





Abstract
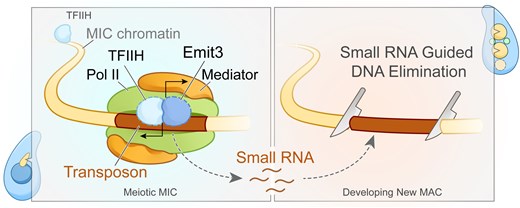
Transposable elements (TEs) pose threats to genome stability. Therefore, small RNA-mediated heterochromatinization suppresses the transcription and hence the mobility of TEs. Paradoxically, transcription of noncoding RNA (ncRNA) from TEs is needed for the production of TE-targeting small RNAs and/or recruiting the silencing machinery to TEs. Hence, specialized RNA polymerase II (Pol II) regulators are required for such unconventional transcription in different organisms, including the developmental stage-specific Mediator complex (Med)-associated proteins in the ncRNA transcription from TE-related sequences in Tetrahymena. Yet it remains unclear how the Pol II transcriptional machinery is assembled at TE-related sequences for the ncRNA transcription. Here, we report that Pol II is regulated by Emit3, a stage-specific TFIIB-like protein specialized in TE transcription. Emit3 interacts with the TFIIH complex and localizes to TE-dense regions, especially at sites enriched with a G-rich sequence motif. Deletion of Emit3 globally abolishes Pol II-chromatin association in the meiotic nucleus, disrupts the chromatin binding of Med, and impairs the TE-biased localization of TFIIH. Conversely, Emit3’s preferential localization to TE-rich loci relies in part on Med-associated proteins. These findings suggest that Emit3, TFIIH, and Med-associated proteins work together to initiate Pol II ncRNA transcription from TE-dense regions, possibly in a sequence-dependent manner.
Introduction
Transposable elements (TEs) are widespread mobile genetic elements. TE mobilization (i.e. transposition), through either copy-and-paste or cut-and-paste mechanisms, plays a critical role in driving the evolution of the host genome [1–3]. However, transposition can also result in undesired consequences. Uncontrolled transposition in the germline genome is particularly dangerous as it can cause hereditary diseases or even sterility [4, 5]. To guard the germline genome integrity, eukaryotes possess small noncoding (ncRNA)-based mechanisms for suppressing TE mobility [5, 6]. Thereby, precursor RNAs derived from certain TE-dense loci are processed into small RNAs, which form an effector complex with Argonaute/Piwi family proteins to target TE-derived transcripts through base-pairing interactions. Targeted transcripts are either subjected to cleavage or mediate the establishment of repressive chromatin states at TE-containing loci [7]. The TE-dense loci (termed Piwi-interacting RNA [piRNA] clusters in animals) largely consist of degenerated TE fragments and are considered as genetic memories of past TE invasions [8]. RNA polymerase II (Pol II, or its variant in plants) is responsible for the transcription of these TE remnants [9–11].
In general, Pol II-dependent transcription (hereafter referred to as transcription) initiates with a stepwise assembly of general transcription factors (GTFs) and Pol II at a promoter region [12, 13]. First, the transcription factor II D (TFIID) complex binds to the promoter sequences. This is followed by the incorporation of transcription factor II A (TFIIA), which interacts with the TATA-binding protein (TBP)—a component of TFIID—to stabilize the association of TFIID with the DNA. Transcription factor II B (TFIIB) couples Pol II and the TBP-bound promoter by directly interacting with TBP and recruiting the Pol II with TFIIB’s zinc-ribbon [14–16]. Transcription factor II F is recruited to the promoter along with Pol II and sets a stable stage for the final loading of transcription factor II H (TFIIH) and transcription factor II E [13]. Engaged TFIIH first melts DNA at the promoter using Xpb, one of its helicase subunits, then triggers Pol II release from the promoter allowing productive elongation by phosphorylation of the Rpb1 C-terminal domain (CTD) using its regulatory kinase module. Besides GTFs, the Mediator complex (Med) which bridges Pol II and gene-specific transcription factors is also crucial for transcription [17].
The evolutionary arms race between TEs and host genomes has led to the development of various TE-suppressing mechanisms, including piRNA clusters and proteins driving piRNA cluster transcription in animals [6, 18]. Tetrahymena thermophila and other ciliated protists have evolved the ultimate TE-suppressing system, in which TEs targeted by small RNAs are initially transcriptionally suppressed and subsequently eliminated from their somatic genomes [19–23]. Ciliates are single-celled organisms characterized by separate germline micronuclei (MICs) and somatic macronuclei (MACs) within a cell (Fig. 1A). TEs and their remnants reside in the so-called internal eliminated sequences (IESs) that are retained in the MIC but excised from the MAC in Tetrahymena [24]. Unlike the MAC with constitutive transcriptional activity, the MIC is transcriptionally silent until the onset of meiosis when it transcribes ncRNAs [21, 25] (Fig. 1A). Pol II is responsible for this MIC transcription [26]. The transcription is believed to be bidirectional [27] and the resulting double-stranded RNAs (dsRNAs) are enriched at TE-dense regions [10, 28]. The maturation of TE-targeting small RNAs, also known as scan RNAs (scnRNAs), starts with the cleaving of dsRNA with a Dicer-like protein, Dcl1 [29–31]. The resulting short dsRNAs are further processed and bound by the Tetrahymena Piwi protein Twi1 to guide heterochromatinization and subsequent elimination of IESs from the developing progeny MAC [20].
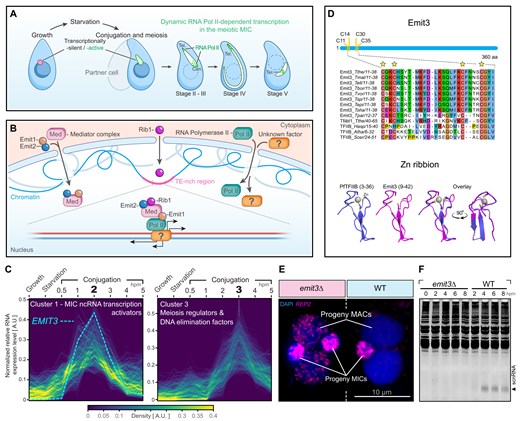
Tetrahymena life cycle and the identification of Emit3. (A) Tetrahymena cells contain two distinct types of nuclei: the somatic macronucleus (MAC) and the germline micronucleus (MIC). The MAC has continuous transcriptional activity, while the MIC remains transcriptionally inactive until meiosis begins. TEs (depicted in magenta) are predominantly retained in the MIC but eliminated from the new MAC. During conjugation, when two cells of different mating types pair, TE-targeting ncRNAs are synthesized in the meiotic MIC. Meiotic prophase is categorized based on the extent of nuclear elongation. In stages II–IV, centromeres (Cen.) and telomeres (Tel.) are clustered at opposite poles of the elongating MIC. By stage V, these structures detach from the MIC termini, although telomeres remain clustered. The ncRNA transcription (indicated by dots) is initially concentrated in IES-rich telomeric and pericentromeric regions during stages II and III. In stages IV and V, transcription is restricted to a narrow telomeric region. (B) Current knowledge about the MIC ncRNA transcription activation mechanism. See the text for details. (C) MIC ncRNA transcription activators and meiosis regulators were grouped into different clusters with respect to the timing of gene expression. Hpm: Hours post mixing. (D) Upper panel: Sequence alignment shows that Emit3 and Tfiibl1 have a conserved zinc-ribbon motif. Tthe: Tetrahymena thermophila; Tmal: Tetrahymena malaccensis; Tell: Tetrahymena elliotti; Tbor: Tetrahymena borealis; Tvor: Tetrahymena vorax; Tsp: Tetrahymena sp.; Tepi: Tetrahymena empidokyrea; Tsha: Tetrahymena shanghaiensis; Tpar: Tetrahymena paravorax; Hasp: Homo sapiens; Atha: Arabidopsis thaliana; Scer: Saccharomyces cerevisiae. Lower panel: The N-terminal region contains a putative zinc-ribbon motif, with the positions of the zinc-binding cysteines indicated. The zinc-ribbon structure of Emit3 is similar to that of Pyrococcus furiosus TFIIB (PfTFIIB), as retrieved from the Protein Data Bank (PDB code: 1pft). Homology modeling of the C-terminal regions is presented in Supplementary Fig. S1. (E) DNA FISH analysis of REP2 IESs (magenta) in emit3Δ and WT cells harvested at 32 h after the induction of conjugation. REP2 IESs are retained in progeny MACs of the mutant. Scale bars: 10 μm. (F) The scnRNA (indicated by an arrowhead) was detected in WT cells but not in emit3Δ cells.
Transcription in the meiotic MIC occurs primarily from A-type IESs (functional equivalents of piRNA clusters) that are enriched in sub-telomeric and pericentromeric regions [32, 33]. The inhomogeneous accumulation of Pol II in the MIC during meiotic prophase, which forms the elongated “crescent” with telomeres and centromeres being clustered at opposite ends reflects this biased transcription [10, 25, 26, 34] (Fig. 1A). The mechanism behind this biased transcription initiation in the meiotic MIC has remained elusive for years. In contrast to the MAC, the meiotic MIC chromatin lacks a methylated histone H3 at lysine 4, a hallmark of active transcriptional chromatin [35]. Furthermore, the MIC ncRNA transcription exhibits bidirectionality and initiates at variable sites across all loci tested, suggesting a noncanonical transcriptional regulation mechanism [27, 33, 36]. Although we recently found two proteins (Emit1 and Emit2) that are required for transcription initiation in the MIC by enabling Med localization to the MIC, and another protein, Rib1, which is needed for the accumulation of the transcription machinery at IES-dense regions (Fig. 1B), additional regulatory proteins need to be identified to comprehensively solve the enigma of ncRNA transcription in meiotic MIC. Specifically, it remains unclear whether Pol II also requires dedicated auxiliary factor (s) for localization to the MIC, for association with the MIC chromatin, and subsequently promoting such noncanonical transcription (Fig. 1B). In this study, we identify a meiotic MIC-specific TFIIB-like protein that plays a critical role in recruiting Pol II to chromatin, thereby facilitating transcription.
