytosol. Indeed, our findings clearly show that PLN ablation interrupts cell-length propagating Ca2+ waves, switching them into non-propagated events, like mini-waves. Thus, these results support the conclusion that PLN ablation increases Ca2+ buffer capacity by decreasing cytosolic Ca2+ and hampers Ca2+ wave propagation resulting in silencing of arrhythmias. Further support to this idea is provided by the experiments in which the SERCA2a inhibitor, CPA, converts non-propagating mini-waves into full propagating Ca2+ waves.
4.4 PLN ablation further increases I/R –induced cardiac damage
In contrast to the beneficial effects on reperfusion arrhythmias, PLN ablation failed to decrease cardiac damage. On the contrary, it exacerbated infarct size and LDH release. The increase in infarct size associated with higher mitochondria damage, observed in SDKO with respect to WT and S2814D hearts, reveals that the enhanced SR Ca2+ uptake by PLN ablation is not capable of preventing the increase in infarct size associated with CaMKII-dependent phosphorylation of RyR2-Ser2814. It also uncovers that ablation of PLN would trigger additional mechanisms responsible for enhanced cardiac damage.
Previous experiments indicated that when SR Ca2+ reuptake is highly augmented by ablation of PLN, post-ischaemic recovery of contractile function was significantly reduced and cardiac infarct size was increased.24 These experiments suggested that the higher ATP consumption of PLNKO hyperactive hearts relative to WTs is the main cause for the increased damage of these hearts.24 In this study, we found that pre-ischaemic total Ht was similar in SDKO and S2814D hearts, making it difficult to attribute the enhanced cardiac damage of SDKO to higher energy consumption in the double mutant hearts. The increased cardiac injury produced by PLN ablation may be at least partially linked to the concomitant loss of HAX-1 from the SR, which may result in increased ER stress and mitochondrial damage. Indeed, it has been recently shown that PLN binds to the anti-apoptotic protein HAX-1 in cardiomyocytes and HAX-1 appears to confer protection from I/R injury.46,47 These pro-apoptotic actions of PLN ablation may partially explain the increase in heart damage of SDKO vs. S2814D and WT hearts. In addition, our results clearly showed that cardiac injury and mitochondrial dysfunction were significantly enhanced in SDKO, when compared to PLNKO hearts (Figures 5 and 6). The greater damage suffered by SDKO vs. PLNKO hearts could be explained by a mechanism linking their unique situation of augmented SR Ca2+ uptake in the presence of a permanent RyR2 Ca2+ leak. Under conditions in which RyR2 are leaky, a persistent and exacerbated SR Ca2+ uptake, would greatly enhance the propensity for SR Ca2+ leak, which may conspire against contractile recovery producing instead, cardiac damage. Indeed, mitochondrial dysfunction could be prevented in SDKO hearts submitted to I/R, when SR Ca2+ leak was inhibited by dantrolene. It could be argued that dantrolene, being a somewhat selective inhibitor of RyR1 may be mediating its protective effects by inhibiting mitochondrial RyR1 rather than SR RyR2. Indeed, it has been shown that stimulation of IP3 receptors with endothelin-1 induces Ca2+ release from the SR and increases mitochondrial Ca2+ via mitochondrial RyR1 rather than RyR2.48 Since reperfusion after ischaemia produced a rapid and transient release of inositol phosphates,49 the possibility of an increase in Ca2+ release through IP3 receptors cannot be excluded. However, biochemical and functional evidence indicates that dantrolene targets a corresponding region of RyR250 and that it effectively affects RyR2-mediated Ca2+-dependent processes under particular stress conditions like I/R injury, CPVT, or CaMKII-dependent arrhythmias.51–54 Moreover, previous results from our own laboratory indicated that inhibiting RyR2 with 1 µM tetracaine, also diminished cell death produced by I/R55 and that Ru360, used to inhibit the permeability transition pore MCU, decreases infarct size and cell death evoked by I/R,11 strongly suggesting that the main Ca2+ mitochondria influx in I/R occurs through the MCU rather than mitochondrial RyR1. Additionally, it is reasonable to expect that a reperfusion-induced mitochondrial damage, exclusively mediated by IP3 release, would cause a similar or higher cardiac damage in PLNKO (with a higher SR Ca2+ content) than in SDKO hearts, which is not the case of the present experiments. Taken together, and considering that we are dealing with RyR2 leaky hearts,16,17 our results support the conclusion that cardiac damage and mitochondrial dysfunction in SDKO hearts mainly occurs through the release of Ca2+ from RyR2. Since it has been shown that IP3 release could facilitate RyR2 activity,56 it is tempting to speculate that a possible IP3 release upon reperfusion in our experiments would contribute to the detrimental mitochondrial effect of RyR2 leak in SDKO hearts. In any case, our results indicate that the failure of the increased SR Ca2+ reuptake to prevent cardiac injury is associated with the inability of this mechanism to prevent mitochondria damage, as revealed by the close correlation between mitochondria dysfunction and infarct size in the different strains (Figure 6E). The higher mitochondria injury, associated with the enhanced SR Ca2+ leak, may be favoured by the close spatial proximity of mitochondria and SR Ca2+ release sites, which would greatly increase the local Ca2+ at the interphase between both organelles,57 impeding the beneficial effect of the increase SR Ca2+ uptake. Moreover, the increased SR Ca2+ leak may activate additional CaMKII-dependent deleterious pathways which could also play a role in the enhanced cardiac damage observed.58,59
