





Abstract
Cockayne Syndrome (CS) is a severe neurodegenerative and premature aging autosomal-recessive disease, caused by inherited defects in the CSA and CSB genes, leading to defects in transcription-coupled nucleotide excision repair (TC-NER) and consequently hypersensitivity to ultraviolet (UV) irradiation. TC-NER is initiated by lesion-stalled RNA polymerase II, which stabilizes the interaction with the SNF2/SWI2 ATPase CSB to facilitate recruitment of the CSA E3 Cullin ubiquitin ligase complex. However, the precise biochemical connections between CSA and CSB are unknown. The small ubiquitin-like modifier SUMO is important in the DNA damage response. We found that CSB, among an extensive set of other target proteins, is the most dynamically SUMOylated substrate in response to UV irradiation. Inhibiting SUMOylation reduced the accumulation of CSB at local sites of UV irradiation and reduced recovery of RNA synthesis. Interestingly, CSA is required for the efficient clearance of SUMOylated CSB. However, subsequent proteomic analysis of CSA-dependent ubiquitinated substrates revealed that CSA does not ubiquitinate CSB in a UV-dependent manner. Surprisingly, we found that CSA is required for the ubiquitination of the largest subunit of RNA polymerase II, RPB1. Combined, our results indicate that the CSA, CSB, RNA polymerase II triad is coordinated by ubiquitin and SUMO in response to UV irradiation. Furthermore, our work provides a resource of SUMO targets regulated in response to UV or ionizing radiation.
INTRODUCTION
The integrity of DNA is continuously challenged by exogenous and endogenous DNA-damaging agents, such as genotoxic chemicals, ionizing radiation (IR), ultraviolet (UV) radiation or reactive oxygen species (ROS) (1). A multitude of cellular mechanisms collectively called the DNA damage response (DDR), ensure efficient responses to genotoxic insults including recognition and repair of DNA lesions. IR induces a set of different types of DNA damage, including oxidized bases, single and double strand breaks (DSBs). The latter are among the most cytotoxic DNA lesions and are repaired by homologous recombination (HR), non-homologous end-joining (NHEJ) and alternative end-joining (Alt-EJ) (2–4).
UV induces cyclobutane pyrimidine dimers (CPD), a photolesion with mild helix- distorting properties and 6-4 photoproducts (6-4PP), a photolesion with strong helix- distorting properties, that both strongly interfere with DNA-transacting processes. In human skin cells, CPDs and 6-4PPs are exclusively removed by nucleotide excision repair (NER). UV-induced photolesions in the transcribed strand of actively transcribed regions are repaired by transcription-coupled NER (TC-NER), whereas CPDs and 6-4PPs localized throughout the genome are repaired by global genome NER (GG-NER) (5). TC-NER and GG-NER differ in their molecular recognition of the DNA lesion, but share the subsequent steps, including lesion verification, excision of 22–30 nucleotides around the lesion and gap filling by DNA synthesis. Proteins that are involved in DNA repair pathways need to be tightly regulated to avoid inappropriate DNA processing. Post-translational modifications like phosphorylation, PARylation, ubiquitination and SUMOylation play pivotal roles in this regulation (6).
Small Ubiquitin-like MOdifier (SUMO) is a 11 kDa protein that can be covalently attached to lysine residues in substrate proteins via an enzymatic cascade, involving a heterodimeric SUMO activating E1 enzyme, a single SUMO conjugating E2 enzyme and a limited number of SUMO E3 ligases (7). SUMOylation is a highly dynamic process due to the presence of SUMO specific proteases that can reverse the SUMOylation of target proteins (8). Mammals express at least three SUMO family members, SUMO1-3, with SUMO2 being the most abundant and essential member (9). Hundreds of target proteins are regulated by SUMOs under both normal and cellular stress conditions (10). The consequences of SUMOylation are specific for different target proteins and can include the alteration of interactions with other proteins, the alteration of enzymatic activity, or affecting substrate stability.
The first link between SUMOylation and DNA repair was revealed in studies on base excision repair (BER), where SUMOylation induces a conformational change in the Thymine-DNA Glycosylase protein and thereby stimulates the repair process (11,12). Furthermore, two SUMO E3 ligases, PIAS1 and PIAS4, accumulate at DSBs. These E3 ligases SUMOylate BRCA1 to induce its activity and SUMOylation is required for the accumulation of different repair components to facilitate repair of DSBs (13).
SUMO and ubiquitin also act together in the DDR, best exemplified by the modification of the homo-trimeric, ring shaped protein Proliferating Cell Nuclear Antigen (PCNA). PCNA encircles DNA where it acts as a processing factor for DNA polymerases and as an interaction platform for proteins involved in DNA metabolism. Mono-ubiquitination of PCNA on lysine 164 upon DNA damage induces the recruitment of polymerases needed for translesion synthesis, whereas SUMOylation on the same lysine inhibits recombination during DNA synthesis by recruiting the anti-recombinogenic helicase Srs2 (14,15). The role of SUMO and ubiquitin crosstalk in DNA repair was further emphasized by the observation that the SUMO-dependent recruitment of RNF4, a well-studied SUMO-targeted ubiquitin ligase (STUbL), to DSBs induces a ubiquitination signal that is essential for efficient repair of DSBs (16,17).
RNF111, another STUbL, was shown to regulate the ubiquitination of XPC in GG-NER. XPC is part of the GG-NER initiating XPC-RAD23-CETN2 DNA damage recognizing complex. RNF111-mediated XPC ubiquitination is required for efficient progression of the NER reaction by stimulating the handover of damaged DNA between XPC and the structure-specific endonucleases XPG and ERCC1/XPF (18–20).
Ubiquitin also plays a pivotal role in the regulation of TC-NER. Two key factors in TC-NER are Cockayne Syndrome (CS) gene products CSA and CSB. CS is an autosomal-recessive disease and patients display severe neurodegeneration and premature aging. CSA- and CSB-deficient cells are both impaired in TC-NER and consequently hypersensitive to UV irradiation. TC-NER is initiated by stalling of RNA polymerase II at lesions, stabilizing the interaction with the SNF2/SWI2 ATPase CSB to facilitate recruitment of the CSA protein. CSA is part of an E3 Cullin ubiquitin ligase complex. CSA was proposed to ubiquitinate and destabilize CSB (21). UVSSA, a more recently identified player in TC-NER, was shown to counteract this CSA-dependent destabilization of CSB by recruiting the deubiquitinating enzyme USP7 (22,23). Additionally, the UV-induced ubiquitination of the elongating RNA polymerase II (RNAPIIo) is dependent on UVSSA (24). Collectively, these data indicate a pivotal role for post-translational modifications during the DNA damage response.
Here. we set out to investigate potential biochemical connections between CSA and CSB. In our hands, CSA did not act as a ubiquitin E3 ligase for CSB in a UV-regulated manner. In contrast, we found that the ubiquitination of RNAPIIo is regulated by the CSA complex in response to UV. Furthermore, we found that CSB is the most strongly regulated substrate for the ubiquitin-like protein SUMO2 in response to UV. Clearance of SUMOylated CSB was dependent on CSA. Our data indicate that SUMOylated CSB, CSA and ubiquitinated RNAPIIo are connected and function together to promote efficient TC-NER.
MATERIALS AND METHODS
Detailed information on antibodies, oligonucleotides, reagents and databases used can be found in Supplementary Table S5.
Plasmids
Expression constructs for CSB K32R, K205R, 2KR and 5KR were generated by site-directed mutagenesis using the pDONR207-CSB wild-type (WT) plasmid as a template. Resulting CSB mutants and WT construct were cloned into pLX303 destination vector for lentiviral transduction (Addgene plasmid #25897), into pBabe-puro-GFP-DEST destination vector (kind gift of Dr Marc Timmers, Freiburg, Germany) for retroviral transduction or into pDEST-EGFP-C1 destination vector for transient transfection, using Gateway cloning technology (Thermo Fisher Scientific). To create a truncated CSB mutant, a cDNA encoding amino acids 1 until 341 of CSB was cloned into pDONR207. For bacterial expression of this CSB fragment, this construct was subsequently cloned into pDEST15 using Gateway cloning technology. Epitope-tagged CSA has been described previously (25).
Cell lines, SILAC labelling and generation of cell lines
U2OS, hTERT1 immortalized RPE1 cells and sv40 immortalized CS1AN (a CSB patient cell line), CS3BE (a CSA patient cell line) and VH10 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FCS and 100 U/ml penicillin and 100 μg/ml streptomycin. For stable isotope labelling by amino acids in cell culture (SILAC), cells were essentially labelled as described before (26). Briefly, cells were grown in medium supplemented with [13C6, 14N4] arginine (referred to as Arg6), [13C6, 15N4] arginine (referred to as Arg10), [2H4, 13C6, 14N2] lysine (referred to as Lys4), [13C6, 15N2] lysine (referred to as Lys8) as indicated.