Materials and methods
Cells and culture conditions
Tetrahymena thermophila cell lines utilized in this study are listed in Supplementary Table S1. The wild-type (WT) strains B2086 and CU428 were obtained from the American National Tetrahymena Stock Center at Cornell University. Knockout strains, including emit1Δ, emit2Δ, rib1Δ, and spo11Δ were generated in previous studies [10, 37] and are maintained in our laboratory. Cells were grown in modified Neff medium, which consists of 0.25% proteose peptone, 0.25% yeast extract, 0.5% glucose, and 33.3 mM FeCl3 (all from Sigma–Aldrich, St. Louis, Missouri), at 30°C without agitation [38]. For conjugation induction, cells of different mating types were initially washed with 10 mM Tris–HCl (pH 7.5), resuspended in the same buffer, and cultured at 30°C for >16 h. Subsequently, equal quantities of the cells were mixed and further incubated at 30°C.
To evaluate the viability of progeny cells generated by conjugation, individual cell pairs were isolated about 10 hours post-mixing (hpm) and placed into separate droplets of Neff medium. These droplets were cultured for an additional 2–3 days, after which they were transferred to 96-well plates with fresh Neff medium, where they were cultured for another 2 days. The number of wells showing cell growth was recorded. Subsequently, 5 μl of these growing cells were transferred into the Neff medium containing either 120 μg/ml of paromomycin (Sigma–Aldrich) or 15 μg/ml of 6-methylpurine (Sigma–Aldrich) and incubated for an additional 3–4 days. Finally, the number of wells with surviving cells was recorded.
RNA-seq analysis
The RNA-seq data of WT cells harvested during vegetative growth, starvation, and different time points of conjugation were generated by previous studies [39–42]. The corresponding raw data were retrieved from the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra), under the accession numbers SRR14473326, SRR14473331, SRR10915514, SRR3470619, SRR3470621, SRR3470623, SRR3145125, and SRR3145127. Raw sequencing reads were first processed using Trim Galore (version 0.6.7) to filter out low-quality reads and those <20 nt (https://github.com/FelixKrueger/TrimGalore). The remaining reads were mapped to thesomatic genome (released in 2021, https://tet.ciliate.org/, [43, 44]) using HISAT2 (version 2.1.0), retaining only uniquely mapped reads with zero mismatches for further analysis. Picard (version 2.25.5) was then employed to remove polymerase chain reaction (PCR) duplicates. Relative gene expression value was determined based on the aligned reads using the Transcripts Per Million (TPM) method [45].
Based on normalized expression data of characterized meiosis genes that were expressed exclusively after the induction of conjugation [46], a threshold of background expression value during growth and starvation stage was determined. Next, genes with their expression lower than this threshold in growth and starvation, but higher than this threshold at any investigated timepoints of conjugation were considered as conjugation-specific genes. Next, these genes were further divided into six clusters based on their gene expression profile similarity, using the Mfuzz package (version 2.56.0, [47]). Clusters were visualized using the pythonpackage PyDLC (version 0.3, https://github.com/clberube/pydlc). Enrichment of meiosis genes, MIC ncRNA transcription regulators, meiosis regulators, and ncRNA processing factors in cluster 1 and 3 were assessed using the Fisher’s exact test. Remote homologs of cluster 1 and 3 proteins were determined by searching against the PDB database (pdb70_from_mmcif, version 2022-03-19) using HH-suite3(version 3.3.0) [48].
Somatic gene knockin and knockout
Somatic gene knockin and knockout strains were generated by introducing recombinant DNA plasmids into Tetrahymena cells via particle bombardment [38]. The plasmids were generated as follows:
To label Emit3 with a human influenza hemagglutinin (HA) epitope, the EMIT3 coding region, along with its 3′ untranslated region (3′-UTR) fragments, was amplified from WT Tetrahymena genomic DNA using primer sets #1 & #2, #3 & #4, and #5 & #6 (Supplementary Table S1). In this process, the HA-coding sequence was fused before the stop codon of EMIT3. Next, the neo4 cassette was obtained by digesting the pNeo4_SmaI plasmid DNA with SmaI [49], while the pBluescript SK (−) vector fragment was excised from the same plasmid by NotI digestion. The five DNA fragments mentioned above were then ligated using the NEBuilder® HiFi DNA Assembly kit (New England Biolabs, Ipswich, MA) to generate the pEmit3-HA-neo4 plasmid. A plasmid for tagging the endogenous P44 with an HA-tag was generated using a similar method with primer sets #7 & #8, #9 & #10, and #11 & #12. To generate a version of the Emit3 tagging plasmid with a cycloheximide resistance cassette, the EMIT3 coding sequence, 3′-UTR sequences, the NotI cut pBluescript SK (−) vector fragment, and the chx cassette that was released from the SmaI site of pChx_SmaI, were fused [49, 50], using the NEBuilder® HiFi DNA AssemblyKit.
P34, P62L, Xpd1, and Xpb2 endogenous HA-tagging plasmids were generated in a slightly different way. Taking P34 as an example, its coding sequence and 3′-UTR sequence were amplified from WT Tetrahymena genomic DNA, using primer sets #13 & #14, and #15 & #16. Meanwhile, the BTU1-EGFP-neo4 fragment was excised from the pEGFP-neo4 plasmid by BamHI and XhoI double digestion [51], and the pBluescript SK (+) vector fragment was obtained from the same plasmid by SacI and KpnI double digestion. Then, all four DNA fragments were ligated using the NEBuilder® HiFi DNA Assembly kit. Next, EGFP-neo4 was replaced with HA-neo4, which was released from the pHA-neo4 plasmid [51], using BamHI and XhoI double digestion. Primers #17-#26 were used for tagging P44, P62, Xpd1, and Xpb2. The Xpb2 coding sequence was synthesized by the ThermoFisher GeneArt DNA synthesis service (Thermo Fisher Scientific, Waltham, MA). The Rpb3, P52, and Tfiibl1 endogenous HA-tagging plasmids were generated in a similar way using primers #27-#38. The major difference is that their coding sequences and 3′-UTR sequences were fused directly with the BTU1-HA-neo4fragment.
An Emit3 rescue plasmid was generated similar to the pEmit3-HA-neo4 plasmid. The major difference is that EMIT3’s 5′-UTR, along with its downstream ORF, was amplified from WT Tetrahymena genomic DNA using primer sets #39 & #2. This was then fused with the 3′-UTR, neo4 cassette, and NotI cut pBluescript SK (−) vector backbone. The resulting plasmid is pEmit3-WT-HA-neo4. The plasmid used for mutating the Emit3 zinc ribbon was generated in asimilar manner. Briefly, an Emit3 zinc ribbon mutation fragment was first synthesized in vitro using the ThermoFisher GeneArt DNA synthesis service. Meanwhile, the 5′-UTR and part of the unmodified EMIT3 coding sequence were amplified from WT Tetrahymena genomic DNA using primer sets #39 & #40 and #41 & #2. These three DNA fragments were first fused by overlapping PCR using primer sets #39 & #2, and then ligated together with the 3′-UTR, neo4 cassette, and the NotI cut pBluescript SK (−) vector backbone using the NEBuilder® HiFi DNA Assembly kit. The resulting plasmid is pEmit3-CS-HA-neo4.
To generate a plasmid for deleting the EMIT3 gene from the Tetrahymena somatic genome, the 5′-UTR and 3′-UTR flanking regions of EMIT3 were amplified from Tetrahymena genomic DNA by PCR using primer pairs #39 & #42 and #43 & #44, respectively. Due to the presence of overlapping sequences, the EMIT3 flanking sequences and the chx cassette, which was released from pChx_SmaI by SmaI digestion, were cloned into the NotI site of pBluescript SK (−) via Gibson assembly. The resulting plasmid is namedpEmit3-chx-ko.
To introduce recombinant plasmids into Tetrahymena, these were first linearized by restriction digestion with an appropriate enzyme, then coated onto 0.6 μm gold microcarriers (Bio-Rad Laboratories, Hercules, CA) or 0.8 μm tungsten particles (Yaoyi, Shanghai, China), and introduced into starved Tetrahymena cells via particle bombardment using the biolistic PDS-1000/He System (Bio-Rad Laboratories) or the GJ-1000 (Scientz, Ningbo, China) [52]. Transformants carrying the neo4 or chx cassette were subsequently selected by culturing in the presence of paromomycin or cycloheximide (Sigma–Aldrich), respectively.
To verify the deletion of EMIT3, candidate knockout strains of different mating types were starved overnight and then mixed to induce conjugation. Total RNA was extracted three hours after mixing, and complementary DNA (cDNA) was synthesized using the SuperScript™ VILO™ cDNA Synthesis Kit (Thermo). For comparison, cDNA from WT cells was prepared in the same manner. The expression of EMIT3 was assessed by reverse transcription PCR using primer pair #45 & #46. The conjugation-specific gene TWI1 was also amplified using primer pair #47 & #48 to serve as a control for loading and developmental stage.
DNA elimination analysis using fluorescence in situ hybridization
To investigate the elimination of REP2 IESs, fluorescence in situ hybridization (FISH) was conducted following a previously established protocol [53]. In summary, Tetrahymena conjugating cells of different genotypes were collected 24 hpm. The cells were fixed and permeabilized simultaneously at 25°C for 30 min using a solution containing 3.7% formaldehyde (Macklin Biochemical, Shanghai, China) and 0.5% Triton X-100 (Sigma–Aldrich). After fixation and permeabilization, the cell pellets were harvested and resuspended in a solution containing 4% formaldehyde and 3.4% glucose (Sigma–Aldrich). These cell suspensions were then spread onto glass slides and left to air-dry in a fume hood for over 16 hours. FISH probes targeting REP2 IESs were generated fromgenomic DNA via PCR using the GoTaq Long PCR Master Mix (Promega, Madison, WI) and primer pair #49 & #50, followed by labeling with Cy3-dUTP (GE Healthcare, Waukesha, WI) with a nick translation kit (Roche, Indianapolis, IN). FISH was carried out as previously described [54]. Fluorescence signals were observed and captured using a Zeiss Z2 microscope (Carl Zeiss, Thornwood, NY). The acquired stacked images were then deconvolved, merged, and converted into 2D images using FIJI software [55]. As the imaging was done with a monochrome camera, grayscale fluorescence signals recorded from different channels were assigned different colors using Photoshop (version CS5, Adobe, San Jose, CA).