4.5 Limitations of the study
We are aware of the limitations that animal models present regarding findings on arrhythmias, infarct size, or mitochondrial damage compared with patients. Therefore, any extrapolation of the present findings to the clinical scenario should be made with caution. In particular, referent to the mitochondrial function experiments, the fact that determinations were made in isolated mitochondria about half an hour after the end of reperfusion (which is the required time period for isolation), and that mitochondria were exposed to a standard buffer, might have affected the results. However, we consider that conclusions are still valid, taking into account that: (i) All models were submitted to the same procedure; (ii) There were not significant differences among groups in mitochondria membrane potential from hearts not submitted to I/R; (iii) The basal mitochondria membrane potential obtained (hearts not submitted to I/R) was about −160 mV, in close relation with reports assessing mitochondria membrane potential with different techniques59; (iv) There was a significant decrease in light scattering in mitochondria from S2814D, SDKO, and PLNKO vs. WT in hearts non subjected to I/R, clearly suggesting an increase in mitochondria Ca2+, which should be expected from the increased SR Ca2+ leak of these mutant hearts; and (v) Significant differences were detected among the different models after hearts were submitted to the I/R protocol, in agreement with our hypothesis.
Furthermore, we recognize that the use of TTX and carbamylcholine could have altered the clinical situation present during I/R. However, it have been shown that both interventions are cardioprotective.60,61 Thus, the use of these drugs in the present experiments would have tended to decrease, if any, the magnitude of our observations in all four groups studied. Moreover, although a recent paper suggests that the mechanical uncoupler blebbistatin, increases AP duration and ventricular fibrillation threshold in perfused rabbit heart,62 several other studies showed that the drug has no significant cardiac electrophysiological effects in a number of species, including mouse cardiac tissue.63–65 These results suggest that the use of the drug could have hardly affected our results.
Finally, in the present experiments, we calculated spark-mediated SR Ca2+ leak. However, previous measurements in isolated myocytes showed that RyR2 mediated Ca2+ leak has a second component not mediated by Ca2+ sparks that should be added to the spark-mediated Ca2+ leak to estimate total RyR2-mediated Ca2+ leak.38 Although the underlying mechanisms of non-spark mediated Ca2+ leak are not completely elucidated, experimental evidence support the concept that, similar to spark-dependent leak, non-spark mediated Ca2+ leak depend on both, SR Ca2+ load and RyR2 sensitization.38 It is, therefore, difficult to expect a higher increase in non-spark mediated Ca2+ leak in S2814D and WT than in SDKO hearts, of such a magnitude that could offset the difference in spark-mediated Ca2+ leak that we observed among them (Figure 3).
In conclusion, our experiments revealed for the first time the opposite roles of increased SR Ca2+ uptake on I/R-induced arrhythmias and cardiac damage and dissected the mechanisms by which these two different effects take place. Whereas the enhanced SR Ca2+ uptake was able to eliminate the propagation of Ca2+ waves, elicited by the exacerbated SR Ca2+ leak of SDKO hearts, it was inefficient to impede the flow of this SR Ca2+ leak to the mitochondria and/or activation of other CaMKII deleterious pathways, aggravating cardiac damage.
Footnotes
Time for primary review: 34 days
Acknowledgements
We wish to thank Luciana Sapia, Omar Vélez-Rueda, Pedro González, Mónica Rando, Solange Bibé, Omar Castillo, and Andrés Pinilla, for their technical assistance.
Conflict of interest: none declared.
Funding
This work was supported by grants from the National Research Council (Argentina), PIP0890 and 0350 and PICT 2014-2524 to A.M., PICT 2015-3210 to C.A.V., UNLPX-642 and UNLPX-795 to A.E.C., National Institutes of Health R01-HL26057 and HL64018 (to E.G.K.) and R01-HL089598, R01-HL091947, R01-HL117641, and R41-HL129570, and the American Heart Association 13EIA14560061 (to X.H.T.W.).
References
Author notes
Carlos A. Valverde, Gabriela Mazzocchi and Mariano N. Di Carlo authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}