U2OS cell lines stably expressing Flag-SUMO2, His10-SUMO2 or His10-ubiquitin were previously described (27,28). U2OS His10-SUMO2-IRES-GFP cells expressing GFP-CSB WT and mutants were generated by infecting cells with retrovirus encoding the different pBabe-GFP-CSB constructs together with a Puromycin resistance gene. Cells were selected for GFP-CSB expression by culturing in medium supplemented with 1 μg/ml Puromycin. CS1AN cell lines co-expressing His10-SUMO2 and tagless CSB WT and mutants were generated by an initial round of infection of cells with lentivirus encoding a His10-SUMO2-IRES-puro construct and Puromycin selection (1 μg/ml) and a subsequent round of infection with lentivirus encoding the different pLX303-CSB mutant constructs. Cells were selected for CSB expression by culturing in medium supplemented with 5 μg/ml Blasticidin. CS1AN cells stably expressing EGFP-CSB WT or mutants were generated by transfecting cells with pDEST-EGFP-CSB constructs also encoding a Neomycin resistance gene. Monoclonal cell cultures were selected with 400 μg/ml G418 (Neomycin) and were selected by flow cytometry based on EGFP expression.
U2OS Flp-In/T-REx cells, which were generated using the Flp-In™/T-REx™ system (Thermo Fisher Scientific), were a gift of Daniel Durocher. These cells were co-transfected with pLV-U6g-PPB containing an antisense guide RNA targeting the CSA/ERCC8 gene (5'-CCAGACTTCAAGTCACAAAGTTG-3') from the Sigma-Aldrich sgRNA library together with an expression vector encoding Cas9-2A-GFP (pX458; Addgene #48138). Transfected U2OS Flp-In/T-REx were selected on puromycin (1 μg/ml) for 3 days, plated at low density after which individual clones were isolated. Knockout of CSA in the isolated clones was verified by sequencing of genomic DNA by nested PCR using the following primers: 5'-CAGTCTGTGTCCAGTTTCTGTG-3', 5'-CATATTTGTTATGTGTTTCTTTGAG-3', 5'-GTACATACATACATACACATTTACCAATAC-3', and 5'-CTGAGAAAAAATGTACCTAAATATTAAG-3', as well as by immunoblot analysis (Rabbit α-CSA/ERCC8, EPR9237, Abcam 137033). The absence of Cas9 integration/stable expression was confirmed by immunoblot analysis (Mouse α-Cas9, 7A9-3A3, #14697, Cell Signaling Technology). CS3BE cells stably expressing His-CSA were generated by infecting cells with lentiviruses encoding CSA and the Blasticidin resistance gene. After infection, cells were selected for expression of CSA by culturing in medium supplemented with 5 μg/ml Blasticidin. RPE1 cell lines immortalized by hTERT1 and expressing inducible shRNA against UBA2/SAE2 were generated by infecting cells with lentiviruses encoding the different shRNA constructs and a Neomycin resistance cassette. Cells were selected for expression of the introduced construct by selection with 400 μg/ml G418.
For the induction of different DNA lesions 50 μM etoposide (Sigma Aldrich) was used in culture medium for 1 h, 0.02% methyl methanesulfonate (MMS) (Sigma Aldrich) was used in culture medium for 1.5 h, 2 mM hydroxyurea (HU) (Sigma Aldrich) was used in culture medium for 2 or 24 h. Cells were treated with 4 Gy of IR and 20 J/m2 UV-C light and lysed after the indicated recovery times. 100 μM 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside (DRB) (Sigma Aldrich) was used in culture medium for 3 h prior to UV irradiation. 2 μg/ml α-amanitin (HY-19610, MedChemExpress and A2263-1MG Sigma Aldrich) was used in culture medium for 24 h prior to UV irradiation.
Live imaging experiments, UV-C irradiation
Localisation studies of GFP-CSB were performed using UV-C (266 nm) laser-irradiation for local DNA damage infliction (29). Briefly, a 2-mW pulsed (7.8 kHz) diode-pumped solid-state laser emitting at 266 nm (Rapp OptoElectronic) was connected to the confocal microscope Leica TCS SP5 AOBS with an Axiovert 200M housing adapted for UV by all-quartz optics. By focusing the UV-C laser inside cell nuclei without scanning, only a limited area within the nucleus (diffraction limited spot) was irradiated. Cells were imaged and irradiated through a 100×, 1.2 NA Ultrafluar quartz objective lens. Images obtained prior to and post UV-C laser irradiation were analysed using the LAS AF software (Leica).
Purification of His10 conjugates using Ni-NTA beads
His10-ubiquitin conjugates and His10-SUMO2 conjugates were purified using nickel-nitrilotriacetic acid-agarose beads (Ni-NTA)(Qiagen) as previously described (30). In brief, cells stably expressing His10-SUMO2 or His10-ubiquitin were lysed in 6 M guanidine–HCl pH 8.0. Small fractions of cells were separately lysed in SNTBS buffer (2% SDS, 1% N-P40, 50 mM Tris pH 7.5 and 150 mM NaCl) as input controls. After sonication and addition of imidazole (50 mM) and ß-mercaptoethanol (5 mM), lysates were incubated with pre-washed Ni-NTA beads. After incubation, beads were washed subsequently with buffers 1–4. Wash Buffer 1: 6 M guanidine–HCl, 0.1 M Na2HPO4/NaH2PO4, pH 8.0, 10 mM Tris–HCl pH 8.0, 10 mM imidazole pH 8.0, 5 mM ß-mercapthoethanol and 0.1% Triton X-100 (0.2% Triton X-100 for immunoblotting sample preparation). Wash Buffer 2: 8 M urea, 0.1 M Na2HPO4/NaH2PO4, 10 mM Tris–HCl pH 8.0, 10 mM imidazole pH 8.0, 5 mM ß-mercaptoethanol and 0.1% Triton X-100 (0.2% Triton X-100 for immunoblotting sample preparation). Wash Buffer 3: 8 M urea, 0.1 M Na2HPO4/NaH2PO4, 10 mM Tris–HCl pH 6.3, 10 mM imidazole pH 7.0, 5 mM ß-mercaptoethanol and no Triton X-100 (0.2% Triton X-100 for immunoblotting sample preparation). Wash Buffer 4: 8 M urea, 0.1 M Na2HPO4/NaH2PO4, 10 mM Tris–HCl pH 6.3, 5 mM ß-mercaptoethanol and no Triton X-100 (0.2% Triton X-100 for immunoblotting sample preparation). Elution of sample was performed twice in one bead-volume of 7 M urea, 0.1 M Na2HPO4/NaH2PO4, 10 mM Tris–HCl pH 7.0 and 500 mM imidazole pH 7.0.
Electrophoresis and immunoblotting
To visualize CSB and RPB1 by immunoblotting, either 6% Tris-glycine gels or Novex 3–8% Tris-acetate gradient gels (Thermo Fisher Scientific) were used for electrophoresis. To visualize other proteins, samples were separated on Novex 4–12% Bis–Tris gradient gels (Thermo Fisher Scientific) with MOPS buffer or via regular SDS-PAGE using Tris-glycine gels. Separated proteins were transferred onto Amersham Protran Premium 0.45 NC nitrocellulose blotting membrane (GE Healthcare) using a submarine system. For whole cell lysates, membranes were stained with Ponceau S (Sigma) as loading control. Membranes were blocked with 8% non-fat milk in PBS 0.05% Tween for 1 h, prior to primary antibody incubation.
RNA synthesis recovery assay
Two independent doxycycline (Dox)-inducible shRNAs against UBA2 and a non-targeted control shRNA (31) were stably expressed in RPE1 cells. Cells were seeded in 96-well plates and the knockdown was induced by Dox treatment. Cells were irradiated with UV-C (10 J/m2), and subsequently cultured for the indicated time-periods (0–24 h) to allow RNA synthesis recovery. RNA was labelled for 1 h in medium supplemented with 1 mM EU (Click-iT® RNA Alexa Fluor® 594 Imaging Kit, Life Technologies) according to the manufacturer's instructions. Imaging was performed on an Opera Phenix confocal High-Content Screening System (Perkin Elmer, Hamburg, Germany) equipped with solid state lasers. General nuclear staining (DAPI) and Alexa 594 were serially detected in nine fields per well using a 20× air objective. Three independent experiments were analysed using a custom script in the Harmony 4.5 software (Perkin Elmer) in which nuclei were individually segmented based on the DAPI signal. RNA synthesis recovery was determined by measuring the mean Alexa 594 intensity of all nuclei per field.
Proteomics sample preparation and mass spectrometry
His10-purified samples were supplemented with ammonium biocarbonate (ABC) to 50 mM. Subsequently samples were reduced with 1 mM dithiothreitol (DTT) for 30 min and alkylated with 5 mM chloroacatemide (CAA) for 30 min and once more reduced with 5 mM DTT for 30 min at room temperature. Proteins were digested with Lys C for 3 h in a 1:100 enzyme-to-protein ratio. Subsequently the peptides were diluted 4-fold with 50 mM ABC and trypsin digested overnight in a 1:100 enzyme-to-protein ratio.