Immunostaining of proteins and double-stranded RNA
Indirect immunostaining of Tetrahymena proteins was performed following a previously established method, with minor modifications [37, 56]. In brief, Tetrahymena cells were fixed and permeabilized simultaneously at 25°C for 30 min using 3.7% formaldehyde, and 0.5% Triton X-100. Fixed and permeabilized cell pellets were then resuspended in 4% formaldehyde and 3.4% glucose, and spread onto glass slides. After air-drying the slides in a fume hood, they were used for protein immunostaining.
For pre-fixation detergent treatment, cells were initially exposed to 1% Triton X-100 with 0.37% formaldehyde on ice for 25 min. After this, formaldehyde was added to bring the final concentration to 3.7%. The cells were then harvested by centrifugation and resuspended in an appropriate amount of fixative containing 4% formaldehyde and 3.4% glucose. The cell suspension was then applied to a slide and air-dried in a fume hood for ∼16 h. After drying, the cells were rehydrated with 1× phosphate-buffered saline (PBS) containing 0.1% Tween-20 (referred to as PBSW; Tween-20 was obtained from Sigma–Aldrich), blocked using a buffer containing 0.1% Tween-20, 3% bovine serum albumin, 10% normal goat serum, and 1× PBS, and subsequently incubated with primary antibodies and fluorescence-conjugated secondary antibodies, all diluted in the blocking buffer. After thorough rinsing with PBSW, the nuclei were stained with 10 ng/ml DAPI (4′,6′-diamidino-2-phenylindole) and mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Immunostaining of Twi1 and Rpb3 was carried out using custom anti-Twi1 (diluted 1:5 000) and anti-Rpb3 (diluted 1:200) antibodies [57, 58]. The HA epitope was stained using either a rabbit monoclonal anti-HA antibody (Clone C29F4, 1:500 dilution; Cell Signaling Technology, Danvers, MA) or a mouse monoclonal anti-HA antibody (Clone 6E2, 1:100 dilution; Cell Signaling Technology). To visualize telomeric repeats and Emit3-HA from the same cell, telomere repeats were first labeled by FISH, using Cy3-conjugated oligo probe containing Tetrahymena telomeric repeats (Supplementary Table S1, oligo #51). Next, HA-tagged protein was labeled by indirect immunostaining, using the anti-HA rabbit monoclonal antibody and a fluorescein isothiocyanate-conjugated goat anti-rabbit IgG secondary antibody. Double-stranded RNA (dsRNA) was detected using a mouse monoclonal anti-dsRNA antibody (Clone J2, 1:1000 dilution, Nordic-MUbio, Fargo, ND), as described previously [10, 28, 59]. For each slide, 20–50 cells were examined. In cases where all cells exhibited consistent cytological patterns, a representative image of a single cell was selected and included in the figure. When cells exhibited heterogeneous cytological patterns, representative images along with quantitative data were provided to illustrate the heterogeneity.
Immunoblotting, immunoprecipiation, and mass spectrometry analysis
To analyze protein expression via western blotting, approximately 1.0 × 106 cells were collected, and protein was precipitated on ice using 10% trichloroacetic acid (Sigma–Aldrich). The precipitated proteins were then resuspended in 1× SDS–PAGE sample buffer (62.5 mM Tris–HCl [pH 6.8], 5% sucrose, 0.015% bromophenol blue, 2% SDS, and 5% 2-mercaptoethanol) and boiled at 95°C for 5 min. Next, the proteins were separated by SDS–PAGE, transferred to a polyvinylidene fluoride (PVDF) membrane (GE Amersham, Fairfield County, CT), and detected using appropriate antibodies. Detection of HA-tagged proteins was achieved with a rabbit monoclonal anti-HA antibody (Clone C29F4, Cell Signaling Technology, 1:2000 dilution). A mouse monoclonal anti-α-tubulin antibody (Clone DM1A, LabVision, Waltham, MA, 1:10 000 dilution) was used to probe for constitutively expressed α-tubulin, while a custom rabbit polyclonal anti-Twi1 antibody (1:10 000 dilution) was used to detect the conjugation-specific protein Twi1.
Protein immunoprecipitation was conducted following a previously established protocol with minor adjustments [10]. In summary, 200 ml of WT meiotic cells (7.5 × 105 cells/ml), or modified meiotic cells expressing endogenous Emit3-HA or Emit3-CS-HA were collected and resuspended in 10 ml of lysis buffer containing 30 mM Tris–HCl (pH 7.5), 20 mM KCl, 150 mM NaCl, 2 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.1% Triton X-100, and 1× cOmplete protease inhibitor cocktail (Roche). The cells were homogenized using a Dounce homogenizer (Kimble Chase, Vineland, NJ), until complete homogenization was achieved. The homogenate was clarified by centrifugation and filtered through a 0.45 μm PVDF syringe filter (Merck Millipore, Billerica, MA) to remove insoluble debris. The resulting lysate was incubated with pre-equilibrated EZview HA agarose beads (Sigma–Aldrich) for 2 h at 4°C. The beads were subsequently washed three times with lysis buffer (excluding PMSF and the protease inhibitor cocktail) to minimize nonspecific protein binding. Bound proteins were eluted with 400 μl of 250 μg/ml HA peptide (dissolved in a solution containing 30 mM Tris–HCl [pH 7.5], 20 mM KCl, 150 mM NaCl, 2 mM MgCl2, and 0.1% Triton X-100). The protein in the eluent was precipitated with 10% tricarboxylic acid and 100% acetone, then boiled in SDS–PAGE sample buffer, and loaded onto an SDS–PAGE gel. After the protein samples had completely migrated into the gel, the electrophoresis was stopped, and the gel was Coomassie stained. Areas with Coomassie staining were excised from the gel, subjected to in-gel tryptic digestion, and then analyzed by mass spectrometry at the Max Perutz Laboratory Mass Spectrometry Facility (Vienna, Austria). The detailed mass spectrometry procedures, along with the raw data, have been submitted to the ProteomeXchange Consortium and are available through the PRIDE repository under the dataset identifier PXD057568 [60]. High-confidence protein–protein interactions were detected using SAINTexpress (version 3.6, [61, 62]). Only interactions with a Bayesian False Discovery Rate (BFDR) of 0.05 or lower, with a spectral count of over three in the sample and fewer than three in the control sample, were considered reliable protein–proteininteractions.
MIC isolation and chromatin occupancy profiling using cleavage under targets and tagmentation
MIC isolation from meiotic cells of different genotypes was conducted following a published method [63]. The purity of the MICs was evaluated by fluorescence microscopy, and their quantity was determined using a hemocytometer. A total of five million purified MICs, with <0.1% MAC contamination, were used for the Cleavage Under Targets and Tagmentation (CUT&Tag) experiment [64]. To facilitate the association between MICs and Concanavalin A beads, the MICs were pre-equilibrated in a buffer containing 150 mM NaCl, 0.5 mM spermidine, 0.1% bovine serum albumin, and 10 mM HEPES (pH 7.4). CUT&Tag was then conducted using BioMag®Plus Concanavalin A beads (Bangs Laboratories, Fishers, IN), a commercial Hyperactive Universal CUT&Tag Assay Kit for Illumina (Vazyme, Nanjing, China), and an anti-HA rabbit monoclonal antibody (Clone C29F4, Cell Signaling Technology). As a control, the same number of MICs purified from untagged WT meiotic cells were processed in parallel. CUT&Tag libraries were generated using indexed primers [65] and the KAPA HiFi HotStart Real-Time Library Amplification Kit (Roche) and subsequently sequenced on a NovaSeq 6000 platform (Illumina, San Diego, CA).
Raw sequencing data were first processed with Trim Galore (version 0.6.7) to eliminate sequencing adapters and low-quality reads. The cleaned reads were then aligned to the MIC genome (GenBank ID: GCA_016584475.1) using Bowtie2 (version 2.3.5.1, [66]). Reads mapping to ribosomal RNAs (rRNAs), transfer RNAs (tRNAs), small nuclear RNAs (snRNAs), and the mitochondrial genome were excluded, ensuring that only uniquely mapped reads to the MIC genome were retained for further analysis. PCR duplicates were removed using Picard (version 2.25.5) with its default parameters (http://broadinstitute.github.io/picard). Normalized CUT&Tag signal tracks were generated using BamCoverage, applying the reads per genome coverage (RPGC) method [67]. The CUT&Tag signal track was visualized using IGV (version 2.8.9, [68]) as well as pyGenomeTracks (version 3.8, [69]). Significantly enriched CUT&Tag peaks were identified from the aligned reads using MACS3 (version 3.0.0, [70]). The top 10% of peaks were submitted to the MEME suite for motif identification using the DREME package [71, 72]. A pileup plot of the CUT&Tag signal track at and around the sites of interest was generated using deepTools2 (version 3.5.1, [67]). Next-generation sequencing data generated by this study can be retrieved from the Sequence Read Archive (PRJNA1182059).
Small RNA sequencing data analysis
The WT small RNA sequencing data from meiotic cells was obtained from the Gene Expression Omnibus (accession number: GSM3032632; [73]). The spo11Δ meiotic small RNA sequencing libraries (two biological replicates) were prepared as previously described, and sequenced on HiSeq-2000 (Illumina) [73]. Raw reads were processed with Cutadapt (version 2.4) to remove adapter sequences [74]. Following adapter trimming, low-quality reads and those <26 nt or >32 nt were filtered out using Trim Galore (version 0.6.10, https://github.com/FelixKrueger/TrimGalore, [33]). Reads matching rRNAs, tRNAs, snRNAs, and mitochondrial RNAs were discarded. The remaining reads, trimmed to their first 25 nt, were aligned to the MIC genome (GenBank ID: GCA_016584475.1) using Bowtie (version 1.3.1), retaining only uniquely and perfectly mapped reads for subsequentanalysis.
Small RNA read coverage was calculated using BamCoverage and normalized by Bins Per Million (BPM) mapped reads, similar to TPM normalization in RNA-seq (version 3.5.1, [67]). Coverage profiles were visualized using either Integrative Genomics Viewer (version 2.8.9, [68]) or pyGenomeTracks (version 3.8, [69]). Peaks and peak summits of scnRNA coverage were identified using MACS3 (bdgpeakcallcommand, version 3.0.0; https://macs3-project.github.io/MACS/).
Results
Emit3 is a sexual reproduction stage-specific TFIIB
Given that the known essential components for TE-targeting MIC ncRNA transcription in Tetrahymena are predominantly expressed during meiotic prophase [10, 29, 30, 75], we hypothesized that other genes regulating this process are also expressed similarly. To identify such genes, we performed transcriptome analysis using the published RNA-seq dataset. The 2950 genes specific for the conjugation (i.e. the sexual reproduction stage) were clustered into six subgroups by the Mfuzz algorithm according to their gene expression profiles (Supplementary Table S2) [47]. We found that the known genes required for the MIC ncRNA transcription were significantly enriched within cluster 1 (Fig. 1C, P-value = 0.00175, Fisher’s exact test). Expressions of these genes peak at 2 h after the induction of conjugation. On the other hand, the known meiosis regulators and DNA elimination factors are enriched in cluster 3 (Fig. 1C, P-value = 1.54e-12, Fisher’s exact test), which showed an expression peak at 3 h after the induction of conjugation. Thus, we believe genes required for the MIC ncRNA transcription have a higher chance of being clustered in cluster 1.
One of these genes, EMIT3 (Enable Micronucleus Transcription 3, TTHERM_000492589) caught our attention, as it encodes a protein similar to TFIIB, which has a conserved role in recruiting Pol II to the core promoter in other eukaryotes. Protein structure modelling revealed that Emit3 possesses a structurally conserved TFIIB zinc-ribbon at its N-terminus (Fig. 1D), which is known to be crucial for recruiting Pol II to promoters in yeast and human TFIIB [15, 76, 77]. The zinc-ribbon residues are highly conserved among Emit3 proteins across different Tetrahymena species, indicating their importance as a structural element. Furthermore, Emit3 has a C-terminal structure similar to the TFIIB B-core (Supplementary Fig. S1A), although its associated B-reader and B-linker structures, known to be important for transcription start site selection and promoter opening in yeasts, are only partially retained (Supplementary Fig. S1B and C) [78].
Emit3 also shows sequence similarity to the constitutively expressed Tetrahymena protein encoded by gene TTHERM_00637498 (BLASTp; E-value = 3e-8, identity 24.02%), hereafter referred to as Tfiibl1 (TFIIB-like 1). Tfiibl1 exhibits higher similarity to human TFIIB than Emit3 and contains all conserved linker elements found in TFIIB (Supplementary Fig. S1C). Collectively, these observations suggest that Tfiibl1 is a canonical TFIIB, while Emit3 is likely its divergent, conjugation-specific variant.
Emit3 is required for meiotic MIC ncRNA transcription
To examine the function of Emit3, we generated knockout mutants lacking EMIT3 in their somatic genome (Supplementary Fig. S2A). While this resulted in a noticeable prolongation of meiotic prophase compared to WT cells, the following meiotic and post meiotic processes appeared largely normal (Supplementary Fig. S2B). However, over 75% of emit3Δ cells arrested before the separation of conjugation partners (Supplementary Fig. S2B), a phenotype commonly associated with failure in DNA elimination that leads to progeny demise [29–31]. Indeed, we found that, unlike in WT cells, the Tetrahymena non-LTR retrotransposon (REP2, [79]) was not eliminated from the progeny MACs in the mutants (Fig. 1E), and the progeny cells were not viable (Supplementary Table S3).
Given that IES elimination is governed by scnRNAs produced during meiotic prophase, we then investigated scnRNA biogenesis in the mutants. Our analysis of total RNAs by gel electrophoresis failed to detect scnRNA in cells lacking EMIT3 (Fig. 1F). We then examined the precursor MIC ncRNA transcripts. To do this, we examined ncRNAs derived from a well-characterized MIC-specific sequence (the M element) using strand-specific Northern blotting. Since the transcripts from this locus are heterogeneous in length [29, 30], smears were detected in the total RNA extracted from WT, dcl1Δ, and rib1Δ meiotic cells (collected at 4 hpm; Fig. 2A). Consequently, the accumulation of dsRNAs in the WT meiotic MICs was detected by immunostaining with an anti-dsRNA antibody (Fig. 2B) [28]. In emit3Δ, no transcript was detected from either strand of the M element (Fig. 2A) and dsRNA was undetectable in the meiotic MIC (Fig. 2B). These observations indicate that EMIT3 is crucial for the MIC ncRNAtranscription.
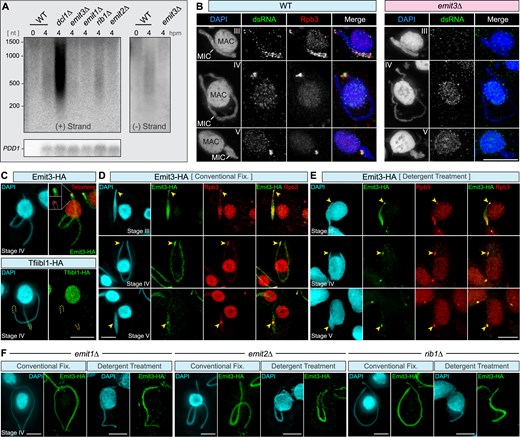
Emit3 localizes to meiotic MIC and is essential for MIC ncRNA transcription. (A) Strand-specific Northern blotting was used to investigate transcripts from (+) and (−) strands of an MIC-specific DNA locus, known as the M-element. Transcripts were detected in WT and rib1Δ cells, and were accumulated in dcl1Δ cells due to the lack of ncRNA cleavage. In contrast, transcripts were not detected in emit1Δ, emit2Δ, or emit3Δ mutants. The expression of the conjugation-specific PDD1 gene was analyzed as a control for loading and developmental stages. (B) Immunostaining of dsRNA (and Rpb3) in WT and emit3Δ cells. Scale bar: 10 μm. (C) Upper panel: Costaining of endogenously expressed Emit3-HA (by immunostaining) and telomeric repeats (by FISH) in a stage IV meiotic cell. Lower panel: Immunostaining of endogenously expressed Tfiibl1-HA in a stage IV meiotic cell. (D) Costaining of Emit3-HA and Rpb3 (an RNA Pol II-specific subunit) was performed to assess their localization. Emit3 and Rpb3 colocalize in the meiotic MIC of Tetrahymena cells, fixed under a mild condition (i.e. conventional fixation). (E) Costaining of Emit3-HA and Rpb3 was performed to assess their localization. Emit3 and Rpb3 colocalize in the meiotic MIC, fixed under a harsh condition (i.e. pre-fixation detergent treatment, which removes nucleoplasmic proteins not tightly associated with chromatin). For technical details, see “Materials and methods” section. (F) Investigation of Emit3 localization in rib1Δ, emit1Δ, and emit2Δ mutants, which exhibit disrupted MIC ncRNA biogenesis. Scale bars: 10 μm.
Emit3 colocalizes with RNA polymerase II in meiotic MIC
To study the localization of Emit3, we generated strains that expressed C-terminally HA-tagged Emit3 under the control of its endogenous promoter (Supplementary Fig. S3A). Consistent with the messenger RNA (mRNA) expression profile of EMIT3 (Fig. 1C), Emit3-HA protein was detected specifically during conjugation by western blotting (Supplementary Fig. S3B). Immunofluorescent staining using an anti-HA antibody detected Emit3-HA in the MIC exclusively at the meiotic prophase (Fig. 2C), indicating that Emit3 has a specialized role in the meiotic MIC. We also produced Tetrahymena strains expressing Tfiibl1-HA fusion protein from its endogenous locus (Supplementary Fig. S3C) and found that Tfiibl1-HA was detected exclusively in the MAC (Supplementary Fig. S4A). These findings support the idea that Tfiibl1 and Emit3 act as the general and conjugation-specific TFIIB, respectively. The former most probably controls mRNA transcription in the MAC, while the latter regulates the MIC ncRNAtranscription.
Simultaneous immunostaining of Emit3-HA and Rpb3, a specific component of Pol II, revealed that Emit3 colocalizes with Pol II in the meiotic MIC. Specifically, in the elongating MIC, Emit3 accumulated at the telomere-clustered “head” regions and the pericentromeric “trunk” regions at the opposite tips of the elongated MIC (Fig. 2D). As the MIC fully elongated, Emit3 was progressively concentrated at the “head” region (Fig. 2C and D, and Supplementary Fig. S4B). After detergent treatment prior to fixation to remove chromatin-unbound nuclear proteins [80], Emit3 as well as Rpb3 remained in the MIC, and their preferential localization at centromere- and telomere-proximal regions became more pronounced (Fig. 2E). This suggests that Emit3 associates with chromatin, and its interaction is especially stable in Pol II-bound regions, particularly in TE-rich regions proximal to centromeres and at clustered telomeres.
Our previous study identified three Med-associated proteins—Emit1, Emit2, and Rib1—that are essential for MIC ncRNA transcription (Fig. 1B) [10]. To investigate whether Emit3’s specific localization pattern in the meiotic MIC depends on Emit1, Emit2, and Rib1, we introduced the Emit3-HA expressing construct into the knockout mutants lacking either EMIT1, EMIT2, or RIB1. Immunostaining of these strains showed that knockout of any of these genes caused Emit3 to distribute more uniformly in the MIC, than in WT cells (Fig. 2F) while it remained on chromatin after detergent treatment. This altered distribution of Emit3 is reminiscent of the changes observed in Pol II localization in emit1Δ, emit2Δ and rib1Δ cells [10]. These findings indicate that Emit3 colocalizes with Pol II in the meiotic MIC, and that its biased accumulation at TE-enriched regions depends on the Med-associated proteins Emit1, Emit2, and Rib1.
Emit3 is required for the association of Emit1, Pol II, and Mediator complex with chromatin
Since the Med-associated proteins Emit1, Emit2, and Rib1 are crucial for MIC ncRNA transcription [10], we examined if Emit3 plays any role in their localizations. These proteins were expressed and localized individually with an HA-tag in WT and emit3Δ cells. As depicted in Fig. 3A–C, Emit1, Emit2, and Rib1 exhibited a similar nonhomogeneous localization pattern in WT meiotic MICs. We found that Emit1 and Emit2 lost their nonhomogeneous pattern in emit3Δ. Moreover, after removing chromatin unbound fractions, Emit1 was no longer detected on chromatin (Fig. 3A). By contrast, Emit2 was still detected in the meiotic MIC after detergent treatment but lost its biased accumulation pattern at specific regions (Fig. 3B). As for Rib1, instead of its relatively broad localization at the pericentromeric region in stage III meiotic MICs in the WT (Fig. 3C) [10], it occupied the periphery of the MIC in the absence of Emit3, especially in stage IV-V. Furthermore, multiple Rib1 spots were found at one terminus of the elongated MIC and along the emit3Δ MIC, which were colabeled by FISH for telomeric repeats. This indicates that EMIT3 is required for telomere clustering and that Rib1 localizes to the telomeres. Nonetheless, because the loss of SPO11, which also impairs the telomere clustering (Fig. 3D) [37], did not cause any noticeable defect in the scnRNA biogenesis (Fig. 3E), the reduced telomere clustering in emit3Δ cells is not the cause of the defective MIC ncRNA transcription.
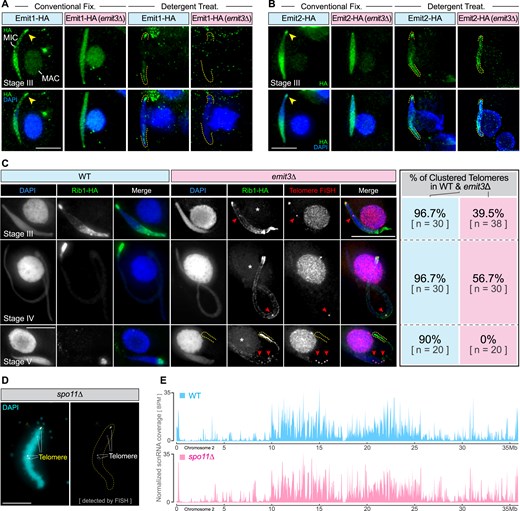
Emit3 is required for the localization of MIC ncRNA transcription regulators. (A) Immunostaining of endogenously expressed Emit1-HA in WT and emit3Δ cells that were fixed under different conditions. Arrowheads indicate eye-catching telomeric clustered regions in the MIC. Dashed lines indicate MIC termini. Scale bar: 10 μm. (B) Immunostaining of endogenously expressed Emit2-HA in WT and emit3Δ cells that were fixed under different conditions. Arrowheads indicate the telomeric clustered regions in the MIC. Dashed lines indicate MIC termini. Scale bar: 10 μm. (C) Immunostaining of endogenously expressed Rib1-HA in WT and emit3Δ cells. Telomeric repeats were stained using FISH. Arrowheads denote telomeric foci. Scale bar: 10 μm. (D) FISH staining of telomeric repeats in spo11Δ meiotic MIC. Scale bar: 10 μm. (E) The abundance profiles of scnRNAs on chromosome 2 of the meiotic MIC in WT and spo11Δ cells. BPM: Bins per million mapped reads.
Because TFIIB is required for recruiting Pol II to the core-promoter region, we then examined if knocking out EMIT3 impairs Pol II’s association with ncRNA production regions in the meiotic MIC, thereby disrupting transcription. We generated strains expressing Rpb3-HA in WT and emit3Δ cells and then studied Rpb3 localization by immunostaining using an anti-HA antibody. We found that Rpb3’s localization pattern was abnormally homogeneous (Fig. 4A) and Rpb3 was no longer present on MIC chromatin after removing the soluble fraction (Fig. 4B), although its presence in the MAC remained unaffected (Fig. 4A, B). This suggests that while Emit3 is not required for the import of Pol II into the meiotic MIC, it is necessary for Pol II’s association with the meiotic MIC chromatin (Fig. 4C).
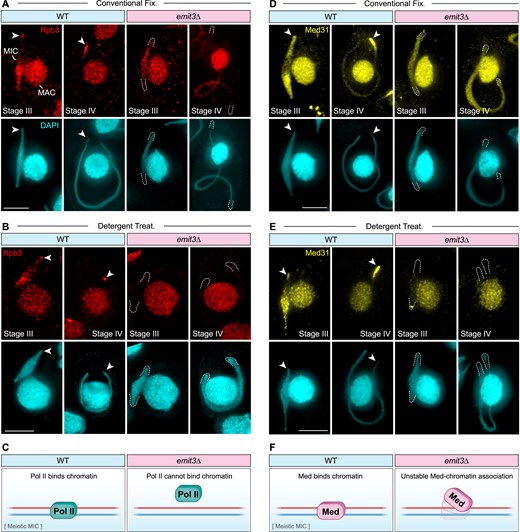
Emit3 is required for the association of the Pol II transcription machinery with meiotic chromatin. (A-B) Immunostaining of the endogenously expressed Pol II subunit, Rpb3, in WT and emit3Δ cells that were fixed under different conditions. Arrowheads indicate the telomeric clustered region in the MIC. Dashed lines indicate MIC termini. Scale bar: 10 μm. (C) A schematic diagram summarizing the Rpb3 immunostaining results. (D-E) Immunostaining of the endogenously expressed Med subunit, Med31, in WT and emit3Δ cells that were fixed under different conditions. Arrowheads indicate the eye-catching telomeric clustered region in the MIC. Dashed lines indicate MIC termini. Scale bar: 10 μm. (F) A schematic diagram summarizing the Med31 immunostaining results.
The Med’s association with meiotic chromatin was also examined using WT and emit3Δ strains expressing C-terminally HA-tagged Med31, an evolutionarily conserved Med component, from its endogenous promoter. As shown in Fig. 4D, Med31-HA displayed a Pol II-like, nonhomogeneous distribution pattern in WT cells but showed more homogeneous distribution in emit3Δ cells. After removing the chromatin-unbound fraction, Med31-HA maintained a nonhomogeneous localization pattern at both ends of the elongating meiotic nucleus and at one end of the fully elongated meiotic nucleus in WT cells. In contrast, Med31-HA was detected only at the telomere-clustered region of the elongating MIC in emit3Δ cells (Fig. 4E and Supplementary Fig. S4C). Notably, in fully elongated emit3Δ meiotic nuclei, Med31-HA became undetectable in the meiotic nucleus, although its association with MAC chromatin was not obviously affected. This premature disassociation of Med31-HA from meiotic chromatin in emit3Δ cells suggests that Emit3 is crucial for maintaining the Med’s association with meiotic chromatin (Fig. 4F). Summing up, Emit3 is required for the association of Emit1, Pol II and Med with meiotic chromatin and for the localization of Rib1 and Emit2 at specific regions of the meioticchromatin.
Emit3 interacts with TFIIH and is required for the preferential accumulation of TFIIH at meiotic MIC telomeric chromatin
To gain further insight into the mechanism of Emit3-directed MIC ncRNA transcription, we aimed to identify its interacting partner proteins through immunoprecipitation (IP), followed by mass spectrometry. Emit3-HA IP was performed with cells collected 3.5 hours after the induction of conjugation, the stage when most MICs were undergoing meiosis and becoming transcriptionally active [10, 33]. Parallel mock IP experiments were conducted using untagged WT cells harvested at the same stage. Proteins that were significantly enriched in the Emit3-HA IP samples were analyzed using SAINTexpress [61, 62] (Supplementary Table S4). Notably, those include orthologs and homologs of several core subunits of the TFIIH complex (Fig. 5A, Supplementary Table S4), including P34 (encoded by the gene TTHERM_00420200), P44 (TTHERM_00152050), P52 (TTHERM_00313290), a homolog of the helicase subunit Xpb (Xpb2, TTHERM_00400821, Supplementary Table S5, and Supplementary Table S6), and a highly divergent P62-like protein (P62L, TTHERM_00590380, Supplementary Fig. S5 and Supplementary Table S6).
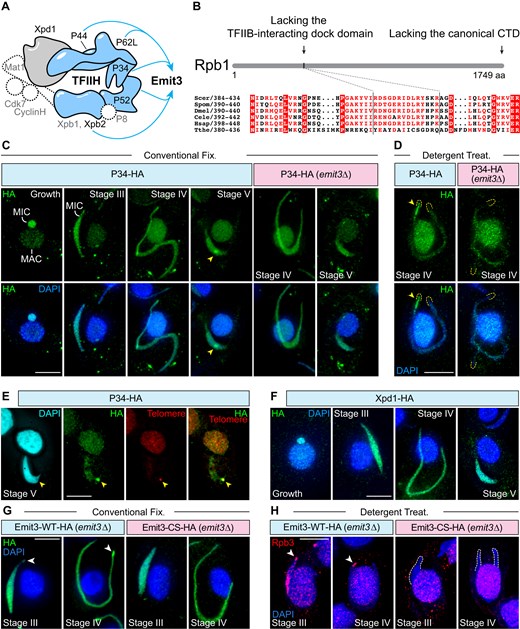
Emit3 interacts with TFIIH through a zinc-ribbon and this interaction is required for recruiting Pol II to the MIC chromatin. (A) Interactions (depicted in blue) between Emit3 and TFIIH components were identified through immunoprecipitation. Components indicated by dashed lines are not present in Tetrahymena. The shape and arrangement of the complex subunits are modeled based on the human TFIIH core complex (PDB code: 6nmi). (B) Tetrahymena Rpb1 lacks both the conserved TFIIB-interacting dock domain and the canonical carboxy-terminal domain (CTD) that contains phosphorylatable heptad repeats. (C-D) Immunostaining of the endogenously expressed TFIIH complex subunit P34-HA in WT and emit3Δ cells that were fixed under different conditions. Arrowheads indicate the telomeric clustered region in the MIC. Dashed lines indicate MIC termini. Scale bars: 10 μm. (E) Costaining of P34-HA (using immunostaining) and telomeric repeats (using FISH) in a stage V meiotic cell. Arrowheads indicate the telomere-clustered region in the MIC. Scale bar: 10 μm. (F) Immunostaining of another endogenously expressed TFIIH complex subunit, Xpd1. Scale bar: 10 μm. (G) Immunostaining of endogenously expressed Emit3-WT-HA (i.e. unmodified Emit3) and Emit3-CS-HA (i.e. zinc ribbon mutated Emit3). Arrowheads indicate enriched Emit3-WT-HA at a pole of the elongated meiotic MIC. Scale bar: 10 μm. (H) Immunostaining of Rpb3 in Emit3-WT-HA and Emit3-CS-HA cells that were detergent-treated prior to fixation. Scale bar: 10 μm.
The ortholog of another TFIIH component, Xpd1 (TTHERM_00316410, Supplementary Table S5), which is involved in TFIIH-dependent nuclear excision repair in other organisms [81], was not copurified with Emit3. Therefore, Xpd1 either does not form a stable complex with Emit3 or is not involved in regulating the MIC ncRNA transcription. Other conserved TFIIH subunits, P8 and the kinase module subunits (Ccnh1 and Mat1), are absent in the current Tetrahymena protein sequence database (version 6, released in October 2024; Supplementary Table S5) [43, 82]. Additionally, none of the proteins that copurified with Emit3 contain a protein kinase domain. The absence of kinase module orthologs is consistent with the lack of conserved phosphorylatable heptad repeats at the C-terminal region of Tetrahymena Rpb1 (Fig. 5B) [83]. Altogether, these findings suggest that Emit3 forms a complex with the core subunits of TFIIH, which is unexpected as a stable interaction between TFIIB and TFIIH has not been detected in yeasts andhumans.
Localizations of all Emit3-associated TFIIH subunits were analyzed by generating strains expressing C-terminally HA-tagged fusion proteins from their endogenous promoters. Consistent with the constitutive expression of their encoding mRNAs (http://tfgd.ihb.ac.cn/), these proteins were detected in both vegetative and conjugating cells (Fig. 5C and Supplementary Fig. S6). In vegetative cells, most of the Emit3 interacting TFIIH subunits were found in both the transcriptionally active MAC and the transcriptionally inert MIC, the latter of which lacks Pol II [10, 26]. This suggests that Tetrahymena TFIIH may have additional functions beyond transcription, potentially including DNA repair, as observed in other organisms [84]. In contrast, Xpb2 was detected only in the transcriptionally active somatic nucleus of vegetatively growing cells (Supplementary Fig. S6). In conjugating cells, all Emit3-assoicated TFIIH subunits exhibited an accumulation at one end of the meiotic MIC, which became more pronounced after removing the chromatin-unbound fraction (Fig. 5D and Supplementary Fig. S6). P34, a representative TFIIH subunit, colocalized with telomeres (detected by FISH), showing that some TFIIH is accumulated at telomere-clustered regions in the meiotic nucleus (Fig. 5E). Xpd1, which was not copurified with Emit3, was detected in both MIC and MAC before and after the induction of conjugation, but it did not show accumulation at telomere-clustered regions (Fig. 5F) indicating that Emit3-associated TFIIH and Xpd1-containing TFIIH localize differently (Fig. 5A and Supplementary Table S4). Altogether, these findings confirm that TFIIH subunits are present in the meiotic nucleus. Given that telomere-clustered regions are enriched with dsRNAs (Fig. 2B) [10], the specific accumulation of Emit3-associated TFIIH subunits at these regions—compared to noninteracting subunits—strongly suggests their involvement in the MIC ncRNA transcription.
To investigate the role of Emit3 in the biased localization of its associated TFIIH subunits, we next generated emit3Δ strains expressing C-terminally HA-tagged P34 from their endogenous promoters and compared their localization to that in WT cells. We found that P34 no longer accumulated in the telomere-clustered region of meiotic MICs (Fig. 5C and Supplementary Fig. S6), showing a similar localization with Xpd1 (Fig. 5F). Removal of the chromatin-unbound P34 fraction using detergent showed that P34 still associated with meiotic chromatin (Fig. 5D), likely reflecting the Emit3- and transcription-independent function of TFIIH in the MIC. The localization of other Emit3-associated TFIIH subunits was also investigated and their localizations were similarly affected by the loss of Emit3, mirroring the pattern observed for P34 (Supplementary Fig. S6). Since Emit3’s preferential localization to the MIC chromatin is dependent on Med-associated proteins (Fig. 2F), we further examined the localization of P34 in the emit2Δ mutant, where Med is unable to localize to the MIC [10]. Our results revealed that P34 also displayed an abnormal, homogeneous localization pattern in the absence of EMIT2 (Supplementary Fig. S6). In summary, the above findings indicate that Emit3 and Med jointly promote the accumulation of TFIIH in the telomere-clustered region of the meiotic nucleus, where ncRNA is highly transcribed.
The zinc-ribbon of Emit3 is essential for recruiting Pol II to MIC chromatin
The zinc-ribbon at the N-terminus of TFIIB in both budding yeasts and humans directly interacts with the conserved dock motif of Rpb1, which is crucial for recruiting Pol II to a promoter [14, 15, 85]. Since Emit3 is necessary for Pol II’s association with MIC chromatin (Fig. 4B), we investigated whether the zinc-ribbon of Emit3 is involved in this association. To address this, we disrupted the zinc-ribbon by substituting the four zinc-binding cysteines with serines (Emit3-CS) and expressed a C-terminally HA-tagged version (Emit3-CS-HA) under the endogenous EMIT3 promoter in emit3Δ cells. For comparison, we expressed a similar C-terminal HA tagged WT Emit3 (Emit3-WT-HA) in emit3Δ cells as a control (Supplementary Fig. S3D).
As described for HA-tagged Emit3 in WT cells (Figs. 2D), Emit3-WT-HA, which rescued emit3Δ, accumulated at the telomere-clusters and the pericentromeric trunk regions of the elongated MIC (Fig. 5G). In contrast, Emit3-CS-HA does not display this distinct localization. This indicates that the zinc-ribbon is necessary for the specific chromatin localization of Emit3, and the B-core domain alone is insufficient to support IES-biased ncRNA production. However, the zinc-ribbon is not required for chromatin association of Emit3, as Emit3-CS remained in the MIC after the removal of the soluble fraction (Supplementary Fig. S4D). IP-MS analysis of Emit3-CS showed that the zinc-ribbon is necessary for the interaction between Emit3 and most of the Emit3-associated TFIIH complex subunits, except P62L (Supplementary Table S4). Consistent with this, in Emit3-CS cells, Pol II was absent from the meiotic MIC chromatin (Fig. 5H) and scnRNA levels were reduced to a background level (Supplementary Fig. S4E). In summary, the zinc-ribbon is essential for Emit3’s localization to specific sites on MIC chromatin, its interaction with most TFIIH components, and maintaining Pol II’s association with meiotic chromatin. Thus, Emit3’s role in promoting ncRNA production is reminiscent of the canonical TFIIB function.
Emit3 is enriched at MIC genome regions with a G-rich motif, where Pol II accumulates and ncRNA transcription preferentially occurs
The colocalization of Emit3 with Pol II, combined with the requirement of Emit3 for recruiting the Pol II transcription machinery to MIC chromatin, led us to hypothesize that Emit3 may associate with MIC ncRNA transcription sites. To explore Emit3’s occupancy on meiotic chromatin, we purified MICs from meiotic cells expressing Emit3-HA from its endogenous promoter (Supplementary Fig. S7). These purified MICs were subjected to CUT&Tag (Cleavage Under Targets and Tagmentation) with an anti-HA antibody. As a control, we performed mock CUT&Tag using an equivalent number of MICs purified from WT meiotic cells. To compare Emit3 occupied sites to the active ncRNA transcription regions on meiotic chromatin, we utilized published RNA-seq data of small RNAs isolated from WT meiotic cells to generate scnRNA coverage profiles across the MIC chromosomes.
We found that the occupancy profile of Emit3 closely mirrors the scnRNA abundance profile (Fig. 6A). Locally, Emit3 was preferentially enriched at scnRNA production sites(Fig. 6A). To determine if this binding to scnRNA production sites is a general feature of Emit3, we calculated the average coverage profile of Emit3 at the meiotic scnRNA production sites and compared it to an equivalent number of random sites. As shown in Fig. 6B, Emit3 was enriched at scnRNA production sites, whereas no such enrichment was observed at random sites. Additionally, we examined the chromatin occupancy of Emit3 in the meiotic MIC under conditions where either Rib1 was absent, or the zinc-ribbon was mutated. As illustrated in Fig. 6C and D, Emit3’s enrichment at scnRNA production regions was markedly reduced (but not abolished) in both rib1Δ and Emit3-CS cells, consistent with the cytological evidence of Emit3 re-distribution in these cells (Figs 2F and 5G, and Supplementary Fig. S4D). In summary, these results strongly imply that Emit3 preferentially associates with scnRNA production regions.
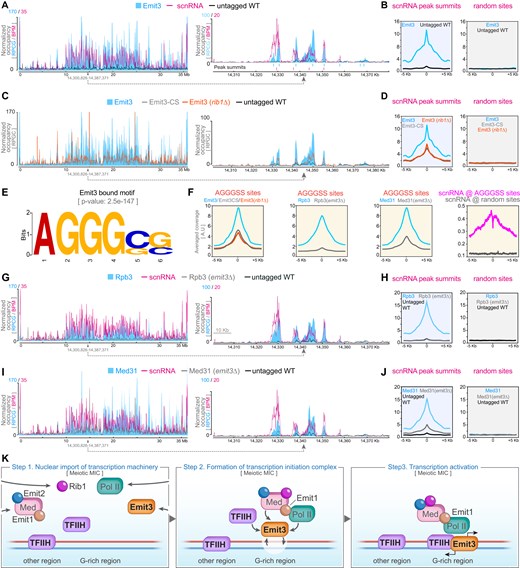
Chromatin occupancy analysis revealed that Emit3 preferentially associates with meiotic scnRNA production regions. (A) Occupancy of Emit3 and the abundance profile of scnRNAs on the meiotic MIC chromosome 2 (left panel) and a representative region of chromosome 2 (right panel). Blue vertical lines indicate Emit3-HA peak summits, while magenta vertical lines mark scnRNA peak summits. BPM: Bins per million mapped reads. RPCG: Reads per genome coverage. (B) Profile visualization of Emit3 CUT&Tag reads distribution around scnRNA peak summits and an equivalent number of random sites in the MIC. (C) Chromatin occupancies of Emit3 (in WT and rib1Δ cells) and Emit3-CS (with a mutated zinc-ribbon) on the MIC chromosome 2. (D) Profile visualization of CUT&Tag reads distribution around the summit of scnRNA peaks, compared to an equivalent number of random sites in the MIC. (E) A consensus motif identified from the Emit3-bound sequences. (F) Profile visualization of CUT&Tag reads for Emit3 (in WT and rib1Δ cells), Emit3-CS, Rpb3 (in WT and emit3Δ cells), and Med31 (in WT and emit3Δ cells), alongside meiotic scnRNA reads. The data are centered around MIC sites containing the Emit3-bound AGGGSS motif. (G) Chromatin occupancies of Rpb3 (in WT and emit3Δ cells) on the MIC chromosome 2. (H) Profile visualization of Rpb3 CUT&Tag reads (in WT and emit3Δ cells) around scnRNA peak summits and an equivalent number of random sites in the MIC. (I) Chromatin occupancies of Med31 (in WT and emit3Δ cells) on the MIC chromosome 2. (J) Profile visualization of Med31 CUT&Tag reads (in WT and emit3Δ cells) around scnRNA peak summits and an equivalent number of random sites in the MIC. (K) Schematic summary of the MIC ncRNA transcription activation mechanism. Pol II, Med, and their regulators are imported to the MIC during meiosis. Subsequently, Emit3 mediates the recruitment of the Pol II transcription machinery to the G-rich regions of meiotic MIC chromatin. The engagement of Emit1, Emit2, Rib1, Med, and possibly also TFIIH stabilizes the transcription initiation complex and activates ncRNA transcription.
Given that conventional TFIIB in other organisms binds to consensus DNA motifs within core promoters, we investigated whether Emit3-bound DNA sequences exhibit any distinct sequence features. To explore this, we selected the top 10% of the most enriched Emit3-bound DNA sequences (corresponding to 800 sequences) for motif identification. This analysis identified a significantly enriched consensus motif, AGGGSS (where S represents C or G), within these sequences (Fig. 6E, P-value: 2.5e-147). This motif does not share sequence similarity with the humans TFIIB recognition elements [86], consistent with the divergence between Emit3 and human TFIIB at the BRE binding regions (Supplementary Fig. S1B). Further computational analysis revealed 19 006 AGGGSS motif-containing sites in the MIC genome. These sites are preferentially located in regions with abundant scnRNA reads (Supplementary Fig. S8A). On average, the motif was identified with a frequency of 2.4 instances per region. Additionally, Emit3 CUT&Tag reads exhibit strong enrichment at and around these sites (Fig. 6F and Supplementary Fig. S8B). Such enrichment was notably reduced in rib1Δ as well as Emit3-CS cells. These findings suggest that Emit3 preferentially associates with the AGGGSS motif, and that Rib1 (or processes associated with its presence) and the zinc-ribbon domainfacilitate this interaction.
In addition to Emit3, we examined Pol II and Med occupancies in WT and emit3Δ meiotic MICs. As shown in Fig. 6G–J, both Rpb3-HA and Med31-HA were highly enriched at regions with abundant scnRNA in WT meiotic nuclei, indicating effective CUT&Tag performance. Remarkably, both chromosome-wide and regional coverage analyses, as well as coverage profile analysis, revealed that the Rpb3 CUT&Tag signal was reduced to background levels in emit3Δ cells (Fig. 6H). On the other hand, the Med31 CUT&Tag signal was reduced, but not abolished in response to the EMIT3 deletion (Fig. 6J). Altogether, these chromatin profiling data revealed that the Pol II transcription machinery preferentially occupies the MIC ncRNA production regions, and this association is dependent on Emit3.
We further examined the distribution of scnRNA reads at sites with the AGGGSS motif. Their distribution at equivalent numbers of random sites was also investigated for comparison. As illustrated in Fig. 6G and Supplementary Fig. S8B, scnRNA reads were preferentially accumulated at sites with this motif over random sites. Notably, 60.4% of scnRNA peaks (median length: 2.82 kb) contained at least one AGGGSS motif. An enrichment analysis using the Fisher’s exact test confirmed a significant enrichment of the AGGGSS motif within scnRNA peaks (P-value < 0.001). It is worth noting that, although scnRNA reads were preferentially accumulated around the AGGGSS motif, they were less enriched at the motif and its closely proximal regions (Fig. 6F and Supplementary Fig. S8C). Given that the AGGGSS motif is bound by the TFIIB-like Emit3 and Pol II transcription machinery (Fig. 6F) and transcript termini are likely not fully represented in scnRNAs [87], the twin peaked pattern of scnRNA reads at AGGGSS motifs may indicate these are sites for transcription initiation. Additionally, the nearly symmetric distribution of scnRNA reads around the motif suggests that transcription at these sites is likely bidirectional, which is also consistent with a previous investigation of ncRNA transcription at a specific MIC locus [27].
Overall, these data suggest that Emit3 preferentially binds to ncRNA transcription regions within meiotic chromatin, likely at transcription initiation sites characterized by a G-rich motif. Furthermore, Pol II and Med appear to depend on Emit3 for recruitment to, or stabilization at these ncRNA transcription regions of meiotic chromatin, reflecting Emit3’s analogous role to the classical TFIIB in recruiting Pol II to the core-promoters (Fig. 6K).
Discussion
A specialized TFIIB-like protein is required for noncanonical Pol II-dependent transcription
In this study, we identified a formerly uncharacterized TFIIB-like protein, Emit3, whose disruption led to a complete loss of MIC ncRNA transcription, resulting in abnormal transposon retention in the new MAC and progeny cell death (Figs 1 and 2, and Supplementary Table S3). Although the phenotype of emit3Δ is similar to that of emit1Δ and emit2Δ with respect to defective MIC ncRNA transcription, detailed analyses demonstrated that Emit3 promotes the MIC ncRNA transcription through a distinct mechanism. Emit1 and Emit2 are associated with the Med and facilitate transcription by enabling the Med to localize to the meiotic MIC [10]. In contrast, while Emit3 also localizes to the meiotic MIC, it does not obviously interact with the Med (Supplementary Table S4), which can still localize to the meiotic MIC in the absence of Emit3 (Figs 4E, and 6I and J). However, Emit3 deletion results in a significant reduction in Med association with MIC chromatin. Remarkably, although Pol II can localize to the meiotic MIC independently of Emit3, its association with MIC chromatin is nearly abolished in emit3Δ mutants (Figs 4B, and 6G and H). Similarly, Emit1, which also interacts with Pol II subunits, depends on Emit3 for its association with MIC chromatin (Fig. 3A). In contrast, while Emit1, Emit2, and Rib1 are required for the biased localization of Emit3, Pol II, and Med in the meiotic MIC (Fig. 2F) [10], the chromatin association of Emit3, Pol II, and Med occurs independently of Emit1, Emit2, and Rib1 (Fig. 2F) [10]. Collectively, these findings suggest that Emit3 plays a fundamental role in activating the MIC ncRNA transcription by promoting the engagement of Pol II and Med with the meiotic MIC chromatin. In contrast, Emit1, Emit2, and Rib1 interact with the Med, playing a crucial role in the activation and facilitation of biased transcription at TE-rich regions (Fig. 6K).
Based on sequence similarity analysis using PSI-BLASTP, Emit3 homologs were identified exclusively in Tetrahymena species (Supplementary Table S6). Nevertheless, Emit3 bears structural similarities to TFIIB (Fig. 1D and Supplementary Fig. S1), a GTF essential for recruiting Pol II to the core promoter through its zinc-ribbon domain [88]. Consistently, a mutation in Emit3’s zinc-ribbon domain resulted in a chromatin-binding defect for Pol II similar to that observed in emit3Δ mutants (Fig. 5H). This suggests that, akin to canonical TFIIB, Emit3 also relies on its zinc-ribbon to recruit Pol II to MIC chromatin. However, unlike canonical TFIIB, Emit3 lacks several conserved structural elements in the linker region downstream of the zinc ribbon (Supplementary Fig. S1C). Notably, it lacks most of the B-reader element, which, while not critical for the Pol II pre-initiation complex formation and transcription, is important for transcription start site selection in budding yeasts [78]. Interestingly, in the parasitic protist Trypanosoma brucei, transcription is often bidirectional and initiated from multiple sites within a broad region. This was suggested to be due to the lack of a conserved B-reader element and other structural features in T. brucei TFIIB, which are typically required for selecting transcription start sites and determining transcriptional directionality [89–94]. Thus, while the presence of the B-reader element in the MAC-specific TFIIB-like protein (i.e. Tfiibl1, Fig. 2C, Supplementary Figs S1C and 4A) may ensure transcription initiation at discrete sites [95], its absence in Emit3 might contribute to the bidirectional initiation of transcription at variable sites in the meiotic MIC[27, 33].
The ncRNA production regions in the meiotic nucleus are characterized by a G-rich motif
It was reported that MIC ncRNA transcription preferentially occurs at A-type IESs [32, 33]. A prior study excluded transgenerational epigenetic regulation as a determinant for transcription site selection and proposed that transcription at these A-type IESs might be determined by an unidentified feature(s) within or near these sequences [73]. This study shows that Emit3 preferentially binds to a G-rich AGGGSS motif (Fig. 6E). Interestingly, while meiotic scnRNAs accumulate around this motif, they are less enriched at the motif itself. This suggests that Emit3 binds to the G-rich motif either directly or indirectly and initiates transcription bidirectionally from this motif. Indeed, 60.4% of prominent scnRNA production regions feature this motif. However, since 36.71% of AGGGSS motif-containing regions express scnRNA at a very low level (i.e. scnRNA reads <10, Supplementary Fig. S8B), additional unidentified features, such as additional sequence motifs, higher-order DNA configuration, or epigenetic nucleosomal features, may help to determine MIC ncRNA transcription initiation sites.
Although a G-rich motif has not been previously linked to transcription initiation in Tetrahymena, earlier studies have identified some IESs flanked by the AAAAAGGGGG (A5G5) motif. This motif has been shown to be critical for the correct elimination of these IESs [96–98]. Notably, a mutation of the G-rich track of this motif completely abolished elimination of these IESs [96]. Together with our present data we propose that Tetrahymena utilizes such G-rich tracks both to promote MIC ncRNA transcription and to guide elimination of IESs in the new MAC. It is worth noting that the G-rich track bears a strong resemblance to the Tetrahymena telomeric repeat sequence TTGGGG, and IES elimination occurs parallel to the de novo addition of telomeric repeats to the broken chromosomes in the new MAC [99], G-rich tracks could originate from the de novo addition of telomeric repeats to excised IESs or transposons, which then integrated into the MIC genome. Indeed, perfect telomeric repeats have been identified at the ends of transposon-like elements in the MIC genomes of Tetrahymena as well as another ciliate, Oxytricha trifallax [100, 101]. Over time, internal telomeric repeats may be rapidly degenerated, as they pose significant challenges to DNA replication and repair processes [102]. Consequently, only divergent G-rich tracks might be left behind that promote MIC ncRNA transcription and thus facilitate elimination of associated transposons or transposon remnants from the new MAC. Further investigation revealed that the association of Emit3 with the AGGGSS motif is partially dependent on Rib1 (Figs 2F, and 6C and D), a known MIC transcription regulator essential for directing Pol II to TE-rich regions [10]. Moreover, interdependency analyses of localization revealed that Emit3, along with other MIC ncRNA regulators, are mutually dependent for achieving a biased accumulation at scnRNA production regions (Figs 2F, and 3A–C). This suggests the presence of a positive feedback mechanism that enhances the accumulation of ncRNA regulators at transcriptionallyactive sites.
The involvement of Emit3, a specialized GTF (TFIIB), in ncRNA transcription from transposons parallels the recruitment of a specialized subunit of the GTF TFIIA in ncRNA transcription from TE-derived sequences in Drosophila, which depends on both G-rich motifs and H3K9me3 modifications [103–105]. Our findings highlight the shared mechanisms but also the evolutionary diversity in strategies employed to facilitate Pol II-driven ncRNA transcription from otherwise silenced TEs.
Emit3 may rely on the TFIIH complex to activate ncRNA transcription
TFIIB forms a complex network with other transcription factors to initiate Pol II-dependent transcription. Similarly, Emit3 is associated with a broad range of interacting proteins, as evidenced by its coimmunoprecipitation with nearly all subunits of the TFIIH complex (Supplementary Table S4). This includes three TFIIH orthologs (P34, P44, and P52), an Xpb paralog (Xpb2), and a divergent P62-like protein (P62L). Given that Tetrahymena possesses two Xpb paralogs (Supplementary Table S5), Xpb2 is likely the specialized helicase component of TFIIH responsible for promoting transcription initiation, at least in the MIC. Xpd is another helicase subunit of the TFIIH complex. It has an ortholog in Tetrahymena (Xpd1, Fig. 5F). However, Xpd1 was not found to interact with Emit3. Moreover, Xpd1 displays a distinct localization pattern in the meiotic MIC compared to the other Emit3-associated TFIIH subunits (Fig. 5A and C, and Supplementary Fig. S6). These observations suggest that Xpd1 may not play a role in promoting MIC ncRNA transcription. However, given the presence of many helicase-domain-containing proteins in Tetrahymena, our data do not rule out the possibility that a divergent helicase-domain-containing Xpd-like helicase might be involved in activating the MIC ncRNA transcription.
The accumulation of TFIIH components at transcriptionally active MIC chromatin was disrupted in the absence of Emit3 (Fig. 5C and D, and Supplementary Fig. S6). This suggests that Emit3 is crucial for recruiting TFIIH components to the MIC chromatin. TFIIH plays a direct role in promoter opening during transcription initiation in othereukaryotes [106], so the disrupted localization of TFIIH in emit3Δ mutants may impede the stable engagement of Pol II. Because TFIIH is also expected to be involved in mRNAtranscription in the MAC, future studies should explore this hypothesis genetically by developing a strategy to inhibit TFIIH activity specifically in the meiotic MIC, for instance, using a controllable, MIC-specific protein degradationtechnique.
Although Emit3 is essential for recruiting Pol II to MIC chromatin, our experiments did not detect a physical interaction between Emit3 and Pol II through immunoprecipitation. Notably, Tetrahymena Pol II lacks the conserved TFIIB-binding dock motif (Fig. 5B), suggesting that an additional intermediary factor may be required to bridge Emit3 and Pol II. Since EMIT3 was identified through a comprehensive classification of mRNA expression profiles as one of the genes coclassified with the known MIC ncRNA transcription regulators (e.g. EMIT1, EMIT2, and RIB1, Fig. 1C and Supplementary Table S2), further functional characterization of the other coclassified genes may help to identify the missing components necessary for thisinteraction.
So far, all known Tetrahymena MIC ncRNA transcription regulators are associated with the transcription initiation step. However, studies in Paramecium tetraurelia have demonstrated that developmental-stage-specific transcription elongation regulators are essential for its MIC transcription [102, 107–109], revealing that regulation can also occur during the elongation phase. The apparent differences in MIC transcription regulation strategies could arise from lineage-specific adaptations or incomplete characterization of regulatory factors, rather than fundamentally distinct strategies. Future identification of elongation regulators in Tetrahymena—or initiation regulators in Paramecium—may reconcile these differences and provide deeper insights into the evolution of MIC ncRNA transcriptional control inciliates.
Acknowledgements
We thank the Campbell lab (Max Perutz Labs, Vienna) for sharing reagents. Mass Spectrometry analyses were performed by the Max Perutz Labs Mass Spectrometry Facility using the VBCF instrument pool. We thank Markus Hartl and his team for their support in proteomics analysis. We thank Dr. Yongqiang Liu (Ocean University of China), Ms. Shirin Strohmeier, and Mr. Lucas Günther Scholl (both from the Max Perutz Labs) for their technical support. Moreover, we appreciate the computing resources provided by IEMB-1, a high-performance computing cluster operated by the Institute of Evolution & Marine Biodiversity. We thank the Equipment Sharing Platform managed by the Institute of Evolution & Marine Biodiversity for allowing us to use the high-speed centrifuge (XPN-100).
Author contributions: Conceptualization: M.T.; Data curation: X.C., M.T., Z.Z., and G.D.; Formal analysis: X.C., Z.Z., T.N., G.D., and M.T.; Funding acquisition: M.T., J.L., K.M., and G.D.; Investigation: X.C., M.T., J.L., Z.Z., X.W., C.A., and Y.L.; Methodology: X.C. and M.T.; Project administration: M.T.; Supervision: M.T.; Resources: M.T., J.L., K.M., and G.D.; Visualization: M.T., M.L., Z.Z., and X.C.; Writing – original draft: M.T.; Writing – review & editing: M.T., K.M., X.C., J.L., and G.D.
Supplementary data
Supplementary data is available at NAR online.
Conflict of interest
The authors declare no competing interests.
Funding
This work is supported by the Science & Technology Innovation Project of Laoshan Laboratory (LSKJ202203202), the National Natural Science Foundation of China (32370451), the Fundamental Research Funds for the Central Universities (202241003), the Natural Science Foundation of Shandong Province (ZR2022JQ13), and by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement (101024333) to M.T., the Austrian Science Fund (I5960-B2) to G.D., Labex EpiGenMed Advanced Grant from Agence Nationale de la Recherche (ANR-10-LABX-12-01) and Equipes 2022 Grant from Fondation pour la Recherche Médicale (EQU202203014651) to K.M., and the Austrian Science Fund (P31606-B20) to J.L. Funding to pay the Open Access publication charges for this article was provided by the Natural Science Foundation of Shandong Province.
Data availability
The detailed masss pectrometry procedures, along with the raw data, have been submitted to the ProteomeXchange Consortium and are available through the PRIDE repository under the dataset identifier PXD057568. Next generation sequencing data generated by this study can be retrieved from the Sequence Read Archive (PRJNA1182059). The small RNA-seq and CUT&Tag datasets can be visualized using the web-based Integrative Genomics Viewer (https://igv.org/app/), using files deposited in Zenodo (https://doi.org/10.5281/zenodo.14233432).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments