





Abstract
In C4 plants, the pyruvate (Pyr), phosphate dikinase regulatory protein (PDRP) regulates the activity of the C4 pathway enzyme Pyr, phosphate dikinase (PPDK) in a light-/dark-dependent manner. The importance of this regulatory action to C4 pathway function and overall C4 photosynthesis is unknown. To resolve this question, we assessed in vivo PPDK phospho-regulation and whole leaf photophysiology in a CRISPR–Cas9 PDRP knockout (KO) mutant of the NADP-ME C4 grass green millet (Setaria viridis). PDRP enzyme activity was undetectable in leaf extracts from PDRP KO lines. Likewise, PPDK phosphorylated at the PDRP-regulatory Thr residue was immunologically undetectable in leaf extracts. PPDK enzyme activity in rapid leaf extracts was constitutively high in the PDRP KO lines, irrespective of light or dark pretreatment of leaves. Gas exchange analysis of net CO2 assimilation revealed PDRP KO leaves had markedly slower light induction kinetics when leaves transition from dark to high-light or low-light to high-light. In the initial 30 min of the light induction phase, KO leaves had an ∼15% lower net CO2 assimilation rate versus the wild-type (WT). Despite the impaired slower induction kinetics, we found growth and vigor of the KO lines to be visibly indistinguishable from the WT when grown in normal air and under standard growth chamber conditions. However, the PDRP KO plants grown under a fluctuating light regime exhibited a gradual multi-day decline in Fv/Fm, indicative of progressive photosystem II damage due to the absence of PDRP. Collectively, our results demonstrate that one of PDRP’s functions in C4 photosynthesis is to ensure optimal photosynthetic light induction kinetics during dynamic changes in incident light.
Introduction
Approximately 30 million years ago (MYA), the C4 photosynthetic mechanism emerged from the ancestral C3 photosynthesis. This entailed evolution of leaf chlorenchyma into two distinct photosynthetic cells, mesophyll and bundle sheath, the recruitment of PEP carboxylase (PEPC) for primary CO2 fixation, and the recruitment of preexisting enzymes to form the circular C4 metabolic pathway (Christin et al., 2013). As diagrammed (Figure 1A), the C4 pathway drives a biochemical CO2 pump that concentrates fixed CO2 in bundle sheath cell chloroplasts, thereby dramatically reducing O2 fixation by Rubisco and loss of carbon due to photorespiration (Hatch, 1987). In the pathway, C4 pyruvate (Pyr) phosphate dikinase (PPDK) fulfills a cardinal role by catalyzing the regeneration of the primary CO2 acceptor phosphoenolpyruvate (PEP) from substrates Pyr, ATP, and Pi, in the stroma of leaf mesophyll-cell chloroplasts (Hatch, 1987; Figure 1A). Although its catalysis is freely reversible, in C4 plants, the PPDK reaction is kept in the PEP-forming direction by the rapid removal of reaction byproducts pyrophosphate (PPi) and AMP by the abundant pyrophosphatase and adenylate kinase known to exist in the mesophyll chloroplast (Figure 1A) (Ashton et al., 1990). Like the other C4 pathway enzymes, C4 PPDK is active in the light and inactive in the dark (Edwards et al., 1985; Chastain, 2011). In low light, the net activity of the stromal pool of PPDK is low but as incident light rises to ∼1,000 μmol quanta m−2s−1 (∼1/2 full sunlight), it becomes fully active (Hatch and Slack, 1969; Yamamoto, et al., 1974; Usuda et al., 1984; Usuda, 1988). In the dark, the enzyme is rendered inactive by the reversible phosphorylation of an active-site Thr residue (Thr 525 in green millet [Setaria viridis]) (Burnell and Hatch, 1985). The bifunctional PPDK regulatory protein (PDRP) confers both the dark-induced phosphorylation and light-induced dephosphorylation of this residue (Figure 1, A and B). PDRP also functions as the light-/dark-sensing component of the PPDK light regulation cycle (Figure 1B). This functional property of PDRP allows the direction of PDRPs opposing PPDK-activation/PPDK-inactivation reactions to be correctly governed in response to light or dark (Burnell and Hatch, 1985; Chastain, 2011). The mechanism by which this is proposed to occur is complex and involves PDRPs demonstrated high affinity for ADP along with swings in stromal ADP concentration that accompany changes in incident light and dark (Burnell and Hatch, 1985; Chastain, 2011; Hart et al., 2011). In vivo, the bifunctional enzyme activities of PDRP (i.e. protein kinase, protein phosphotransferase) differ with respect to rates of PPDK dephosphorylation and PPDK phosphorylation, with light activation of inactive PPDK being about three-fold faster than dark inactivation of active PPDK. For example, in dark adapted maize (Zea mays) leaves, the stromal pool of inactive C4 PPDK is fully activated in as little as ∼5 min after dark-adapted leaves are exposed to high-irradiance light (>1,000 μmol quanta m−2s−1) (Nakamoto and Edwards, 1983; Roeske and Chollet, 1989; Chastain et al., 2002). However, when illuminated leaves are placed into the dark, PDRP inactivation of PPDK is slower, requiring ∼20–300 min to convert the fully light-activated pool of C4 PPDK into inactive/phosphorylated enzyme (Nakamoto and Edwards, 1983; Roeske and Chollet, 1989; Chastain et al., 2002). Given the pivotal role of PPDK in the C4 cycle, its regulation by PDRP is predicted to be necessary for efficient C4 cycle function (Burnell and Hatch, 1985; Hart et al., 2011, Wang et al., 2021). This prediction is based largely on the observation of how PDRP can rapidly increase or decrease the fraction of active PPDK in the stroma in response to up/down changes in incident irradiance level (Hatch and Slack, 1969). By this regulatory action, PDRP is thought to exert a control on the rate of ATP and Pyr consumed in the PPDK-catalyzed PEP synthesis reaction as part of a feedback mechanism aimed at balancing ATP consumption with the level of ATP supplied via photophosphorylation (Chastain, 2011; Hart et al., 2011). In this capacity, PDRP’s strict regulation of PPDK is thought to maintain C4 cycle “robustness” by preventing transient overconsumption of stromal ATP and Pyr by PPDK during dynamic changes in incident light intensity (Hart et al., 2011). Consistent with this view are the findings of a recent study by Wang et al. (2021). These authors developed a dynamic (nonsteady state) model of NADP-ME-type C4 photosynthesis for identifying factors that limit net CO2 assimilation in fluctuating light. When a simulation of exposing dark-adapted leaves to high light was run on the model, PDRP activity (i.e. rate of re-activation of PPDK) was found to limit photosynthetic rates in the initial phase of the light induction curve. Whereas these simulations were run for dark → high-light and low-light → high-light transitions, the model suggests that PDRP exerts a similar transient, regulatory control of C4 photosynthesis each time the incident light falls and rises due to sunflecks or cloud cover over the course of a photoperiod. As such, the authors infer that increasing endogenous levels of PDRP will accelerate the initial phase of the light induction curve and increase the photosynthetic efficiency under the fluctuating light conditions inherent in field crop canopies.
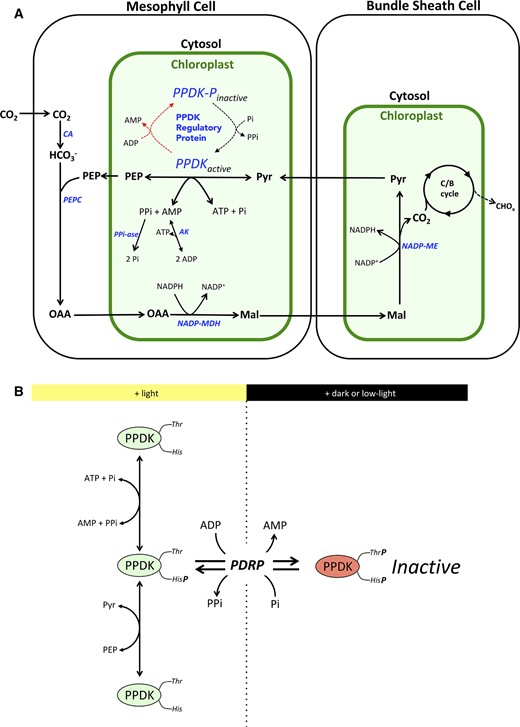
Overview of the NADP-ME C4 metabolic cycle with emphasis on the roles of PPDK and PDRP. A, Context view of reversible phosphorylation of PPDK by PDRP in the C4 cycle. In the light, PPDK fulfills a cardinal role in the C4 metabolic pathway by regenerating the primary CO2 fixation substrate PEP from Pyr. The linked action of stromal pyrophosphatase (PPi-ase) and adenyl kinase (AK) exert a substantial influence on the direction of catalysis of both PDRP and PPDK activities. CA, carbonic anhydrase; PEPc, PEP carboxylase; NADP-MDH, NADP malate dehydrogenase; NADP-ME, NADP malic enzyme;OAA, oxaloacetic acid; Mal, malic acid; C/B, Calvin Benson cycle. B, PDRP regulates PPDK activity by reversible phosphorylation of an active-site Thr located one residue removed from the central catalytic His. Only the His-P PPDK catalytic intermediate is amenable to phosphorylation/inactivation by PDRPs protein kinase function.
Another often overlooked putative role PDRP may fulfill in C4 cycle function is its regulatory action in the dark, where it maintains the massive stromal pool of PPDK in the inactive state during cessation of C4 cycle activity. This action is assumed to prevent the unregulated turnover of the C4 cycle intermediates Pyr and PEP in the dark which in turn could potentially prolong the light induction phase of photosynthesis (Leegood and Furbank, 1984; Usuda, 1985).
Until recently, testing these hypothetical roles using a reverse genetics approach in a C4 plant has until the present era posed substantial technical challenges to carryout. However, the establishment of S. viridis as a model NADP-ME C4 plant along with the availability of CRISPR–Cas9 gene-editing tools has provided a more facile means to create knockout (KO) mutants for C4 regulators such as PDRP (Brutnell et al., 2010). Notably, because only a single PDRP gene is present in the S. viridis genome (Sevir.2G348700), versus two in maize and three in sorghum (Sorghum bicolor) (Emms et al., 2016; Chastain et al., 2018), it was possible to use CRISPR–Cas9 genome editing to generate a C4 NADP-ME species devoid of PDRP activity without the need to also account for additional paralogs that may have divergent functions (Chastain et al., 2018). Presented here are the results of our initial biochemical and physiological characterization of the KO mutant plants that reveal a role for PDPR in the light induction phase of photosynthesis.
Results
PDRP KO lines lack in vitro and in vivo PDRP activity
A CRISPR–Cas9 system gene-editing protocol based on the Cermák et al. plant genome engineering toolkit (2017) was utilized to knockout the single PDRP gene of the S. viridis genome (Sevir.2G348700). Using this protocol, six T0 plants, each representing an independent transformation event, were recovered via hygromycin selection. Sanger sequencing of the PCR-amplified PDRP exon 1 genomic DNA region revealed each T0 line to be homozygous for the same 68-bp deletion flanked by the CRISPR–Cas9 guide RNAs (Figure 2, A and B; Supplemental Figure S1). Three of the T0 KO lines, named T27, T28, and T29 were selfed to produce segregating T1 generation plants. These were subsequently genotyped by PCR screening for (loss) of the hygromycin phosphotransferase transgene (Zhu et al., 2021), which confirmed the transgene (both Cas9 and the hygromycin selectable marker) had segregated out. Screened homozygous T1 KO plants were propagated to ultimately produce T3 generation seed for generating all PDPR KO plants utilized in this study. As shown in Figure 2B, the CRISPR–Cas9-mediated 68-bp deletion modified the exon 1 reading frame to instead encode 22 missense amino acids before an in-frame stop codon is reached downstream in the first exon. The expression of a truncated polypeptide from this modified locus was not tested. However, all three PDPR KO lines were shown to be devoid of PDRP enzyme activity (Figure 2C), indicating that if any truncated PDRP accumulated, it was nonfunctional. This was established by using a highly sensitive in vitro immuno-based PDRP protein kinase (PPDK phosphorylating) assay in which we did not detect any residual PDRP activity present in desalted crude leaf extracts of the PDRP KO lines (Figure 2C). As shown, wild-type (WT) leaf extract contained appreciable PDRP activity as evidenced by the ADP-dependent phosphorylation of PPDK. The faint band within the WT control minus ADP reaction (Figure 2C, upper) represents minor amounts of endogenous PPDK-ThrP introduced into the assay via the 10-µL WT leaf extract used as the source of PDRP in the in vitro reaction. Notably, this faint band in the minus ADP control lanes of the PDRP KO assays is absent signifying the lack of endogenous PPDK-ThrP in PDRP KO leaf extracts (and absence of PDRP activity). This observation was more keenly shown in immunoblots of leaf extract isolated from illuminated and dark-adapted leaves probed with PPDK-ThrP antibody (Figure 3). In this experiment, leaves of WT and two of the PDRP KO lines, T27 and T28, were first dark adapted for 4 h and then illuminated for 30 min under high irradiance. For WT plants, we found that after 4-h dark, endogenous PPDK had become highly phosphorylated (Figure 3, A and B, upper). When WT leaves were then exposed to high light for 30 min, nearly all the PPDK-ThrP present in the dark-adapted leaves was dephosphorylated into the active dephosphorlyated PPDK. In contrast, we were unable to detect any PPDK-ThrP signal in leaf extracts from the two PDRP KO lines, irrespective of dark or light treatment of the leaves, again demonstrating the absence of PDRP activity in the PDRP KO plants. When the anti-phospho-PPDK probed blots were stripped and reprobed with anti-PPDK antibody (for detecting total PPDK), the PPDK band intensities appear to be equivalent between WT versus KO leaves (Figure 3, A and B, lower), suggesting that PPDK levels are not likely effected by loss of PDRP. Finally, representing a third line of evidence that PDRP is absent in the KO plants was the loss of in vivo light-/dark regulation of leaf PPDK activity (Figure 4). Using a rapid extract and assay method that preserves in situ PPDK activation state, we show that PPDK activity in dark-adapted PDRP KO leaves was essentially of the same level as in illuminated leaves. In contrast, the PPDK activity in dark-adapted WT leaves was largely suppressed. Approximately 8% of the PPDK activity remaining in dark-adapted WT leaves likely represents the PPDK activity originating from the cytosolic PPDK isoform, which is not light-/dark regulated since PDRP is plastid localized (Chastain et al., 2018). Notably, PPDK-specific activity in the PDRP KO leaves in both light- and dark-exposed leaves was comparable to the high PPDK-specific activity recorded for the illuminated WT leaves.
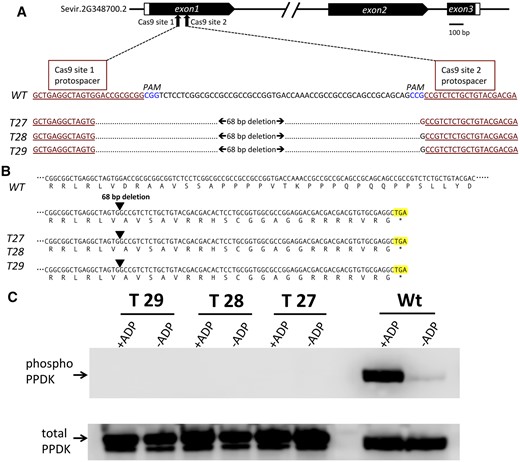
CRISPR–Cas9 edited S. viridis PDRP gene knockout mutants lack PDRP enzyme activity. A, Schematic diagram of the S. viridis PDRP gene model (Sevir.2G348700.2) showing Cas9 target sites within the first exon. Shown directly below is an alignment of exon 1 WT sequence with the corresponding CRISPR–Cas9 edited DNA sequence of KO lines T27, T28, and T29 with the resultant 68-bp deletion. CRISPR–Cas9 target protospacer sequences are underlined and highlighted in red. PAM sequences are highlighted in blue. B, CRISPR–Cas9 deleted PDRP exon 1 sequence shifts the reading frame of exon 1 in the KO lines to encode missense codons until a stop codon (highlighted) is encountered 66-bp downstream from the deletion site. C, In vitro PDRP protein kinase assays of desalted leaf extracts. PDRP knockout lines T27, T28, T29, and WT were assayed for ADP-dependent PDRP protein kinase (PPDK phosphorylating) activity using an immuno-based assay with recombinant maize C4 PPDK as substrate. Upper part shows immunoblots of terminated reaction aliquots probed with anti-phospho-PPDK antibody. Control assays (-ADP) omitted ADP in the reaction mix. Lower part serves as a lane load control and is the same blot stripped and reprobed with anti-PPDK polyclonal antibody. The lower molecular mass band evident in T27, T28, T29 lanes (lower, C) is most likely attributable to the presence of a higher proteolytic background in the leaf extract used in the assays and is apparently unrelated to genotype. Additional assay sets also show the occurrence of this minor band in WT lanes (see Supplemental Figure S2). Results identical to those of the single experiment shown in the upper panel were obtained in three independent assays.
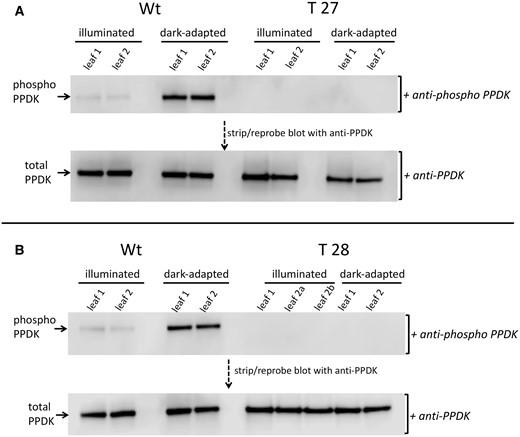
Immunoblot detection of PPDK and phospho-PPDK in leaf extracts isolated from dark-adapted or illuminated leaves of PDRP knockout lines T27, T28, and WT. Leaves were dark-adapted for 2 h and then illuminated for 30 min at 1,300 µmol quanta m−2s−1 as described in “Materials and methods.” Shown are separate immunoblots for WT versus T27 (A) and WT versus T28 (B). Upper parts are immunoblots of duplicate leaf extracts probed with anti-phospho-PPDK antibody. The T28 immunoblot (B) contained three separate leaf samples. Lower parts show the same immunoblots stripped and reprobed with anti-PPDK antibody. Each lane contained 2-µg total soluble protein.
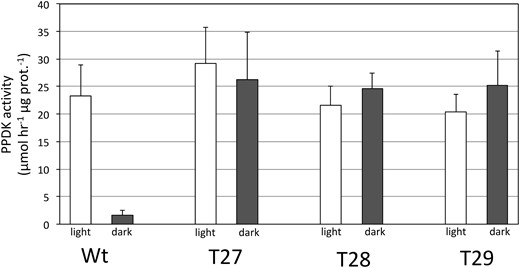
PPDK enzyme activity in desalted leaf extracts isolated from dark adapted and illuminated leaves. T27, T28, T29, and WT. Leaves were dark adapted for 2 h and then illuminated for 30 min at 1,300 µmol quanta m−2s−1 as described in “Materials and methods”. PPDK activity is shown as percentage of illuminated WT leaves (WT PPDK activity = 1.04 µmol min−1mg prot−1). Values shown are the means ± se, n = 3.
Growth phenotype under standard growth chamber conditions
The initial PDRP KO transformants and subsequent progeny lines through the T3 generation were regenerated and maintained in 2% CO2 as a precaution that loss of PDRP would impair growth and development thus jeopardizing recovery of PDRP KO plants. Germination and growth in normal air were not assessed until homozygous T3 seed was harvested from high-CO2-grown plants. Interestingly, when germinated and grown under standard growth chamber conditions (normal air conditions, ∼700-μmol quanta m−2s−1 light, 16-/8-h day/night, 31°C/22°C photoperiod), all three independent PDRP KO lines were visually indistinguishable from WT plants in terms of growth and vigor (Figure 5).

WT and PDRP knockout plants at the 3-to 4-week stage after germination and growth under standard growth chamber conditions. Plants were germinated and grown under standard growth chamber conditions (normal air conditions, ∼700-μmol quanta m−2 s−1, 16-h/8-h day/night, 31°C/22°C photoperiod). Additional, details provided in “Materials and methods.”
Effect of irradiance level on PPDK activation state in WT and PDRP KO leaves
As described above, the level of extractable PPDK activity in illuminated C4 leaves increases proportionally with increasing incident light intensity to a maximum level at ∼1/2 sunlight (∼1,000 μmol quanta m−2s−1) (Hatch and Slack, 1969; Yamamoto, et al., 1974; Usuda et al., 1984; Usuda, 1988). It had been generally assumed that this irradiance-level dependence of PPDK activity is due exclusively to the phospho-regulatory actions of PDRP and not any other agent (Chastain and Chollet, 2003; Chastain, 2011). We examined this assumption by measuring extractable PPDK activity in T27 and WT leaves illuminated at low to high irradiances (Figure 6). Here PPDK activation state was determined from rapid leaf extracts prepared from dark-adapted 3-cm mid-leaf sections that had been floated on dH2O and illuminated from above. For WT leaves, the PPDK activation state (PPDK specific activity) was shown to increase with successively higher incident irradiance until reaching a maximum level at around 600 μmol quanta m−2s−1. In contrast, the PPDK activation state in the T27 leaves at all irradiance levels and in dark was comparable to the highest light activated state of PPDK in WT leaves.
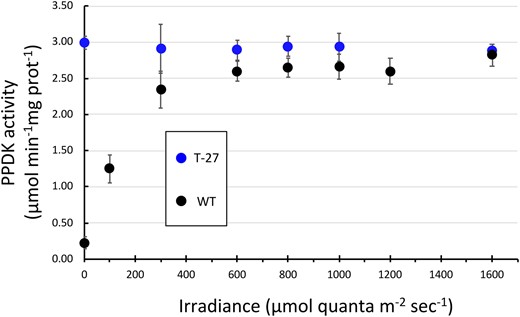
Leaf PPDK activation state in WT and T27 leaves as a function of increasing irradiance. PPDK enzyme activity was measured in leaf extracts from leaves that had first been dark adapted for 30 min and then illuminated for 15 min at the designated irradiance level. Data points for T27 leaves at 100 and 1,200 μmol quanta m−2s−1 were omitted due to insufficient number of replicates. Values shown are the means ± se, n = 3. Differences between genotypes for data points 600, 800, 1,000, and 1,600 μmol quanta m−2 s−1 are not significant (P > 0.05, two-tailed Student’s t test).
Effect of PDRP loss on photosynthetic CO2 assimilation and gas exchange properties of leaves
We examined the effect of PDRP loss on net CO2 assimilation (Anet) of S. viridis leaves via gas exchange analysis of leaves during a time course of illumination. Specifically, plants were removed from the growth chamber and allowed to acclimate in the dark for 30 min prior to logging data. Following this acclimation period, Anet was recorded every minute over a 120-min time course of changing intervals of high light and low light. Specifically, Anet was first logged for a 30 dark period and followed by illumination with high light (1,500 μmol quanta m−2s−1) for 30 min then transitioned to low light (100 μmol quanta m−2s−1) for 30 min. After the initial ∼10-min light induction phase, we observed lower rates of Anet for the T27 leaves versus WT leaves (Figure 7A) and at the end of the 30-min high light illumination period, rates of Anet were ∼15% lower that WT. When leaves were then transitioned into lower irradiance (100 μmol quanta m−2s−1), rates of Anet for both WT and T27 declined rapidly but Anet was still higher in the WT versus T27 leaves (Figure 7A). To investigate if the lower Anet of T27 leaves persisted longer than 30 min, we repeated this experiment by extending the initial high-irradiance illumination period to 90 min. As shown in Figure 8A, at ∼30 min into the illumination period, the T27 leaves exhibited the same ∼15% lower Anet versus WT as in Figure 7A. However, as the illumination period proceeded beyond 30 min, Anet for T27 and WT gradually converged toward the end of the 90-min illumination, suggesting that loss of PDRP in the T27 leaves greatly prolongs the duration of the photosynthetic induction phase (Figure 8A). To see if the lower Anet in the T27 leaves could be attributed to limitations to stomatal CO2 diffusion, we made corresponding measurements of stomatal conductance (gs) (Figure 8B). Interestingly, around 15 min into the illumination period, gs was lower in T27 leaves but increased to WT levels after an additional 20 min of illumination (Figure 8B). The corresponding measurements of the internal leaf CO2 concentration (Ci) were found to be comparable to WT throughout the 90-min illumination period (Figure 8C). Thus, the transiently lower gs observed for T27 (Figure 8B) is unlikely to impact the rates of Anet we observed for this genotype (Figure 8A).
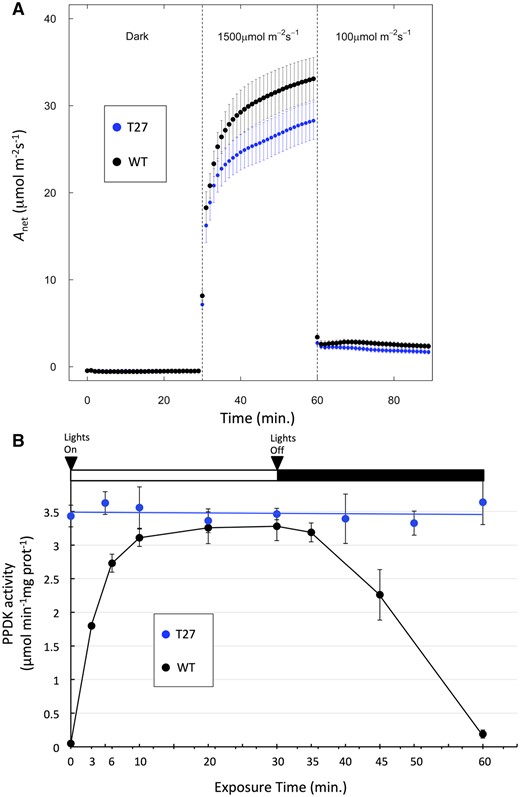
Light activation and dark deactivation of photosynthetic CO2 assimilation and PPDK activity in WT and T27 leaves. A, Net CO2 assimilation (Anet) in WT and T27 leaves during a dark → high light → low light transition. Anet values are the means ± se, n = 11, WT; n = 10, T27; differences between WT and T27 for measurements at 40–59 min are statistically significant (P < 0.05, two-tailed Student’s t test). B, PPDK light activation/dark deactivation in WT and T27 leaves during dark→light→dark transitions. Leaves used for PPDK light activation curve were first dark adapted for 2 h prior to illumination at 1,300 µmol quanta m−2s−1. Anet measurements and PPDK enzyme activity were conducted separately using leaves grown under identical conditions. Values shown for PPDK enzyme activity are the means ± se, n = 3.
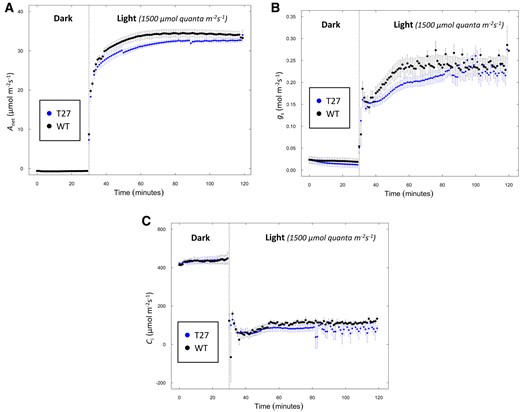
Simultaneous measurement of net CO2 assimilation, gs, and intercellular CO2 in WT and T27 leaves. A, Net CO2 assimilation (Anet), (B) gs, and (C) intercellular CO2 (Ci) from 30 min of dark into 90 min of light. Anet, gs, Ci values are the means ± se, n = 7, WT; n = 8, T27; differences in Anet between WT and T27 for measurements at 45–60 min are statistically significant (P < 0.05, two-tailed Student’s t test). Differences in gs between WT and T27 for measurements at 48–60 min are statistically significant (P < 0.05, two-tailed Student’s t test).
To ascertain any link between the loss of PDRP and the reduced Anet in T27 leaves shown in Figures 7, A and 8, A, we assessed PPDK activation state in leaf extracts of similarly high-light-illuminated leaves (1,300-μmol quanta m−2s−1 versus 1,500-μmol quanta m−2s−1, Figure 7A). Here PPDK activation state was determined from rapid leaf extracts prepared from 3-cm mid-leaf sections that had been floated on dH2O and illuminated from above. As shown in Figure 7B, at the onset of illumination, the WT PPDK activity increased rapidly from nil to the nearly fully light-activated level in around 6 min. In contrast, PPDK enzyme activity in the T27 KO leaves remained unchanged from the high initial activity in dark-adapted leaves to the end of the 30-min illumination period (Figure 7B). When WT leaves were transitioned into the dark, inactivation of PPDK was shown to be slower versus light activation of PPDK, requiring ∼30-min dark for full inactivation, as is typical for other C4 species (Nakamoto and Edwards, 1983; Roeske and Chollet, 1989; Chastain et al., 2002). When illuminated T27 leaves were transitioned back into dark, PPDK activation state, sampled alternately from WT leaves at 10, 20, and 30 min into the dark period, was shown to remain high and unchanged (Figure 7B).
Whole leaf Pyr levels
Since PPDK activity in the PDRP KO leaves was found to be maximally high and constant in the dark (Figure 7B), we speculated that PPDK-mediated PEP synthesis in mesophyll stroma was also active in the dark. As a consequence, we thought it possible that the mesophyll stromal pool of Pyr would decline to abnormally low levels as it is converted to PEP once leaves were dark adapted. To examine this possibility, we assessed whole leaf Pyr levels at end of a 2-h dark adaptation period and at 3 min after the onset of illumination. It should be noted that while these measurements represent Pyr in the combined bundle sheath and mesophyll chlorenchyma, whole leaf measurements in maize have been shown to qualitatively mirror dark/light changes of Pyr in isolated mesophyll cells (Usuda, 1985; Roeske, 1989). For dark-adapted leaves, we found Pyr levels in T27 leaves to be ∼40% of WT leaves (Figure 9). When these dark-adapted leaves were exposed to high light for 3 min, Pyr in the T27 leaves was shown to remain relatively unchanged from dark levels while decreasing in the WT leaves.
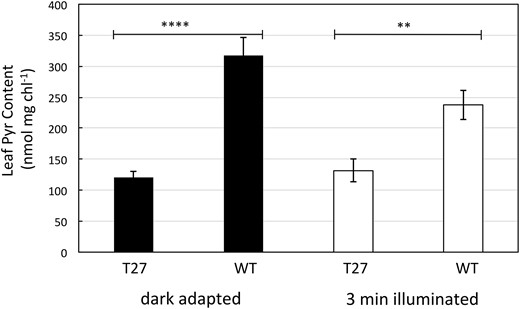
Leaf Pyr content of dark adapted and 3 min illuminated WT and T27 leaves. Leaves were dark adapted for 2 h prior to illumination for 3 min at 1,300 µmol quanta m−2s−1. Values are the means ± se, n = 8 except for dark adapted T27 where n = 6. Asterisks indicate statistically significant differences by two-tailed Student’s t test: *P < 0.05; **P < 0.01; ****P < 0.0001.
Whole leaf PS II efficiency during growth under a constant or fluctuating light photoperiod
We sought to assess how the loss of PDRP affects C4 leaf photophysiology under rapidly changing incident light conditions that are more typical of field grown plants (e.g. transient disruptions of direct sunlight by clouds or lightflecks on lower canopy leaves). For this purpose, we measured PS II efficiency via pulse-amplitude-modulation (PAM) chlorophyll fluorescence (Fv/Fm) in T27 and WT plants growing under a fluctuating light photoperiod versus plants grown under steady high light or steady low light photoperiod as described previously (Schneider et al., 2019). In this experiment, the fluctuating light regime consisted repeating cycles of 1 min at 1,200 µmol quanta m−2 s−1 and 4 min at 300 µmol quanta m−2 s−1 over a 16-h photoperiod. Steady light-treated plants were grown under constant illumination of 850 µmol quanta m−2 s−1 or 90 µmol quanta m−2 s−1 per 16-h photoperiod. At the onset of the experiment, the initial measurements at Day 0 showed Fv/Fm in both WT and T27 leaves to be approximately the same (∼0.71, Figure 10). After 7 days under the fluctuating regime, Fv/Fm declined markedly in T27 leaves to 0.37 at Day 7. For WT fluctuating light-treated leaves, Fv/Fm was shown to decline through Day 3 to 0.64 before increasing to 0.68 at Day 7. Under the steady high light regime Fv/Fm was shown to decline in T27 leaves from 0.71 at Day 0–0.59 at Day 3 before recovering to 0.68 at Day 7. The Fv/Fm of steady high light-treated WT leaves was largely unchanged over the course of the run ending at 0.68 at Day 7. The sharp decline of Fv/Fm in fluctuating light-treated T27 leaves from 0.70 to 0.37 is therefore directly attributable to the loss of PDRP since a similar decline in fluctuating light-treated WT leaves or steady high light-treated T27 leaves was not observed. We also ran an additional 7-day SL experiment at very low light (90 µmol quanta m−2 s−1). Under the low steady light regime, Fv/Fm values remained relatively constant for T27 and WT from Day 0 (∼0.70) through Day 7 (∼0.73), with nearly identical Fv/Fm values between genotypes at each daily time point (Figure 10).
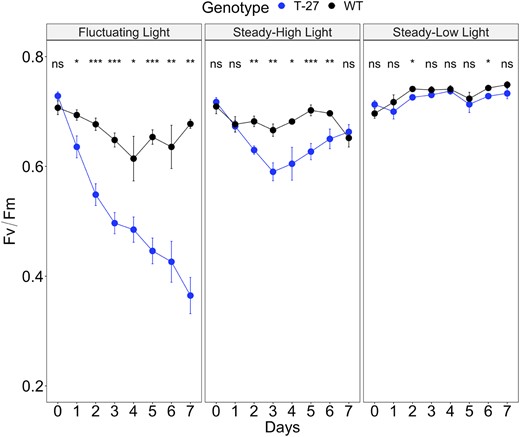
Effect of a fluctuating light photoperiod on Fv/Fm. Shown are daily measurements of Fv/Fm in WT and T27 KO leaves during 7-day growth under a fluctuating light photoperiod versus a control steady high light or low light photoperiod. The fluctuating light regime consisted of repeating cycles of 1 min at 1,200 and 4 min at 300 µmol quanta m−2 s−1 per 16-h/8-h light/dark photoperiod. For the steady high light and steady low light controls, plants were grown under a 16-h/8-h light/dark photoperiod of 850 µmol quanta m−2 s−1 and 90 µmol quanta m−2 s−1, respectively. Time points are the means ± se, n = 6–8. A three-way repeated measure ANOVA for Genotype × Light treatment × Day (see Supplemental Table S2) and post-hoc Tukey’s test was used to examine differences in Fv/Fm between genotypes within growth Light treatment across 7 days (Supplemental Table S3). Where P-values ns, not significant, * ≤0.05, ** ≤0.01, and *** ≤0.001
Discussion
As a strict regulator of a cardinal C4 cycle enzyme, PDRP has long been viewed as an essential regulatory component of the C4 photosynthetic process in important crop species such as maize and sorghum (Burnell and Hatch, 1985; Edwards et al., 1985; Hart et al., 2011). However, we show in this study that when PDRP is eliminated from a closely related NADP-ME C4 species, that is, the model C4 grass S. viridis, there is no apparent effect on growth and vigor when plants are cultivated under standard growth chamber conditions where irradiance was maintained at a constant ∼1/3 full sunlight (700 μmol quanta m−2 s−1) per 16-h per photoperiod. In hindsight, this observation should not be surprising given that the plants were grown under a steady (e.g. nonfluctuating) irradiance of 700 μmol quanta m−2 s−1, which is above the ∼600-μmol quanta m−2 s−1 threshold where the stromal PPDK pool becomes maximally activated (Figure 6). At or above this illumination level, PPDK activity in the WT leaves is essentially at the same high level of the T27 leaves (2.6-µmol min−1mg prot−1 for WT and 2.9-µmol min−1mg prot−1 for T27 [Figure 6]). Importantly, as incident light remains above 600 μmol quanta m−2 s−1, PDRP can exert no further regulatory influence (i.e. the stromal pool of PPDK is largely in the dephosphorylated state). Thus, any effect on growth and vigor due to loss of PDRP in the T27 plants would be obscured when PPDK activity is similarly high in both WT and T27 leaves at the irradiance of 700 μmol quanta m−2 s−1 used for the growth experiment shown in Figure 5. Only under sub-saturating irradiances (e.g. <600 μmol quanta m−2 s−1), would we expect PDRP to influence C4 pathway function. At this range of irradiances, PDRP can effectively adjust the level of PPDK activity in WT leaves to match the level of irradiance incident on leaves (Figure 6) (Nakamoto and Edwards, 1983; Roeske and Chollet, 1989; Chastain et al., 2002). Furthermore, PDRPs regulatory action (or lack thereof) would also be obscured when plants are grown under a constant versus fluctuating light photoperiod. As stated in the introduction, under sub-saturating irradiances PDRP can increase or decrease the net activity of the stromal PPDK pool in response to changes in incident light in rapid fashion on a time scale of minutes (Figure 7B; Hatch and Slack, 1969; Nakamoto and Edwards, 1983; Roeske and Chollet, 1989; Chastain et al., 2002). Thus, the true impact of how the loss of PDRP in T27 leaves may affect plant growth and vigor is more likely to be revealed when plants are grown under dynamic lighting which cycles into lower irradiances (e.g. <600 μmol quanta m−2 s−1) where the stromal PPDK pool is partially activated by PDRP.
Although the effect of dynamic light on growth and vigor in the KO plants was not explored in this study, two net assimilation experiments presented here strongly suggest that the KO plants would fix less carbon per photoperiod and have reduced growth relative to WT under dynamic lighting conditions. These experiments, shown in Figures 7, A and 8, A, reveal that the PDRP KO plants have a prolonged photosynthetic induction phase relative to WT when leaves are transitioned from dark to light or from low light to high light. In the experiment shown in Figure 8A, the rate of photosynthesis in the KO leaves was shown to lag behind (i.e. increase at a slower rate) compared to WT leaves shortly after the onset of illumination. Later in the illumination period, the CO2 assimilation rate of the PDRP KO leaves continued to rise and nearly match the rate of the WT leaves at ∼2 h into the illumination period. A plausible explanation for the slower photosynthesis induction kinetics is that in dark or low light, and prior to the onset of highlight illumination, the mesophyll stromal Pyr pool in the T27 is reduced to an abnormally low level. Since PPDK in leaves of the PDRP KO plants was shown to be fully active in the dark or low light (Figures 6 and 7, B), it seems likely that most of the stromal Pyr may be converted into PEP by the action of the highly active stromal PPDK pool. Moreover, as Pyr is converted to PEP in dark or low light, the Pyr in the stroma would likely remain depleted since import from the cytosol by the plastid Pyr/H+ transporter is inactive in the dark and likely less active in low light as flux is pH gradient dependent (Flügge et al., 1985; Ohnishi and Kanai, 1987, 1990). How would an abnormally lower mesophyll stromal Pyr level cause the prolonged photosynthetic induction phase we observed in the PDRP KO leaves? We propose that as leaves are first exposed to light, the low Pyr level would limit the supply of PEP produced by PPDK thus limiting the rate of primary carboxylation by PEPc relative to WT. Consistent with this hypothesis are our measurements of whole leaf Pyr levels, which showed Pyr levels in dark-adapted KO leaves were ∼60% lower than WT (Figure 9). Whereas whole leaf Pyr levels are not a direct measure of mesophyll cell Pyr, previous studies in maize that examined Pyr levels in both whole leaves and isolated mesophyll cells found Pyr status in leaves mirrors light-/dark-induced changes in Pyr levels in isolated mesophyll cells (Nakamoto and Edwards, 1983; Roeske and Chollet, 1989). Likewise, an initially lower stromal Pyr pool can also account for why CO2 assimilation rates of the KO leaves were shown to eventually recover to WT at ∼2 h into the illumination period (Figure 8A). The 2-h point likely represents the time required for newly fixed carbon to completely “refill” the steady-state stromal Pyr pool as well as other downstream C4 metabolite pools. At this point, net CO2 assimilation is no longer limited by the rate of primary carboxylation (e.g. at the 2-h point) but by the secondary carboxylation rate (i.e. Rubisco) (von Caemmerer, 2000). Previous studies on light induction in C4 plants support this speculation by linking mesophyll stromal Pyr levels to faster light induction kinetics (Leegood and Furbank, 1984; Usuda, 1985). Wang et al. also provide in silico evidence that initial, dark-level Pyr levels can limit the speed of light induction. Using their nonsteady-state model of NADP-ME-type C4 photosynthesis to run simulations of light induction, it was revealed that the rate of activation of PPDK by PDRP imposed a substantial limitation on Anet in the initial phase of light induction. When PDRP levels were incrementally increased in subsequent simulation runs, rates of Anet correspondingly rose until the limitation on Anet shifted from PPDK activation rate (e.g. PDRP level) to Rubisco activity and gs, respectively. This result implied that Pyr levels in dark-adapted leaves are normally high enough to fuel additional PEP synthesis in the event that PPDK can be activated at a faster rate at the onset of incident light (e.g. by increasing PDRP concentration). However, our findings suggest this observation holds only if PDRP maintains PPDK in the phosphorylated/inactivated state in dark or prolonged low light (as was assumed in the model simulations). Otherwise, the absence of PDRP would lead to lower Pyr levels in the dark (e.g. Figure 9) thus impeding light induction more severely than those shown simulated in the model. Thus, we propose that one of PDRPs roles in C4 photosynthesis is aimed at stabilizing the stromal Pyr pool during the dark or in periods of prolonged decreases in incident light intensity. Presumably, this benefits C4 photosynthesis by ensuring that primary CO2 carboxylation is not Pyr limited during photosynthetic induction or re-induction as leaves transition from low light to high light.
Moreover, in a dynamic light environment, the slower photosynthetic induction kinetics we observed for the T27 plants (Figures 7, A and 8, A) would not be limited to just at the onset of the photoperiod but may occur frequently throughout the photoperiod during up/down changes in irradiance, such as occurs with sunflecks within a canopy or transient cloud cover (Slattery et al., 2018). Under a dynamic light scenario, the slower light induction kinetics of the PDRP KO leaves (Figure 7A) would be expected to reduce total fixed carbon over the course of a photoperiod relative to WT. For example, as shown in Figures 7, A and 8, A, transitioning from dark to high light or low light to high light, rates of Anet in the PDRP KO plants are ∼15% lower than WT during the first 30 min of illumination. Integrating this loss of potential CO2 assimilation over the course of a growing season should translate into a substantial reduction in fixed carbon accumulation leading to proportionally lower biomass and growth relative to WT (Acevedo-Siaca et al., 2020). Future experiments focusing on growth of the mutant plants in dynamic light environments or under field conditions will be needed to confirm this prediction.
We did, however, conduct a short 7-day dynamic lighting experiment. Here we assessed the effect of a fluctuating light photoperiod on Fv/Fm (i.e. photosystem II efficiency) in S. viridis seedling leaves (Figure 10). Over the 7-day period, we observed a significant decline in Fv/Fm in the fluctuating light exposed T27 leaves linked strictly to the absence of PDRP. It is unlikely the reduction in Fv/Fm induced by the fluctuating light regime is linked to the slower photosynthesis induction kinetics we observed for the T27 plants (Figures 7, A and 8, A) since at Day 0, the Fv/Fm of the T27 and WT leaves are approximately the same (Figure 10). In other words, the decline in Fv/Fm appears only after a multi-day exposure to the fluctuating light regime, while the slower light induction kinetics can be observed in leaves grown under normal illumination. In this case, it is more likely the decline in Fv/Fm in the T27 leaves is attributable to cumulative photodamage. This could be the result of the repetitive, brief 1-min phases of high light the leaves are exposed to over the 16-h photoperiod. We speculate that during these flashes, the supply of PEP for primary carboxylation becomes unstable and limiting due to the unregulated PPDK activity. In turn, this impairs the capacity of the photosynthetic apparatus to utilize the excitation energy for photochemistry. If true, this suggests PDRPs regulation of PPDK may be important for protecting the photosynthetic apparatus from damage.
Our finding that extractable PPDK is constitutively high in the KO leaves (Figure 7B) does not necessarily mean that C4 PPDK activity is unconstrained in vivo. Other physio-biochemical factors besides PDRP-mediated phosphoregulation may contribute to regulation of PPDK activity in the chloroplast stroma. These include competitive inhibition by substrates and light-/dark-induced changes in stromal pH (Jenkins and Hatch, 1985). With respect to the latter, light-/dark changes in stromal pH can strongly influence the direction of catalysis. For example, at the alkaline pH of an illuminated chloroplast (pH 8.0–8.3), the Pyr to PEP reaction is strongly favored over the reverse, PEP to Pyr reaction. At pH 6.5–7.0, the pH of the stroma in the dark, the reverse reaction is strongly favored. However, because of the abundant pyrophosphatase and adenyl kinase activity in C4 mesophyll compartment, the PEP to Pyr reaction is unlikely to be operating, even at neutral pH (Ashton et al., 1990).
One notable insight comes from our observation that the peak PPDK activity of high-light-illuminated WT leaves was approximately the same as the high PPDK activity found in KO leaves (Figure 6). It was previously reported that much of the PPDK in C4 leaves remain in the inactive, phosphorylated form at high irradiances (Chen et al., 2014). However, assuming that the high PPDK activity we recorded for T27 leaves represents a maximum of potential PPDK activity localized in the stroma (e.g. the PPDK activity recorded at 1,000 µmol quanta m−2 s−1, Figure 6), our results indicate that nearly all of the PPDK in high light-illuminated WT leaves is in the dephosphorylated, active form. This interpretation is also supported by the phospho-PPDK immunoblot analyses shown in Figure 3. Here the comparison is the faint Thr-457 phosphorylated PPDK signal in the highlight-illuminated WT leaf extract lanes versus the prominent phospho-PPDK signal in dark-adapted WT leaf extract lanes. Additionally, the lack of any immunologically detectable phosphorylated PPDK in the dark-adapted or high-light-illuminated leaf extract lanes for T27 is consistent with previous speculation that the conserved PDRP regulatory target Thr within the PPDK active site is amenable to phosphorylation only by PDRP and not by any other endogenous protein kinase (Chastain, 2011).
Concluding remarks
To date, only one other C4-pathway regulatory enzyme, PEPC kinase (PpcK), has been examined for its importance or impact to C4 photosynthesis using a reverse genetics approach (Furumoto et al., 2007). This study created RNAi PpcK knockdown mutants of Flaveria bidentis that essentially lacked light-induced phosphorylation of PEPC at the PpcK N-terminal target Ser residue. The phosphorylation of this residue reduces feedback inhibition of PEPC by malate and is therefore predicted to enhance C4 photosynthesis (Chollet et al., 1996). An extensive analysis of the photosynthetic properties of the knockdown mutants did not reveal any physiological or biochemical effects on C4 pathway function due to the loss of PEPc phospho regulation. Furthermore, growth and development of the mutant lines were identical to WT when plants were grown under summer glass house conditions. It was concluded that PpcK regulation of PEPc is not essential for efficient C4 PS under the optimal conditions of growth (Furumoto et al., 2007). In this regard, our finding that under steady light conditions the loss of PDRP had no apparent effect on growth and vigor of the S. viridis KO plants mirrors this study. However, unlike the PpcK knockdown mutants, we were able to demonstrate how the loss of PDRP in C4 leaves translates into marked physiological changes (i.e. slower light induction kinetics, reduction in Fv/Fm in fluctuating light) and biochemical (i.e. lower leaf Pyr levels in the dark) in KO leaves. Thus, we conclude that PDRP is more strictly integrated into C4 pathway regulation than PpcK and has a potentially greater regulatory impact in terms of ensuring higher diurnal and seasonal rates of net carbon fixation, ostensibly for C4 leaves that are subject to dynamic light.
Materials and methods
CRISPR/Cas9 gene-editing, transgenic plant regeneration, and genotype analysis
A CRISPR–Cas9 system gene-editing protocol based on the Cermák et al. plant genome engineering toolkit (2017) was utilized to KO the single PDRP gene of the green millet (S. viridis) genome (Sevir.2G348700). Two single-guide RNAs (sgRNA) were designed near protospacer adjacent motif (PAM) sites in the first exon at 279–301 bp and at 361–380-bp downstream from the ATG start codon (Figure 2A) using a web-based CRISPR tool (http://crispor.tefor.net/). Using protocols as described in Cermak et al. (2017), oligonucleotides, encoding the upstream and downstream sgRNA sequence sites (Supplemental Table S1), were synthesized and inserted into the module vectors pMOD_B2518 and pMOD_C2517, respectively, via an Esp3I restriction site. These were subsequently digested with AarI restriction enzyme and assembled together with AarI cut pMOD_A1110, the Cas9 gene carrying module vector, and AarI cut pTRANS_250d, the plant transformation backbone. This final assembled “destination” construct was transformed into Agrobacterium tumefaciens strain AGL1 and used for callus transformation of S. viridis accession ME034v at the DDPSC Tissue Culture facility. Procedures for transformation, regeneration, and recovery of transgenic PDRP KO lines followed the protocols of Brutnell et al. 2010. Six T0 plants, including the lines used in this study (named T27, T28, and T29), were recovered via hygromycin selection. Using genomic DNA isolated from the lines, a 757-bp PDRP exon 1 region PCR product was amplified using primers positioned upstream (AGCTGCATCATCGAAATTGCCGTC) and downstream (AGGCTCTCGATGAGCTGATGCTTT) of the CRISPR–Cas9 target sites. This PCR product was directly sequenced via Sanger sequencing to confirm CRISPR–Cas9 editing of the target sequences in each T0 line. Sequencing primers used to generate sequence were designed to bind upstream and downstream of the CRISPR–Cas9 target sites within the 757-bp PCR product. As shown in Figure 2B and Supplemental Figure S1, each T0 line was found to be homozygous for the same 68-bp deletion produced by the action of CRISPR–Cas9 editing. To confirm the loss of the transgene (Cas9 and the hygromycin selectable marker) during segregation, T1 generation plants were screened using PCR primers targeting the hygromycin phosphotransferase transgene (Fwd: AGCTGCATCATCGAAATTGCCGTC, Rev: AGGCTCTCGATGAGCTGATGCTTT) (Zhu et al., 2021). Primers targeted to a C2H2-type zinc finger protein in the S. viridis genome (Fwd: CAGCAAGCCGCCTATATGGAG, Rev: CAGCAAGCCGCCTATATGGAG) that generates a ∼500-bp PCR product served as a positive PCR control for the screen.
Genomic DNA for PCR genotyping was extracted from S. viridis leaves using a CTAB DNA extraction protocol modified from Fulton et al. (1995) as follows. Leaf tissue samples (50–100 mg) were placed into 1.8-mL screw cap tubes along with two to three steel balls (one-eighth-inch Grade 1000 Type 430 [Abbott Ball]). The leaf material was then finely ground by first immersing the tubes briefly into liquid N2 and then shaking in a paint shaker (Harbil, model no. 5G-HD) for 2 min with the cycle of freezing and shaking repeated twice more. Genomic DNA was extracted from the ground leaf material by the addition of 500-µL CTAB extraction buffer (125-mM Tris, pH 7.5, 150-mM Sorbitol, 0.83-M NaCl, 23-mM EDTA, 0.83% CTAB (w/v), 0.83% Sarkosyl (w/v), 1% polyvinylpyrrolidone (PVP) (w/v) and incubating at 65°C for 30 min while mixing contents by hand or vortex several times. After incubation, the tubes were cooled to room temperature and an equal volume of chloroform:isoamyl alcohol (24:1) was added to the extraction mixture. The tubes were then vortexed vigorously for 30 s and centrifuged at 13,000g for 10 min, 4°C. The upper aqueous layer was then transferred to a new tube and combined with 0.7 vol. of ice-cold 100% isopropanol and gently mixed to precipitate the DNA. The precipitated DNA was collected by centrifugation (13,000g, 10 min, 4°C). The DNA pellet washed twice with ice-cold 70% (v/v) ethanol. Air dried DNA pellets were resuspended with 100-µL H2O prior to use.
Directly following transformation, T0 and T1 plants were grown in controlled environmental chambers with the following conditions: photosynthetic photon flux density (PPFD) of 500-μmol quanta m−2 s−1 photosynthetically active radiation (PAR) 16-h photoperiod, 28°C day temperature, 24°C night temperature, and 2% CO2. Screened homozygous T1 KO plants were propagated to produce T3 generation seed for generating all PDPR KO plants utilized in this study. Most experiments in this study focused on the T27 PDRP KO line due to the identical genotype and phenotype observed in all three recovered lines.
Plant material
WT S. viridis (accession ME034v) was used for all experiments. Transgenic PDRP KO plants used for experiments were derived from T3 seed. Seeds were pretreated in 5% liquid smoke over night at 30°C prior to sowing in order to increase the germination rate (Sebastian et al., 2014). Plants used for the gas exchange and the fluctuating light experiments were grown at Washington State University in Pullman Washington in environmental growth chambers (BioChambers Inc., Winnipeg, Manitoba, Canada) with a day and night length of 16 h/8 h at 31°C and 22°C with ambient air at 50% relative humidity. PPFD at plant height was 1,000-µmol quanta m−2 s−1 PAR from high-pressure sodium and metal-halide lamps and the chamber maintained a relative humidity of ca. 40%. Plants for gas exchange were grown individually in 1-L pots. Plants used for the fluctuating light experiments were grown in smaller 215-mL pots. All plants were grown in a Sunshine mix LC-1 soil (Sun Gro Horticulture, Agawam, MA, USA) and were irrigated as needed, while fertilizing every 4 days with Peters 20-20-20 (2.5-g L−1). All other experiments utilized plant material similarly cultivated at Minnesota State University except growth chamber lighting was provided by fluorescent lamps at a PPFD of 700-μmol quanta m−2 s−1 PAR at plant height. The Fv/Fm experiments shown in Figure 9 utilized 7- to 8-day-old seedlings at the two-leaf stage. All other experiments in this study utilized preflowering plants at the 3–4 week stage.
In vitro PDRP protein kinase assays
Assays of the PDRP protein kinase activity in crude leaf extracts (Figure 3C) was performed using a previously established immuno-based procedure (Chastain et al., 2018) including the following modifications. Briefly, 190-µL assay reactions containing 70 µg of affinity-purified recombinant maize (Z. mays) PPDK (fully activated, nonphospho form), 50-mM Bicine-KOH, pH 8.3, 10-mM MgCl2, 5-mM DTT, 2-mM NH4(SO4)2, 1-mM ADP, 0.2-mM ATP, 0.2-mM P1,5-di(adenosine-5')-pentaphosphate were incubated at 30°C for 10 min prior to initiating the reaction with leaf extract to ensure that recombinant PPDK monomers were fully tetramerized. Preparation of crude leaf extract was carried out as follows. About 200 mg of leaf material was excised from the middle portion of a fully expanded leaf that had been illuminated for 15 min at ∼800 µmol quanta m−2 s−1. Immediately after harvesting, the leaf material was homogenized in a 4°C prechilled mortar and pestle in 2-mL ice-cold extraction buffer (50-mM Tris pH 8.3, 5-mM MgCl2, 0.1-mM EDTA). The leaf homogenate was centrifuged for 30 s and the clarified supernatant desalted by passage through a Zeba protein desalting column (Thermo-Scientific, Waltham, MA, USA). The assays were initiated by addition of 10 µL of desalted leaf extract (∼3-µg mL−1 total protein) and incubated at 30°C for 30 min. The assays were terminated by the addition of an equal volume of 2× concentrated SDS–PAGE sample buffer. Aliquots of the terminated reaction were used to generate immunoblots that were subsequently probed with an anti-phospho-PPDK antibody (Chastain et al., 2018). For the control assays, ADP was omitted from the reaction mixture. Soluble leaf extract protein was quantified using a modified Coomassie dye-binding method (Coomassie Plus, ThermoFisher Scientific) with crystalline BSA as standard.
Immunoblot detection of PPDK and phospho-PPDK in leaf extracts
Leaf material used for immunoblot analysis of PPDK and phospo-PPDK was pretreated in dark- or high light as follows. Leaf sections (3 cm) were excised from the middle portion of a fully expanded leaf and floated adaxial side up on dH2O in an open container and maintained at 30°C. The leaf segments were then dark adapted for 2 h after which a set of segments were removed and flash frozen in liquid nitrogen. The remainder of the segments was then illuminated from above with a red/blue spectrum LED lamp at a PPFD of 1,300-µmol quanta m−2s−1 (PAR). After 30 min of illumination, the leaf segments were flash frozen in liquid nitrogen and stored in at −80°C for later preparation of leaf extract. Leaf extracts were prepared by homogenizing frozen leaf segments in a prechilled mortar and pestle with 1.5–2.0-mL ice-cold extraction buffer consisting of 50-mM Tris pH 8.0, 2-mM EDTA, 1-mM PMSF, and ThermoFisher Halt protease inhibitor cocktail plus ∼30-mg insoluble PVP. After a 5-min centrifugation at 16,000g and 4°C, aliquots of the clarified supernatant were combined with an equal volume of 2× concentrated SDS–PAGE sample buffer and stored at −20°C until used for generating immunoblots. Procedures for SDS–PAGE and immunoblotting were as described previously and include the following modifications (Chastain et al., 2008, 2018). For SDS–PAGE, 10% or 12% Bolt Bis–Tris precast acrylamide gels (Thermo-Fisher Scientific) were used. The primary antibodies used in immunoblot procedures were: an affinity-purified (rabbit) polyclonal antibody raised against maize recombinant C4 PPDK (Chastain et al., 2008); an affinity-purified (rabbit) polyclonal antibody raised against a phosphopeptide corresponding to the Thr-phosphorylation domain of maize C4 PPDK (Chastain et al., 2008); A goat anti-rabbit alkaline phosphatase conjugate (Promega, Madison, WI, USA) was used as the secondary antibody in all immunoblot procedures. For signal detection, immunoblots were incubated with the chemiluminesence substrate CDP-Star and digitally scanned using an LI-COR C-Digit blot scanner (LI-COR Biosciences, USA).
PPDK enzyme assays
Leaf material used for PPDK enzyme assays consisted of 3-cm leaf sections excised from the middle portion of a fully expanded leaf and floated adaxial side up in a tank of dH2O maintained at 30°C prior to dark/light treatment as specified in Figures 4, 6, and 7, B. At specified intervals, sets of dark- or light-treated leaf sections were removed and flash frozen in liquid nitrogen and subsequently stored at −80°C for later extraction and assay. Leaf extracts used for PPDK enzyme assays were prepared by homogenizing leaf segments in a prechilled mortar and pestle with 1.5–2.0-mL ice-cold extraction buffer consisting of 50-mM Tris pH 8.0, 5-mM MgSO4, 5-mM DTT, 0.1-mM EDTA, 10-mg mL−1 blue dextran, 1-mM PMSF, and ThermoFisher Halt protease inhibitor cocktail. After a 5 min, 4°C centrifugation at 16,000g the clarified supernatant was desalted by passage through a Zeba protein desalting column (Thermo-Fisher Scientific). PPDK activity in the extracts was assayed in the PEP-forming direction using a coupled, PEPc/malate dehydrogenase-based spectrophotometric assay method adapted for 96-well plate reader format (Ashton et al., 1990). The assay mix consisted of 50-mM Tris, pH 8.3, 8-mM MgSO4, 5-mM DTT, 5-mM (NH4)2SO4, 2.5-mM KHPO4, 10-mM KHCO3, 2-mM Pyr, 1-mM glucose-6-P, 0.50-mM NADH, 1-mM ATP, 0.5–1.0-U mL−1 PEPc, 2.5-U mL−1 MDH. Prior to initiating assays, leaf extracts were incubated at 30°C for 10 min to ensure the inactive PPDK monomers and dimers were fully oligomerized into the active PPDK tetramer. Assays were initiated by addition of leaf extract (∼10 µg of protein) and incubated at 30°C for the duration of the 8-min assay. Assays without ATP were carried out in parallel to serve as controls.
Gas exchange measurements
T27 and WT plants grown under ambient CO2 and a PPFD of 850-µmol quanta m−2 s−1 (PAR) for 4 weeks were used to measure the response of net CO2 assimilation (Anet) to changes in photosynthetic quanta flux density (PPFD). The gas exchange measurements were made with a LI-6800 portable photosynthesis system (LI-COR Biosciences, Lincoln, NE, USA) equipped with a Multiphase flash fluorometer circular chamber (6800-01A). Two leaves from an individual plant were dark adapted for 30 min in the leaf chamber and then Anet was logged every min for 30 min in the dark and then under the following light conditions: 1,500 µmol quanta m−2 s−1 for 30 min, then 100 µmol quanta m−2 s−1 for 30 min. All measurements were made at a leaf temperature of 30°C, a sample CO2 concentration of 400 mbar, and a VPD of ca. 1.5. An auto program was used to collect gas exchange parameters at 1-min intervals from leaves of separate T27 and WT individual plants. A similar experiment was conducted on both T27 and WT plants from the same growth conditions where leaves were first acclimated for 30 min in the dark LI-COR 6800 leaf chamber and then Anet was measured with an auto program for 30 min in the dark and then at 1,500 µmol quanta m−2 s−1 for 120 min, logging every minute.
Leaf Pyr content
Pyr concentrations were determined from dark-adapted or light-exposed 3-cm leaf sections excised from the middle portion of fully expanded leaves. Leaf sections were first floated adaxial side up in a tank of dH2O maintained at 30°C. After 2-h dark, a set of leaf sections were removed and flash frozen in liquid nitrogen. The remainder of the leaf sections were then illuminated from above with a red/blue spectrum LED lamp at a PPFD of 1,300-µmol m−2s−1 (PAR) for 3 min and then flash frozen in liquid nitrogen. All flash frozen leaf sections were stored at −80°C for later use. Extracts for Pyr analysis were prepared from frozen leaf sections as follows with all steps were carried out in a 4°C cold room. Leaf sections (∼100 mg) stored at −80°C were re-introduced into liquid nitrogen for subsequent homogenization in a mortar and pestle prechilled with liquid N2. Leaves were ground to a powder and then rapidly homogenized in 0.5-mL extraction buffer (50-mM Tris pH 8.0, 2-mM EDTA). An aliquot of the homogenate (150 µL) was immediately deproteinized by addition of 15 µL of concentrated trichloroacetic acid (TCA) provided by a sample deproteinizing kit obtained from Biovision Inc. (cat. #K823) per kit instructions. An additional aliquot of the homogenate was taken for determination of total chlorophyll (Wintermans and DeMots, 1965) for use in the quantitation of leaf Pyr as nmol Pyr per mg chl in each extracted leaf sample. After centrifugation to remove the TCA precipitated protein, the extract was neutralized to a pH of 6.5–7.0 by addition of 8 µL of concentrated KOH provided from the deproteinizing kit. Pyr was assayed in neutralized extracts using a colorimetric assay method obtained in kit form from Sigma-Aldrich (Cat #MAK332) using kit instructions. Recovery of exogenously added Pyr subjected to the same extraction protocol was 105% ± 13% (n = 3).
Fluctuating light experiments
Seeds were germinated and grown for 7–8 days until the two-leaf stage in environmental growth chambers as described in “plant material” above. Plants were then moved to either a fluctuating light treatment (1 min at 1,200 µmol quanta m−2 s−1 and 4 min at 300 µmol quanta m−2 s−1) or to a steady high light (850 µmol quanta m−2 s−1) or low light (90 µmol quanta m−2 s−1) treatment each under a 16-h/8-h light/dark photoperiod, for 7 days (Schneider et al., 2019). For each light growth treatment, eight biological replicates per genotype (T27 and WT) were simultaneously grown, with each plant in an individual per pot and randomly shuffled under their growth light treatment. Each plant was measured daily around the same time with a PAM fluorescence imaging system (Maxi imaging-PAM, IMAG-K7 from Walz GmbH, Effeltrich, Germany). Plants were dark adapted before PAM measurements of Fv/Fm that was averaged over the entire canopy for each individual plant as previously described by Schneider et al. (2019). Initially up to four plants were measured simultaneously but as the plants grew larger one plant at a time was measured.
Statistical analysis
Statistical differences between genotypes for the data shown in Figures 6–9 were analyzed by Student’s two-tailed t test (assuming unequal variances) using an EXCEL data analysis software package (version 16.54). Statistical analysis for data presented in Figure 10 was performed using R software (version 4.1.0, R Foundation for Statistical Computing, Vienna, Austria). Normality and equal variances were tested before running a four-way repeated measure ANOVA with main factors of Genotype × Light treatment × Day × Batch. There was not a main effect due to the repeated experiments (Batch); therefore, we present only a three-way ANOVA using the main factors of Genotype x Light treatment x Day (see Supplemental Table S2) with a post-hoc Tukey’s test to examine differences in Fv/Fm between genotypes within the same growth Light treatment across 7 days (Supplemental Table S3).
Accession numbers
Sequence data from this article can be found in the GenBank data libraries under the following accession numbers: (XM_034724397, Sevir.2G348700) and (XM_034732529, Sevir.36253900).
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Aligned Sanger sequenced PDRP exon 1 regions of S. viridis T0 KO plants.
Supplemental Figure S2. In vitro PDRP protein kinase assays of desalted leaf extracts.
Supplemental Table S1. Primers used in this study.
Supplemental Table S2. Results of a three-way repeat measure for ANOVA analysis of Figure 10 data.
Supplemental Table S3.Figure 10 ANOVA for Genotype × Light treatment × Day post-hoc Tukey’s test.
Z.W., P.H., Q.Z., J.R., and T.B. collectively generated the transgenic S. viridis knockout lines. K.O. carried out gas exchange measurements. K.O. and H.H.K. analyzed Fv/Fm values. N.N.A., C.C.J., A.M., A.N.R., and C.H. collectively carried out the biochemically related analyses. C.J.C. wrote the manuscript with input from A.B.C.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plphys/pages/general-instructions) is Chris J. Chastain ([email protected]).
Acknowledgments
The authors wish to acknowledge the skillful assistance of Jenna R. Wegsheid in carrying out the PPDK enzyme assays shown in Figure 4.
Funding
This research was in part funded by the Office of Biological and Environmental Research in the DOE Office of Science (SC0018277) to A.B.C.
Conflict of interest statement. None declared.
References
Author notes
Present addresses for Hans-Henning Kunz: Department of Plant Biochemistry, LMU Munich, Munich, 82152, Germany.
BASF Corp., Morrisville, North Carolina, 27560, USA.
McClintock LLC, St. Louis, Missouri, 63131, USA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}