Mass spectrometry
Samples were acidified and subsequently desalted and concentrated on triple-disc C18 reverse phase StageTips (32). Peptides were eluted twice, with 40% and 60% acetonitrile (ACN) in 0.1% formic acid, respectively. Peptides were vacuum centrifuged until all liquid was evaporated and re-suspended in 0.1% formic acid. Peptides were analysed by mass spectrometry using a Q-Exactive Orbitrap (Thermo Fisher Scientific) coupled to an EASY-nLC system (Proxeon).
Processing of mass spectrometry data
MaxQuant (version 1.5.3.30) was used to analyse RAW data. The MaxQuant output protein groups table was further analysed using Perseus software (version 1.5.3.1). Data were filtered by removing ‘reverse identified’, ‘only identified by site’ and ‘potential contaminants’. LFQ intensities were log2 transformed. The following groups were compiled from the three biological replicates: U2OS 1 h IR, U2OS 1 h UV, U2OS-His10-SUMO2 mock treated, U2OS-His10-SUMO2 1 h IR, U2OS-His10-SUMO2 6 h IR, U2OS-His10-SUMO2 1 h UV, U2OS-His10-SUMO2 6 h UV.
Protein groups that had at least three valid values in at least one group were selected for further analysis. Missing values were imputed using Perseus software by normally distributed values with a 1.8 downshift (log2) and a randomized 0.3 width (log2) considering the whole matrix. Subsequently, two-sample t-tests were conducted between different experimental conditions (two-sided). Proteins that showed a log2 difference of >0.66 and a P-value of <0.05 at least in one His10-SUMO2 purified condition compared to the U2OS parental control were selected as SUMO2 target proteins. IR-induced SUMO2 targets 1 h or 6 h post-IR needed a log2 difference of >0.66 and a P-value of <0.05 compared with His10-SUMO2 mock treated control and parental U2OS 1 h IR conditions. UV-induced SUMO2 targets 1 h or 6 h post-UV needed a log2 difference of >0.66 and a P-value of <0.05 compared with His10-SUMO2 mock treated control and parental U2OS 1 h UV condition.
Identification of proteins that bind to SUMOylated CSB
Escherichia coli strain BL21 was co-transformed with a plasmid encoding the GST-CSB N-terminus (aa 1–341) and a plasmid encoding the SUMO2 conjugation machinery (33). Expression of transgenes was induced by 0.5 mM IPTG at 25°C overnight. Bacteria were harvested by centrifugation, washed twice with icecold PBS before resuspending in PBS, 0.5 M NaCl, 1 mM PMSF and cOmplete™ mini protease inhibitor cocktail (Sigma). Cells were lysed by sonification and the addition of 10% Triton X-100. Lysates were cleared by centrifugation at 13 000 rcf and supernatant was incubated with glutathione Sepharose beads (GE Healthcare) for 2 h at 4°C. After incubation, beads were washed twice with PBS, 0.5 M NaCl,1 mM PMSF and cOmplete™, mini protease inhibitor cocktail (Sigma) and washed three times with 50 mM Tris pH 7.5, 0.5 M NaCl. Each sample was equally divided over two new reaction tubes and one half was treated with 10 μg of recombinant SENP2 overnight at 4°C. Beads were washed three times with 50 mM Tris pH 7.5 and 0.5 M NaCl, then washed twice with 50 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP-40, 1 mM MgCl2, 20 mM NEM and cOmplete™, mini protease inhibitor cocktail (Sigma) before incubation with cell lysates.
For the preparation of the cell lysates, CS1AN cells were irradiated with UV-C at 20 J/m2 and lysed 1 h after UV treatment in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP-40, 1 mM MgCl2, 20 mM NEM and cOmplete™, mini protease inhibitor cocktail (Sigma)). Cells were sonicated and treated with 500 U/ml benzonase for 1 h at 4°C. Cleared cell lysate was incubated with previously prepared glutathione Sepharose containing SUMOylated GST-CSB, de-SUMOylated GST-CSB or GST only for 2 h at 4°C. After incubation, beads were washed four times with lysis buffer, subsequently three times in 50 mM fresh ammonium bicarbonate (ABC) and bound proteins were trypsinized with 2 μg trypsin overnight at 37°C.
Flag-SUMO2 and Flag-Ubiquitin immunoprecipitation
Flag-SUMO2 and Flag-Ubiquitin conjugates were enriched by anti-Flag immunoprecipitation as described previously (27).
RESULTS
Identification of SUMOylated proteins in response to IR- or UV-induced DNA damage
We set out to identify dynamic SUMO2 target proteins that are involved in the DDR, employing an unbiased quantitative proteomics approach that we previously developed (30). We have focussed on SUMO2 since this is the most abundant mammalian SUMO family member (9). Proliferating U2OS cells stably expressing His10-SUMO2 were irradiated with either 20 J/m2 UV-light or 4 Gy of IR or mock treated (untreated control). Moreover, parental U2OS cells were irradiated with either UV or IR as control for a-specific binding to the Ni-NTA beads. Parental U2OS cells were lysed after 1 h recovery upon DNA damage induction and U2OS His10-SUMO2 cells were lysed after 1 or 6 h recovery upon DNA damage induction. SUMOylated proteins were subsequently purified, trypsin digested and peptides were analysed by mass spectrometry (Figure 1A). We verified the efficiency of SUMO enrichment by immunoblotting (Figure 1B).
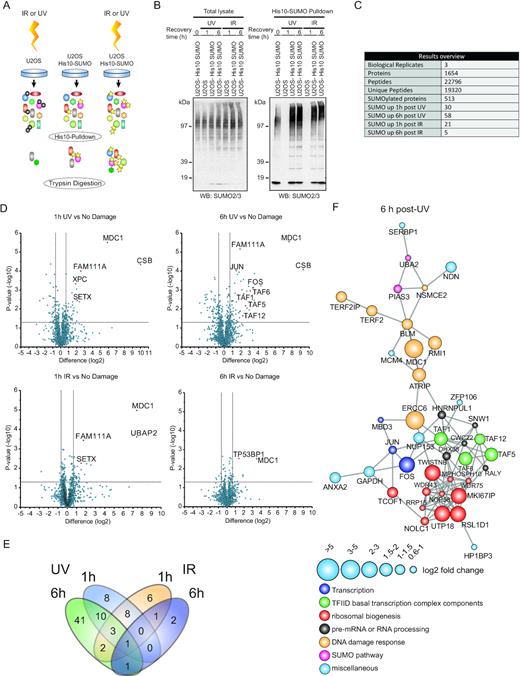
Identification of SUMOylated proteins in response to IR- or UV-induced DNA damage. (A) Experimental set-up. U2OS cells stably expressing His10-SUMO2 or parental cells were irradiated with ultraviolet light (UV) (20 J/m2) or ionizing radiation (IR) (4 Gy) or were mock treated. Cells were lysed 1 h or 6 h after DNA damage induction and SUMOylated proteins were purified by means of Ni-NTA pulldown. Purified proteins were trypsin digested and peptides were analysed by mass spectrometry. (B) Total lysates and His10-SUMO2 purified fractions were analysed by immunoblotting using a specific antibody against SUMO2/3. (C) The experiment, as described in (A) was performed in triplicate and protein groups were selected. SUMOylated proteins required a minimal fold change of 1.5 with a P-value <0.05 in at least one of the U2OS His10-SUMO2 conditions compared to the parental control. SUMO2 targets after 1 or 6 h IR or UV damage required a minimal fold change of 1.5 with a P-value <0.05 compared to the His10-SUMO2 expressing control that was mock treated. (D) Volcano plots showing all identified proteins. Dashed lines indicate a cut-off of 1.5-fold change (log2 of 0.66) and a P-value of 0.05 (–log10 of 1.3). Selected SUMO2 targets are marked in a red and display a label. (E) Venn diagram showing overlap of SUMO2 targets in different conditions. (F) Protein interaction network based on the STRING database showing connections between SUMO2 targets identified 6 h after UV irradiation with a medium confidence level of 0.4. Increased node size represents increased log2 fold change in LFQ intensities relative to mock treated control. Colours of nodes indicate main functional groups.
Label free quantification of proteins that were identified by mass spectrometry revealed 513 putative SUMO2 targets (Supplementary Table S1). Strikingly, significantly more proteins were modified by SUMO2 in response to UV compared to IR. After 1 h recovery post-UV irradiation we identified 30 proteins that showed increased SUMOylation compared to the mock treated control and this number increased to 58 proteins at 6 h recovery post-UV irradiation. Twenty one proteins showed enhanced SUMOylation after 1 h recovery post-IR and after 6 h recovery post-IR as few as five proteins showed increased SUMOylation compared to the mock treated control (Figure 1C, Supplementary Table S2). The DNA damage-checkpoint protein MDC1 was identified as SUMO2 target protein in all conditions (Figure 1D and E), in line with earlier observations (34) and thus served as a positive control. XPC SUMOylation in response to UV (Supplementary Figure S1) likewise served as a positive control (18).
We also observed that 41 out of 58 proteins that showed increased SUMOylation 6 h after UV irradiation were neither SUMOylated after 1 h recovery upon UV damage nor in response to IR-induced damages (Figure 1E, Supplementary Table S2), showing that they were specifically targeted for SUMOylation at a later time point after UV damage. Our data demonstrate that UV-induced DNA lesions activate a unique and more pronounced SUMOylation response compared to IR, indicating important roles of SUMOylation during cellular responses to UV lesions at early and later stages.
STRING network analysis of SUMO2 targets identified after 6 h recovery upon UV irradiation, revealed interconnected groups of proteins. We identified functional clusters of proteins involved in the DNA damage response, transcription, the SUMO pathway, ribosomal biogenesis and RNA processing. Interestingly, amongst the proteins associated with transcription we identified multiple components of the TFIID basal transcription factor complex (Figure 1F, Supplementary Table S2).
UV-induced SUMOylation of CSB is dependent on transcription and located at the N-terminus
The most dynamic SUMO2 target protein that we identified was CSB, showing a massive ∼1000-fold increase in SUMOylation specifically upon UV-induced damage at both recovery time points, but not in response to IR (Figure 1D). Since CSB is a crucial player in the TC-NER pathway, we chose to investigate the function of CSB SUMOylation in more detail. Since UV is known to strongly inhibit transcription elongation whereas IR hardly does, it is likely that particularly lesion-stalled transcription complexes trigger CSB SUMOylation. We explored a wider range of DNA-damaging agents for their ability to induce CSB SUMOylation and found that SUMOylation of CSB is also induced by methyl methanesulfonate (MMS) and etoposide in addition to UV (Figure 2A). Etoposide is also an inhibitor of transcription elongation and consistently induced CSB SUMOylation (Figure 2A). Furthermore, MMS induced CSB SUMOylation, which could indicate that the concentration and duration of the MMS treatment likewise resulted in stalling of transcription. These agents also induce transcription-blocking DNA lesions that can be repaired by TC-NER (35,36). Other types of DNA lesions like hydroxyurea-induced replication stress or IR-induced double strand breaks (DSBs), did not stimulate the SUMOylation of CSB (Figure 2A).
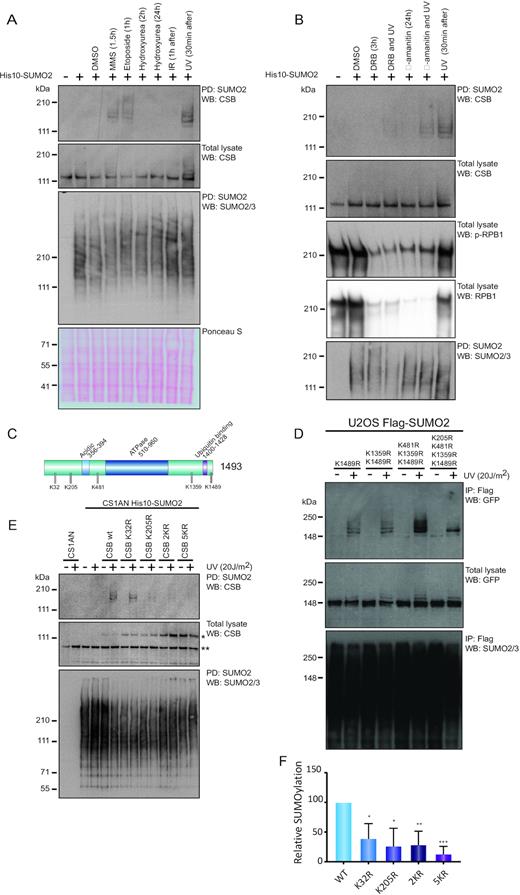
UV-induced SUMOylation of CSB is dependent on transcription and located at the N-terminus. (A) U2OS cells stably expressing His10-SUMO2 were left untreated or were treated with either DMSO, or 0.02% MMS, 50 μM etoposide, 2 mM hydroxyurea, 4 Gy IR or 20 J/m2 UV. Times indicate treatment period of cells with the compound before lysis. For IR and UV treated cells, times represent recovery period after treatment. SUMO2 conjugates were enriched by Ni-NTA pulldown (PD). Total lysates and SUMO2-enriched fractions were analysed by immunoblotting using antibodies against CSB or SUMO2/3. MMS, methyl methanesulfonate; IR, ionizing radiation; UV, ultraviolet light irradiation. (B) U2OS cells stably expressing His10-SUMO2 were treated with DRB or α-amanitin and/or UV irradiation (20 J/m2). Times indicate treatment period with the compound before lysis. For irradiation, these times indicate recovery period after irradiation prior to lysis. For the DRB and UV irradiated sample, cells were treated with DRB 3 h prior to UV irradiation and lysed 30 min after the UV treatment. For α-amanitin and UV irradiated samples, cells were treated 24 h prior to UV irradiation and lysed 30 min after UV treatment. SUMO2 conjugates were enriched by Ni-NTA pulldown. Total lysates and SUMO2-enriched fractions were analysed by immunoblotting using antibodies against CSB, p-RPB1 (S2/S5), RPB1, or SUMO2/3. DRB, 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside. (C) Schematic overview of CSB including known domains and localization of SUMOylation consensus sites (ψKXE). (D) U2OS cells stably expressing Flag-SUMO2 were infected with retroviruses encoding different SUMOylation consensus site mutants of GFP-CSB, as indicated. Cells were treated with UV irradiation (20 J/m2) and lysed after 1 h recovery. SUMO2 conjugates were enriched by Flag IP. Total lysates and SUMO2-enriched fractions were analysed by immunoblotting using antibodies against GFP or SUMO2/3. (E) CS1AN cells stably expressing His10-SUMO2 were infected with lentiviruses encoding different CSB SUMOylation consensus site mutants. Cells were treated with UV irradiation (20 J/m2) and lysed after 30 min recovery. SUMO2 conjugates were enriched by Ni-NTA pulldown. Total lysates and SUMO2-enriched fractions were analysed by immunoblotting using antibodies against CSB or SUMO2/3. * marks the exogenously expressed CSB construct. ** marks the CSB-piggyBac transposable element derived three fusion (CPFP) (57). K32, 205R (2KR); K32, 205, 481, 1359, 1489R (5KR). (F) Quantification of (E). Relative amount of SUMOylated CSB was determined based on immunoblots. Intensities were corrected for exogenous CSB expression levels (see * in E) and protein loading as determined by expression of CPFP (** in E) and Ponceau S stain. Values were normalized to CSB WT SUMOylation. Error bars represent SD of the mean obtained from three independent experiments *P-value <0.05; ** P-value <0.01; *** P-value <0.001 (two-sided).
Subsequently, we tested whether CSB SUMOylation was dependent on active transcription and on the stalling of RNAPIIo at the lesion. To evaluate this, we treated U2OS His10-SUMO2 cells with DRB or α-amanitin, two potent inhibitors of RNAPII (37), prior to induction of DNA lesions by UV irradiation. α-amanitin interferes with a conformational change of RPB1 underlying the transcription mechanism; therefore, it inhibits elongation. DRB inhibits CDK-Activating Kinase (CAK), which is associated with TFIIH, and thereby blocks transcription initiation. DRB treatment reduced phosphorylated RPB1 (p-RPB1) and total RPB1 as expected and the reduction in p-RPB1 in response to α-amanitin is due to a striking reduction in total amount of RPB1 (Figure 2B). We observed that UV-induced SUMOylation of CSB was decreased in cells that were pre-treated with these inhibitors. Blocking either initiation or elongation of transcription did itself not result in CSB SUMOylation (Figure 2B), indicating that transcription and the stalling of RNAPIIo at the lesion are prerequisites for CSB SUMOylation.
In order to investigate the role of CSB SUMOylation during the UV response, we aimed to identify the SUMO target lysines of CSB. CSB is a 1493 aa protein that includes five lysines embedded in SUMOylation consensus motifs, which are characterized by a large hydrophobic residue (ψ) upstream, and a glutamic acid two positions downstream of the lysine (ψKxE) (Figure 2C). We constructed lysine to arginine point mutants to disrupt potential SUMOylation sites in GFP-tagged CSB, starting from the most C-terminal lysine and mutating upstream SUMOylation motifs consecutively. Next, we compared SUMOylation levels of the different CSB mutants after UV irradiation by exogenous expression in U2OS His10-SUMO2 cells. Mutating the three most distal potential SUMOylation sites (K481,1359,1489R) did not strongly affect UV-induced CSB SUMOylation. However, adding the more N-terminal mutation K205R caused a pronounced SUMOylation decrease (Figure 2D). We expected that the residual SUMOylation of the CSB quadruple mutant (K205,481,1359,1489R) was located at position K32. Mutating both N-terminal lysine (2KR) or all five consensus sites (5KR) led to a complete loss of UV-induced SUMOylation (Supplementary Figure S2).
To further investigate the SUMO acceptor sites of CSB, we generated single mutants for K205R and K32R in addition to 2KR (K205R and K32R) and 5KR (K32,205,481,1359,1489R) mutants. These mutants, together with a His10-SUMO2 construct, were expressed in the CSB-deficient patient cell line, CS1AN (Figure 2E). Mutating either one of the N-terminal lysines (K32 or K205) resulted in a significant reduction of CSB SUMOylation when corrected for exogenous expression levels, although uncorrected SUMOylation levels were lower in the K205R mutant than in the K32R mutant. The CSB 5KR mutant showed a pronounced additive reduction in CSB SUMOylation levels (Figure 2E and F). Collectively, our results show that CSB is predominantly SUMOylated at two N-terminal lysine residues and that CSB SUMOylation is dependent on active transcription and the stalling of RNAPIIo at the lesion.
SUMOylation is necessary for efficient recruitment of CSB to UV-damaged DNA and for transcription recovery after UV damage
Next, we aimed to evaluate if SUMOylation is needed for efficient transcription recovery after UV-induced lesions, which is dependent on functional TC-NER. To address this, we made use of RPE1 cells harbouring inducible shRNAs targeting the SUMO E1 subunit UBA2, to reduce global SUMOylation levels (Supplementary Figure S3). We measured the relative amount of RNA synthesis, represented by the incorporation of 5-ethynyl-uridine (EU) into nascent RNA, after UV irradiation in cells with reduced SUMOylation levels. Shortly after irradiation the RNA synthesis dropped due to stalling of elongating RNA polymerases (RNAPIIo). In wild-type human cells with fully functional TC-NER, lesions are efficiently repaired and UV-inhibited RNA synthesis resumes in a time-dependent manner (38), frequently via re-initiation of RNA synthesis (39). Contrary, CS cells with defective TC-NER are incapable of restoring UV-inhibited RNA synthesis. We observed that reduction of SUMOylation, due to the induced knockdown of UBA2, had no impact on the UV-induced inhibition of transcription but merely delayed the recovery of RNA synthesis (RRS) after UV irradiation (Figure 3A), demonstrating a role for SUMOylation in the transcription recovery after UV damage, most likely facilitating the repair of UV-induced lesions or stimulating transcriptional restart.
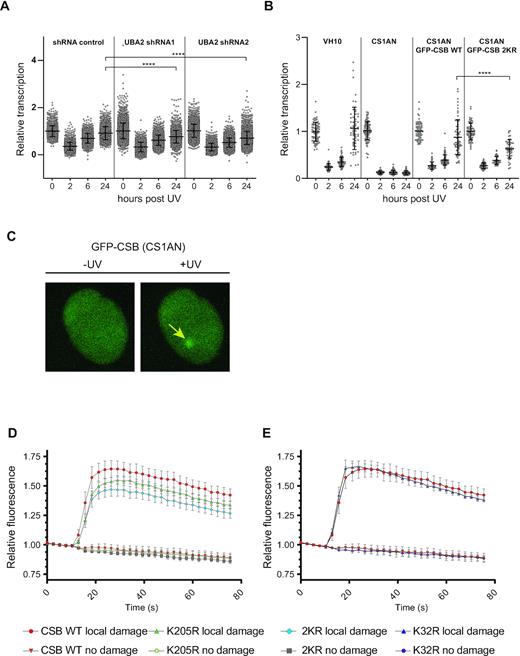
SUMOylation is necessary for efficient transcription recovery after UV damage and for efficient recruitment of CSB to UV-damaged DNA. (A) Two independent doxycycline (Dox)-inducible shRNAs against UBA2 and a non-targeted control shRNA were stably expressed in RPE1 cells. Knockdown was induced by Dox and cells were treated with 10 J/m2 UV. Relative RNA synthesis was measured by incorporation of EdU, prior to UV, and at 2, 6 and 24 h post-UV irradiation. Error bars represent SD of the mean of three independent experiments. Data were analysed by one-way Anova followed by a Tukey's multiple comparison test. **** represents P-value <0.0001. (B) The experiment described in A was repeated, using CSB-deficient patient cells (CS1AN) without rescue, or expressing GFP-tagged CSB wild-type (WT) or 2KR mutant (K32R, K205R) and VH10 cells. Recovery times after UV irradiation are indicated. All datapoints, average RRS and SD for triplicate experiments are shown. Data were analysed by one-way Anova followed by a Tukey's multiple comparison test. **** represents P-value <0.0001. (C–E) Accumulation kinetics of fluorescent intensities at locally induced UV damage of GFP-CSB WT and mutants expressed in CS1AN cells. Relative fluorescence was measured in the local damage area and in a non-treated area in the cell nucleus as shown in C. Results shown are the means of four independent experiments. Error bars represent the SD. Panel D shows kinetics of CSB WT, CSB K205R and CSB 2KR (K32R, K205R). Panel E shows kinetics of CSB WT and CSB K32R.
Subsequently, we tested whether this delay in RRS after UV irradiation upon UBA2 knockdown could be attributed to lack of SUMOylation of CSB. For this purpose, we employed CS1AN cells, the CSB-deficient patient cell line. We re-introduced GFP-tagged wild-type CSB or a double mutant of CSB (K32,205R: 2KR), lacking the main SUMOylation sites (Supplementary Figure S2). Cells expressing CSB 2KR showed a statistically significant reduction in RRS 24 h after UV irradiation (Figures 3B and S4), indicating that CSB SUMOylation contributes to the delay in RRS upon UBA2 knockdown (Figure 3A).
To gain more insight into the role of CSB SUMOylation during the cellular response to UV irradiation, we next investigated the recruitment of the SUMOylation-mutants of CSB to locally induced UV lesions in living cells (Figure 3C). For this purpose, we fused CSBWT, CSBK32R, CSBK205R and CSB2KR to GFP, induced local UV damage using a UV-C laser and measured the recruitment kinetics of GFP-CSB to the damage site (22,29). Four replicates showed a reduced recruitment of the CSBK205R mutant compared to CSBWT (Figure 3D). Unlike the CSBK205R single mutant, the CSBK32R mutant doesn’t seem to have any obvious defect in recruitment to the local damage sites (Figure 3E). Interestingly, the CSB2KR mutant showed a more pronounced impairment of recruitment that might indicate a functional contribution of CSBK32 SUMOylation in the absence of CSBK205 SUMOylation (Figure 3D and E). Collectively, we showed that SUMOylation is needed for efficient transcription recovery after UV irradiation and that decreased CSB SUMOylation impairs the recruitment and/or stabilization of CSB at the damage site.
SUMOylation of CSB influences binding to RNA polymerase associated proteins
Our results described in the previous section could potentially be explained by a SUMOylation-dependent alteration of CSB protein interactions. SUMOylation is a low stoichiometric post-translational modification, making it difficult to study direct protein interactions in vivo. Therefore, we have used a strategy to study SUMOylation-dependent alterations in protein interactions of CSB using an in vitro approach (33). Since the main UV-induced SUMOylation sites were located in the N-terminal part of CSB, we focussed on this part of the protein. We co-transformed E. coli with a plasmid encoding GST-tagged CSB truncation mutant (aa 1–341) and a plasmid encoding the SUMOylation machinery and subsequently purified the SUMOylated truncated CSB protein (SUMO-CSB). To ensure the same amounts of CSB protein in the SUMOylated and non-SUMOylated sample, we equally divided the purified SUMO-CSB sample and deSUMOylated one half by adding recombinant sentrin/SUMO-specific protease 2 (SENP2). SUMOylation and deSUMOylation of CSB were confirmed by immunoblotting (Supplementary Figure S5). We included a third sample with GST-only as negative control (Figure 4B). GST, SUMO-CSB and CSB were subsequently incubated with a lysate of UV-irradiated CS1AN cells. After incubation, beads were washed and purified proteins were trypsin-digested on the beads. Interactomes were analysed using mass spectrometry (Figure 4A and B).
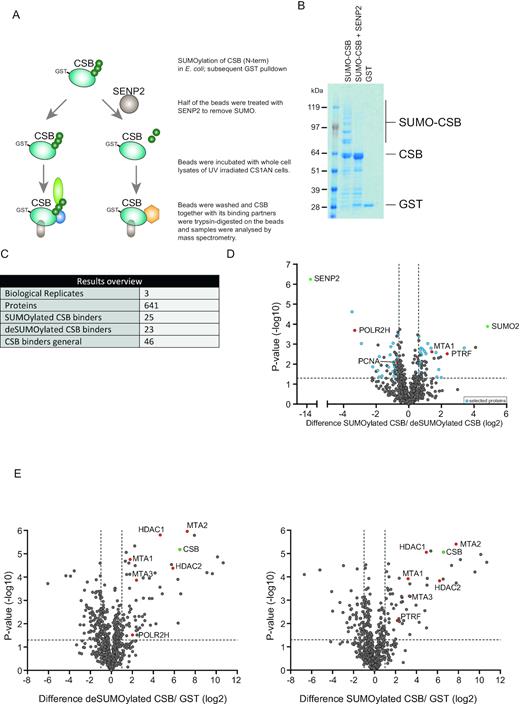
SUMOylation of CSB influences binding to RNA polymerase associated proteins. (A) Schematic overview of experimental set-up. GST tagged truncated CSB protein (aa 1–341) and SUMO machinery were co-expressed in E. coli and subsequently purified with glutathione resin. The resulting sample was split in two equal aliquots and one aliquot was treated with SENP2 overnight. De-SUMOylated and SUMOylated truncated CSB were incubated with lysates of UV-treated CS1AN cells. After incubation and washing, proteins were trypsinized on the resin and peptides were analysed by mass spectrometry. (B) Coomassie stain showing SUMOylated truncated CSB, unmodified truncated CSB and GST control. (C) The table shows a summary of identified proteins. Putative bindings partners are defined as proteins that are significantly different between non-SUMOylated CSB and SUMOylated CSB samples and are also significantly enriched compared to the GST control. (D) Volcano plot showing relative LFQ intensities of proteins in SUMOylated CSB samples compared to deSUMOylated CSB samples. Dashed lines indicate a cut-off of 1.5-fold change (log2 of 0.66) and a P-value of 0.05 (–log10 of 1.3). Putative differential binding partners which are also enriched compared to the GST control are marked in blue. Proteins that function as internal control are marked in green. In text discussed proteins are marked in red. (E) Volcano plots showing relative LFQ intensities of proteins in deSUMOylated CSB samples compared with GST controls (left panel) or SUMOylated CSB samples compared with GST controls (right panel). Proteins that function as internal controls are marked in green. Proteins discussed in the text are marked in red.
We selected differentially binding proteins based on the fold change and P-value of their abundances in the SUMO-CSB sample compared to unmodified CSB. Proteins that showed no differential binding compared to the GST control were excluded. The presence of SUMO2 as the most significant and enriched protein identified, confirmed and validated the approach (Figure 4D). Of the 641 proteins identified, 46 proteins co-enriched with CSB irrespective of SUMOylation, compared with the GST control (Figure 4C and E, Supplementary Table S3). 25 proteins showed increased binding to SUMO-CSB compared to unmodified CSB, whereas 23 proteins showed preferential binding to unmodified CSB compared to SUMO-CSB (Figure 4C and D, Supplementary Table S3). Within the protein group that co-enriched with SUMO-CSB, we identified multiple RNA polymerase-associated factors that were not previously described to bind to CSB, including polymerase I transcript release factor (PTRF). PTRF is involved in termination and re-initiation of RNA polymerase I transcription (40,41). Consistently, CSB is a known regulator of RNA polymerase I transcription (42). Furthermore, we identified MTA1, a member of the NURD complex to have a preference for SUMO-CSB. Other subunits of the NURD complex bound unmodified CSB equally well (Figure 4E).
SUMO-Interaction Motifs (SIMs) enable non-covalent interaction between SUMOylated proteins and readers. These SIMs have been defined as SIMa, [PILVM]-[ILVM]-X-[ILVM]-[DSE](3), SIMb, [PILVM]-[ILVM]-D-L-T or SIMr, [DSE](3)-[ILVM]-X-[ILVMF](2) by Vogt and Hofmann (43). PTRF and MTA1 are missing these SIMs. Of note, this is no formal proof that they cannot bind SUMO.
Interestingly, we found that the RNA polymerase subunit PolR2H (Rbp8) and PCNA have a preference for unmodified CSB. PolR2H might represent the elongating RNAPIIo to which CSB is known to bind. PCNA is a crucial component of TC-NER, as it is responsible for the recruitment of the gap-filling DNA polymerases (44,45). In conclusion, our data suggest that SUMOylation of CSB alters its protein interactions including to RNA polymerase associated proteins, which could contribute to efficient TC-NER.
CSA destabilizes SUMOylated CSB
Another well-described function of SUMOylation is the destabilization of target proteins by recruitment of STUbLs, leading to the subsequent ubiquitination and proteasomal degradation of the SUMO-target protein. Interestingly, the WD repeat protein CSA that is recruited to UV-induced DNA lesions in a CSB-dependent manner (46), was shown to be a substrate receptor of a Cullin/RING (CRL) ubiquitin E3 ligase complex and was previously proposed to target CSB for ubiquitination (21,47). Therefore, we investigated the influence of the CSA–CRL complex on CSB SUMOylation. To this end, we employed U2OS cells stably expressing His-tagged SUMO2 to enable SUMO2 purification. CSA was knocked down using an shRNA-based approach (Supplementary Figure S6). These cells were treated with UV irradiation or were left untreated as indicated. Subsequently, cells were lysed 1, 3 or 6 h after UV irradiation and SUMOylated proteins were enriched by Ni-NTA pulldown. As shown in the middle panel of Supplementary Figure S6, efficient and equal SUMO enrichment was confirmed by immunoblotting. In the fourth panel, CSA knockdown was confirmed by immunoblotting. The top panel shows that one set of CSA knockdown cells showed considerably higher levels of SUMOylated CSB compared to parental U2OS cells and one set of CSA knockdown cells did not.
To further investigate a potential role for CSA in regulating the levels of SUMOylated CSB, we employed a CRISPR-Cas9-based knockout approach (Figure 5A). Deletion of CSA was verified by immunoblotting (Figure 5B). Efficient enrichment of SUMOylated proteins from U2OS cells expressing His10-SUMO2 was confirmed by immunoblotting (Figure 5A bottom panel). This panel also shows accumulation of SUMOylated proteins in response to proteasome inhibition, particularly in lanes 8, 11 and 14. The top panel of this figure shows that SUMOylated CSB could be detected in response to UV treatment. In the absence of CSA, SUMOylated CSB accumulated to a higher extent at all three timepoints, compared to CSA wild-type cells. The reduction in SUMOylation due to the presence of CSA could not be reversed by blocking proteasomal degradation as shown in lanes 8, 11 and 14. In response to MG132 treatment, SUMO and ubiquitin are trapped on the targets that can be no longer degraded and the pools of free SUMO and ubiquitin will concomitantly decrease. Due to these limited pools of free SUMO and ubiquitin, less SUMO and ubiquitin will be available for conjugation to new target proteins. As a result, the SUMOylation and ubiquitination levels of proteins that are not degraded by the proteasome will decrease. This appears to be the case for SUMOylated CSB in response to MG132 at 1 h and 6 h post UV. These results indicate that the CSA–CRL complex regulates the stability of SUMOylated CSB in response to DNA damage directly or indirectly in a proteasomal-independent manner.
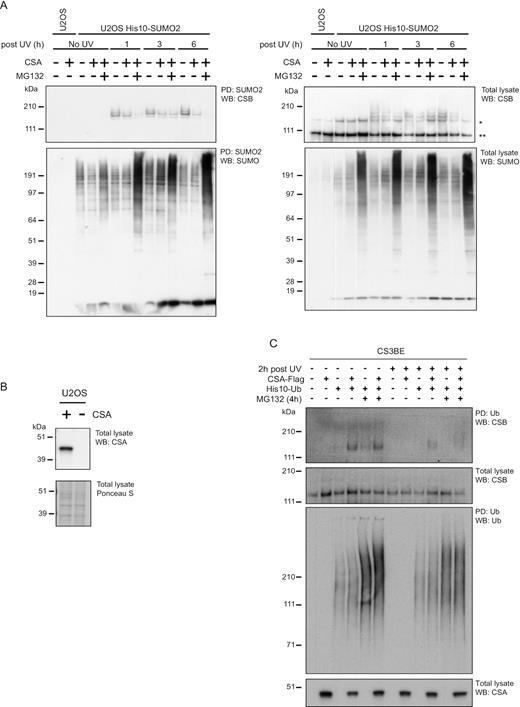
CSA destabilizes SUMOylated CSB. (A) U2OS WT and CSA-deficient cells stably expressing His10-SUMO2 were used to study whether CSA affects the SUMOylation of CSB. These cells were subjected to UV irradiation (20 J/m2) and/or proteasomal inhibition (MG132) as indicated. Cells were lysed 1, 3, or 6 h after UV irradiation. Subsequently, SUMOylated proteins were purified from these lysates by Ni-NTA pulldown. SUMO-enriched fractions (PD) and total lysates were analysed by immunoblotting using antibodies against CSB or SUMO2/3. *marks full-length CSB; ** marks the CSB-piggyBac transposable element derived 3 fusion (CPFP). (B) Immunoblotting to confirm the absence of CSA in U2OS cells established by CRISPR-targeting the CSA gene. (C) CSA-deficient CS3BE patient cells and a derivate cell line that was rescued by introducing CSA-Flag were used to study whether CSA affects ubiquitination of CSB. His10-ubiquitin was stably expressed in these cells as indicated and cells were treated overnight with cycloheximide to prevent new protein synthesis. The next day, cells were UV irradiated (20 J/m2) and/or treated with MG132 as indicated. Ubiquitinated proteins were purified by His10-purification. Total lysates and ubiquitin-enriched (PD) fractions were analysed by immunoblotting using antibodies against CSB, CSA or Ub as indicated.
CSA stimulates ubiquitination of RNA polymerase II but not CSB after UV irradiation
Following the observation that CSA influenced the destabilization of SUMOylated CSB, we tested whether CSA might target SUMOylated CSB for ubiquitination and would therefore act as a STUbL. To evaluate this, we investigated the influence of CSA and UV irradiation on the ubiquitination of CSB. CSA-Flag was reintroduced in the CS3BE cells described above and these cells were engineered to stably express His10-ubiquitin. To decrease background ubiquitination of CSB that could arise from misfolding of this 1493 amino acid long protein during protein synthesis since a significant percentage of newly synthesized proteins are misfolded (48,49), we treated the cells overnight with the translation inhibitor cycloheximide. We next irradiated the cells with UV light, lysed 2 h post-UV irradiation and purified ubiquitinated proteins. Efficient purification of ubiquitin and accumulation of ubiquitinated proteins in response to proteasome inhibition were confirmed by immunoblotting (Figure 5C, third panel). The absence of CSA in CS3BE and presence of CSA in the rescued cells were also confirmed by immunoblotting (Figure 5C, bottom panel). Next, we tested whether CSB ubiquitination was enhanced in the presence of CSA and we confirmed that this was the case (Figure 5C, top panel). However, CSA-dependent ubiquitination of CSB was already detectable in unirradiated cells (Figure 5C, lanes 1–6) and was reduced rather than enhanced upon UV treatment (Figure 5C, lanes 7–12). Inhibiting the proteasome did not result in an accumulation of ubiquitinated CSB, although total ubiquitin conjugates increased as expected (Figure 5C). The destabilization of SUMOylated CSB can therefore not be explained by UV-induced CSA-dependent ubiquitination. Furthermore, ubiquitination of CSB does apparently not cause subsequent proteasomal degradation.
We next hypothesized that the observed UV- and CSA-dependent destabilization of SUMOylated CSB shown in Figure 5A, is an indirect effect of a ubiquitination event of another unknown protein. Therefore, we set out to identify possible UV-induced ubiquitination targets of CSA in an unbiased manner. We used CSA-deficient CS3BE cells stably expressing His10-ubiquitin with or without exogenous expression of CSA-Flag. Cells were stable isotope labelled by amino acids in cell culture (SILAC) as described in the upper panel of Figure 6A. Cells were treated in four different manners, including no UV irradiation, UV irradiation in combination with 1 h recovery (short recovery), or 6 h recovery (long recovery) and 6 h recovery combined with proteasome inhibition. Differential ubiquitination of proteins was analysed by mass spectrometry for each of the four different treatments (Figure 6A). Ubiquitination of CSB was not detected in this screen. Most intriguingly, we identified the largest RNAPII subunit, RPB1, as a differentially ubiquitinated protein 1 h after UV irradiation in a CSA-dependent manner. This ubiquitination of RPB1 was not detected after the 6 h recovery, but stabilized upon proteasome inhibition, indicating a CSA-dependent destabilization of RPB1 (Figure 6A, Supplementary Table S4).
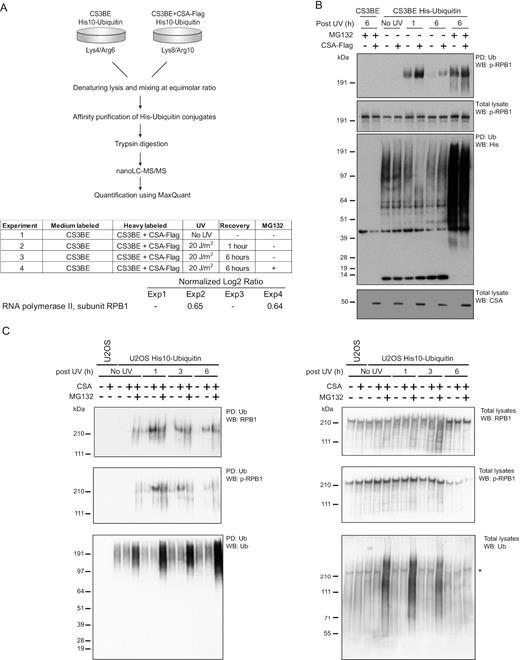
CSA stimulates ubiquitination of RNA polymerase II in a UV-dependent manner. (A) CS3BE cells with or without ectopic expression of CSA-Flag and stably expressing His10-ubiquitin were SILAC labelled and subjected to UV irradiation (20 J/m2) and/or treated with MG132 or were left untreated as indicated. Cells were lysed 1 or 6 h after UV irradiation. Ubiquitinated proteins were purified by Ni-NTA pulldown. Eluted proteins were trypsinized and analysed by mass spectrometry. The table shows an overview of experimental conditions and the log2 medium/heavy ratios of the RNAPII subunit RPB1 in each experiment. (B) CS3BE cells with or without ectopic expression of CSA-Flag and stably expressing His10-ubiquitin were treated with UV irradiation (20 J/m2) and MG132 where indicated and lysed after the indicated recovery times. Total lysates and ubiquitin-enriched fractions (PD) were analysed by immunoblotting using antibodies against p-RBP1 (S2/S5), His or CSA. (C) U2OS w.t. and CSA-deficient cells stably expressing His10-ubiquitin were subjected to UV irradiation (20 J/m2) and/or proteasomal inhibition (MG132) or were left untreated as indicated. Cells were lysed 1, 3 or 6 h after UV irradiation. Ubiquitinated proteins were purified by Ni-NTA pulldown. Ubiquitin-enriched fractions and total lysates were analysed by immunoblotting using antibodies againstRPB1, p-RPB1 (S2/S5) or ubiquitin. *residual p-RPB1 (S2/S5) signal in blot re-probed with ubiquitin antibody.
Subsequently, we carried out experiments to verify our proteomics data, using immunoblotting analysis of His10-ubiquitin-enriched fractions (Figure 6B). In the third panel of Figure 6B, His10-ubiquitin enrichment was confirmed in the stably expressing CS3BE cells as expected. The same panel shows that ubiquitin was stabilized by proteasome inhibition as expected. The bottom panel of Figure 6B confirms the presence of CSA-Flag in the rescued CS3BE cells. The second panel confirms the presence of elongating RPB1 (p-RPB1) in all samples. This is the relevant form of RPB1 in this context. Next, we asked whether p-RPB1 is a substrate for ubiquitination following UV irradiation, and whether that is dependent on CSA and leads to proteasomal degradation. This is shown in the top panel of the Figure. In CS3BE cells lacking His10-ubiquitin, no p-RPB1 is detected, showing correct negative controls. Interestingly, ubiquitination of p-RPB1 is only detected in response to UV damage. Cells expressing CSA have considerably more ubiquitinated p-RPB1. This is unlikely due to loading errors as shown in the second and third panels of this figure. At 6 h post irradiation, the ubiquitination signal of p-RPB1 diminishes, presumably due to degradation of the ubiquitinated p-RPB1. This is supported by the results in lanes 9 and 10 in which the proteasomal inhibitor MG132 is present. In conclusion, these results confirm our proteomics results that CSA regulates ubiquitination of p-RPB1 in response to UV damage. In the absence of CSA, some ubiquitination of p-RPB1 in response to UV damage was still noticeably, indicating that UV-induced ubiquitination of elongating RPB1 in response to UV partly occurs in a CSA-independent manner.
Next, we carried out a similar experiment as described in Figure 6B, now using U2OS cells proficient or deficient for CSA (Figure 6C). This experiment confirms our results obtained in Figure 6A and B, strengthening the conclusion that CSA regulates the ubiquitination of p-RPB1 in response to UV damage. Using the samples shown in Figure 6C and staining them for RPB1 ubiquitination, we noticed that after a short recovery (1 h) upon UV irradiation, RPB1 was ubiquitinated in a CSA-dependent manner in U2OS cells but not subjected to proteasomal degradation as treatment with proteasome inhibitor did not result in an accumulation of ubiquitinated RPB1 (Figure 6C). However, after a longer recovery time of 6 h, proteasome inhibition stabilized ubiquitinated RPB1. Nevertheless, in the total lysate controls, the proteasome inhibitor does not rescue the levels of RPB1 and p-RPB1, indicating that proteasomal degradation of RPB1 at the 6 h timepoint is limited. Collectively, these results indicate that CSA is stimulating the ubiquitination of RNAPII either directly or indirectly upon UV irradiation, leading to proteasomal degradation of a subset of RNAPII only after a longer recovery upon the DNA damage.
DISCUSSION
Link between CSA, ubiquitinated RNAPII and SUMOylated CSB
Mutations in the CSA and CSB genes give rise to Cockayne Syndrome, a severe neurodegenerative and premature aging disorder that is associated with hypersensitivity to UV irradiation, primarily due to defects in TC-NER. CSA is the substrate recognition factor of an E3 Cullin ubiquitin ligase complex. However, it is currently unclear which proteins are targeted for CSA-dependent ubiquitination at sites of DNA damage. CSB has been suggested as a UV-specific target for CSA mediated ubiquitination and subsequent proteasomal degradation (21). Although we observed an increase in CSB ubiquitination in a CSA-dependent manner, this was not induced and even slightly reduced in response to UV irradiation and ubiquitinated CSB was not stabilized in response to proteasome inhibition. Of note, the usage of tagged-ubiquitin constructs precludes the detection of potential linear ubiquitin chains on CSB. Nevertheless, in our hands it does not appear that CSB is a target for UV-dependent degradation via the CSA–CRL E3 ligase complex. However, we did find that the recruitment of this complex is responsible for the destabilization of SUMOylated CSB after UV. Our observation provides a novel link between CSB and CSA, but raises the question how CSA is regulating the destabilization of SUMOylated CSB.
A potential explanation for the destabilization of SUMOylated CSB by the CSA–CRL complex is the ubiquitination of other targets by this complex. We identified the RNAPII subunit RPB1 as a key target for the CSA complex. Ubiquitination of RPB1 directly or indirectly by the CSA complex could potentially induce the dissociation of CSB from chromatin and its translocation to the nucleoplasm where it can be deSUMOylated by SUMO-specific proteases (Figure 7). Intriguingly, RNAPII ubiquitination and degradation is believed to be a ‘last-resort’ response to DNA damage (50). In contrast to CSB, we could observe CSA-dependent UV-induced ubiquitination of the RNAPII subunit RPB1. However, we could only observe a stabilization of the ubiquitinated RPB1 upon inhibition of the proteasome at a later timepoint (6 h) post-UV irradiation, indicating a ubiquitination event that does not immediately lead to degradation. Also we observed that there is a significant residual UV-induced ubiquitination of RNAPII in CS3BE cells lacking CSA, which indicates the presence of other ubiquitin E3 ligases targeting RNAPII. CSA-independent ubiquitination of RPB1 could be regulated by the E3 ligases NEDD4 and the Elongin A,B,C complex (51). These E3 ligases play important roles at the early stage after UV irradiation, within 30 min, and mediate K63-linked ubiquitin chains.
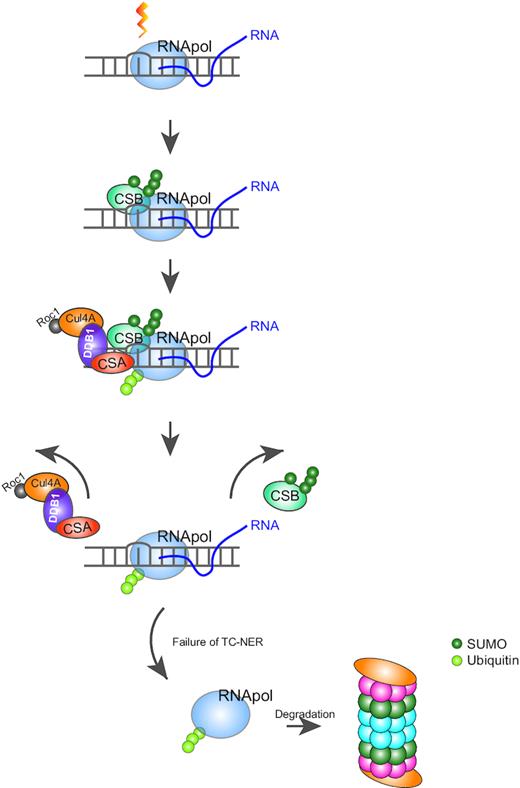
SUMO and ubiquitin cooperate during TC-NER. The processing RNAPII is stalled upon encountering a DNA photolesion with helix distorting property, which is introduced by UV light, MMS or etoposide within an actively transcribed DNA region. In response to the stalled RNA polymerase, CSB is SUMOylated, recruited and stabilized at the lesion site. CSA is subsequently recruited to the site of damage and stimulates ubiquitination of RNAPII directly or indirectly. After a short recovery upon DNA damage, SUMOylated CSB is destabilized by the presence of CSA. The proteasomal degradation of ubiquitinated RNAPII is observed at a late stage (6 h) after UV irradiation, possibly related to failure of repair.
Intriguingly, it was previously found that CSB contains a ubiquitin binding-domain (UBD) that is required for TC-NER (52). If this observation is connected to our findings, CSA-dependent ubiquitination of RNAPII might provide a docking site for the UBD in CSB. Subsequent release or degradation of ubiquitinated RNAPII might result in co-release or degradation of CSB. The UBD domain of CSB is not required for its SUMOylation (53). Whether the UBD domain is required for the clearance of SUMOylated CSB is currently unclear. Follow-up projects could focus on the potential connection between CSB SUMOylation, its UBD domain and RNAPII ubiquitination. An alternative explanation for the observed effect would be that the degradation of other proteins is required for the destabilization of SUMOylated CSB.
SUMO group modification in response to UV
The SUMOylation of CSB was described in a previous publication, which reported lysine 205 as major SUMOylation site (53). We could confirm this finding but additionally found a contribution of lysine 32 to SUMOylation of CSB. To evaluate the functional importance of K32 SUMOylation, we studied the recruitment of WT, the single mutants K205R and K32R and the 2KR CSB mutant to local UV lesions and observed that mutating both N-terminal lysines of CSB impaired the recruitment to the local UV-induced damage more effectively than the single mutation of K205, showing a functional contribution of K32 if K205 is not available for SUMOylation.
We could further confirm that SUMOylation and the two N-terminal SUMOylation sites in CSB contribute to RRS after UV damage. This is in line with the previous publication showing the effect of UBC9 (SUMO E2-conjugating enzyme) knockdown on RRS (53). Because CSB was the only TC-NER protein that we identified in our mass spectrometry screen to be SUMOylated in response to UV, it is likely that CSB SUMOylation contributes to this effect. Interestingly, lesion recognition in GG-NER occurs via XPC, a well-known UV-regulated SUMO target that we confirmed in our screen (18–20). Thus, both branches of NER involve lesion recognition factors that are regulated by SUMO.
Our results linking reduced levels of SUMOylated CSB to the presence of CSA are in contrast to this previous publication (53). This could potentially be due to differences in SUMO-enrichment methodology between both studies. We prefer highly efficient enrichment of His10-tagged SUMOylated proteins using NiNTA beads, which is compatible with the use of strongly denaturing buffers to inactivate endogenous SUMO proteases, whereas the previously published study employed immunoprecipitation, necessitating the use of milder buffer conditions to prevent denaturation of the employed antibodies.
Although we did not identify any other obvious TC-NER proteins as UV-responsive SUMO2 targets, we did identify dynamic SUMOylation in response to UV irradiation of proteins associated with transcription, like the TFIID basal transcription factor components TAF1, TAF5, TAF6 and TAF12. This observation suggests that the transcription response to elongation-blocking DNA lesions is in part controlled through SUMOylation. These results further highlight the SUMO group-modification concept, where modification of a set of targets within a biological pathway is needed for stronger biological effects, as was observed in the context of DSB repair (54). Our results indicate that SUMO co-regulates a considerably larger set of targets in response to UV irradiation compared to IR.
Model
Taken together we suggest a model wherein CSB is SUMOylated and recruited to UV lesion-stalled RNAPII. This association recruits the CSA–CRL complex to the site of damage, where it stimulates the ubiquitination of the RNAPII subunit, RPB1 in a direct or indirect manner. The activity of CSA initiates the release of SUMOylated CSB, but CSA does not act as a STUbL for SUMOylated CSB. Ubiquitinated RPB1 resides at the site of the lesion until the lesion is either repaired or in case repair failed, it would trigger the proteasomal degradation of RPB1. Our data fit well with initial observations in the field on reduced UV-induced ubiquitination of RPB1 in fibroblasts from CS patients (55). Overall our results provide new insights in the cooperative signaling roles for SUMOylation and ubiquitination of TC-NER components. Whereas CSB is a prime SUMO2 target, the stability of SUMOylated CSB and the ubiquitination of RPB1 is dependent on CSA. Combined, these two small modifiers may contribute to RRS, presumably via efficient TC-NER and transcription restart in response to UV irradiation. Furthermore, we identified an extensive set of SUMOylated proteins in response to UV irradiation. Detailed functional analysis of these proteins will improve our understanding of the role of SUMO group modification in the cellular response to UV irradiation.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (56) partner repository with the dataset identifier PXD010609.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Prof. Dr Marc Timmers (Freiburg) for the pBabe-puro-GFP-DEST destination vector and Prof. Jesper V. Olsen (Copenhagen) for mass spectrometry support during the initial phase of the study.
FUNDING
European Research Council (ERC) [310913 to A.C.O.V. and 340988 to W.V.]; the Netherlands Organisation for Scientific Research (NWO) [Gravitation program CancerGenomiCs.nl to W.V. and ALWOP.143 to A.P., 855-01-074 to L.F.M.]; NTC Netherlands Toxicogenomics Center [050-060-510 to L.F.M.]; LUMC research fellowship and NWO-VIDI grant [016.161.320 to M.S.L.]. Funding for open access charge: European Research Council grant [310913].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments