





Abstract
The ALADYN web server aligns pairs of protein structures by comparing their internal dynamics and detecting regions that sustain similar large-scale movements. The latter often accompany functional conformational changes in proteins and enzymes. The ALADYN dynamics-based alignment can therefore highlight functionally-oriented correspondences that could be more elusive to sequence- or structure-based comparisons. The ALADYN server takes the structure files of the two proteins as input. The optimal relative positioning of the molecules is found by maximizing the similarity of the pattern of structural fluctuations which are calculated via an elastic network model. The resulting alignment is presented via an interactive graphical Java applet and is accompanied by a number of quantitative indicators and downloadable data files. The ALADYN web server is freely accessible at the http://aladyn.escience-lab.org address.
INTRODUCTION
The characterization of proteins and enzymes is usually articulated along the logical cascade sequence  structure
structure  function. The current understanding of the connection between the various terms of this tripartite ladder has been much shaped by the availability of quantitative comparative schemes. Indeed, sequence and structure comparative (alignment) methods have been used to clarify the extent to which similarities at the level of primary sequence reverberate at the level of native conformation (1–5). The same methods, in addition, lend naturally to be used to classify proteins and to detect evolutionary relationships among them (6–11).
function. The current understanding of the connection between the various terms of this tripartite ladder has been much shaped by the availability of quantitative comparative schemes. Indeed, sequence and structure comparative (alignment) methods have been used to clarify the extent to which similarities at the level of primary sequence reverberate at the level of native conformation (1–5). The same methods, in addition, lend naturally to be used to classify proteins and to detect evolutionary relationships among them (6–11).
In recent years, computational schemes ranging from atomistic simulations to coarse-grained models (12–17) have aptly complemented single molecule experiments by showing that for several enzymes the native structure gives a specific imprinting to the molecule's internal dynamics. The latter, in turn, can directly impact on the functionality of many, though not all, enzymes by favoring the interconversion between biologically relevant conformers, such as the rest and catalytically potent forms.
Based on this perspective, valuable insight into the structure–function relationship was provided by investigations where the large-scale internal dynamics was compared for proteins with a substantial degree of structural similarity (12,18). The good spatial superposability of the proteins of interest was essential to identify their structurally equivalent amino acids, whose large-scale functional motion could be finally compared (12,19,20).
While this dynamics-oriented comparative scheme is valuable, the necessity to identify structurally equivalent pairs of amino acids prior to measuring their dynamical consistency rules out, a priori, the possibility to detect similar large-scale movements in proteins lacking an overall fold similarity. This limitation can be overcome by alignment strategies that are tolerant from the structural point of view and that directly promote the identification of common internal-dynamics patterns in two proteins. The so-called ‘dynamics-based alignment’ that was recently introduced by some of us (21) is a general quantitative method to perform such comparisons and was used to highlight pervasive funtional-oriented relationships between proteins that differ, according to the CATH classification (10), at the level of topology, and even class or architecture (21,22).
The dynamics-based alignment is now offered, after a major algorithmic redesign, in the form of a web server named ALADYN. With respect to the method originally formulated in (21), the alignment search implemented in ALADYN is more general and efficient. In particular, the constraint that segments of aligned amino acids in the two proteins had to have the same sequence order and directionality has been removed. A more computationally effective scoring function and stochastic optimization of the alignment have also been adopted. The resulting method is therefore much faster than its original formulation: an alignment of two proteins of 250 amino acids is typically returned in ∼1 min on modern workstations.
The online server allows users to submit freely (without registration) jobs that require up to 20 min of CPU time. The allowed CPU limit is sufficient to align two proteins of about 1000 amino acids. Stand-alone LINUX and MAC-OSX versions of the ALADYN executable are made freely available upon request.
MATERIALS AND METHODS
Background
The large-scale structural rearrangements that accompany or assist the biological function of several proteins and enzymes are known to occur along generalized directions corresponding to the lowest energy modes of the system. Due to the collective character of these modes, which entail the concerted displacement of several amino acids, it can be expected a priori that their salient features can be adequately captured using simplified, coarse-grained protein models, such as elastic networks (23–25).
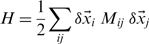
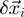 denotes the displacement of the i-th CA centroid from the native position. The lowest-energy modes correspond to the eigenvectors of
denotes the displacement of the i-th CA centroid from the native position. The lowest-energy modes correspond to the eigenvectors of 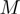 having the smallest, non-zero, eigenvalues. These modes dominate the equilibrium fluctuation dynamics of the system (26). In fact, indicating with
having the smallest, non-zero, eigenvalues. These modes dominate the equilibrium fluctuation dynamics of the system (26). In fact, indicating with 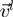 the
the 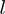 -th eigenvector of
-th eigenvector of 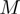 and with
and with 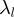 the associated eigenvalue one has that the mean square fluctuation (or mobility) of the i-th CA centroid is,
the associated eigenvalue one has that the mean square fluctuation (or mobility) of the i-th CA centroid is, 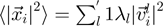 , where the prime indicates that the sum is restricted to non-zero eigenvalues and
, where the prime indicates that the sum is restricted to non-zero eigenvalues and  indicates the displacement of the
indicates the displacement of the 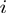 -th centroid entailed by the
-th centroid entailed by the 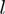 -th mode.
-th mode.Optimal dynamics-based alignment
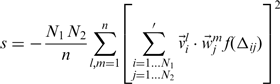
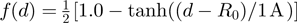 and the indices
and the indices 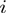 and
and  run respectively over the
run respectively over the  amino acids of the first protein and the
amino acids of the first protein and the 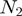 of the second one. The primed sum denotes that score contributions are further restricted to pairs where the square mobility of amino acids
of the second one. The primed sum denotes that score contributions are further restricted to pairs where the square mobility of amino acids 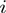 and
and 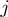 does not exceed by a factor of 4 the average one per amino acid. This restriction is introduced to avoid artifacts resulting from the presence of highly mobile loops or termini. The
does not exceed by a factor of 4 the average one per amino acid. This restriction is introduced to avoid artifacts resulting from the presence of highly mobile loops or termini. The 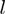 -th lowest energy eigenmodes of the first and second protein is indicated as
-th lowest energy eigenmodes of the first and second protein is indicated as 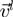 and
and 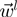 , respectively. As customary, only the
, respectively. As customary, only the  lowest energy modes are considered. The sigmoidal function
lowest energy modes are considered. The sigmoidal function 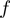 is used to restrict the score contribution to amino acid pairs
is used to restrict the score contribution to amino acid pairs 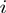 and
and 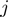 at a distance smaller than the interaction range
at a distance smaller than the interaction range 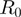 = 7 �: specifically, the effective amino acid distance,
= 7 �: specifically, the effective amino acid distance, 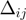 , used in the sigmoidal function, measures the spatial separation of the fragments
, used in the sigmoidal function, measures the spatial separation of the fragments 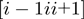 and
and 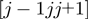 . A priori the latter could be matched with either the same or opposite sequence orientation. For the two cases, the segments distance is defined respectively as:
. A priori the latter could be matched with either the same or opposite sequence orientation. For the two cases, the segments distance is defined respectively as: 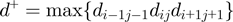 and
and 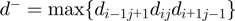 , with
, with 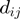 being the Euclidean distance of amino acids
being the Euclidean distance of amino acids 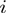 and
and 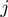 . The most appropriate sequence orientation is chosen a posteriori by setting
. The most appropriate sequence orientation is chosen a posteriori by setting 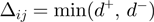 .
.The function in Equation 2 rewards those superpositions of the proteins having high scalar product among the fluctuation modes of every amino acid pair within the cutoff distance. This quantifies the consensus of the fluctuation of regions in spatial proximity.
The minimization of the score,  , over the relative rotations and translations of the two molecules of interest is carried similarly to the MISTRAL structural alignment method (27). The two proteins are first superposed by optimally aligning segments of up to 50 amino acids. This initial superposition is next optimized by minimizing
, over the relative rotations and translations of the two molecules of interest is carried similarly to the MISTRAL structural alignment method (27). The two proteins are first superposed by optimally aligning segments of up to 50 amino acids. This initial superposition is next optimized by minimizing  over the possible relative orientations of the molecules. The list of equivalent amino acids is finally computed using a ‘seed and grow’ search for matching segments (seed threshold equal to 4.5 � and tolerance equal to 5 �) (27,28).
over the possible relative orientations of the molecules. The list of equivalent amino acids is finally computed using a ‘seed and grow’ search for matching segments (seed threshold equal to 4.5 � and tolerance equal to 5 �) (27,28).
Finally, the statistical significance of the returned alignment is computed by comparing its score,  , against a reference probability distribution of alignment scores of unrelated protein pairs. This reference distribution was obtained starting from the representative protein data set of Sierk and Pearson (29). From this set, we randomly picked
, against a reference probability distribution of alignment scores of unrelated protein pairs. This reference distribution was obtained starting from the representative protein data set of Sierk and Pearson (29). From this set, we randomly picked  pairs of non-homologous and structurally dissimilar proteins (differing at the level of CATH topology) and computed the distribution of their alignment scores in dependence of the length of the longest protein of each pair. As customary (29), based on the high level of dissimilarity of these pairs, it is expected a priori that only a negligible fraction of the random alignments will correspond to true positive correspondences; and the distributions are used as ‘gold standard’ for other queries.
pairs of non-homologous and structurally dissimilar proteins (differing at the level of CATH topology) and computed the distribution of their alignment scores in dependence of the length of the longest protein of each pair. As customary (29), based on the high level of dissimilarity of these pairs, it is expected a priori that only a negligible fraction of the random alignments will correspond to true positive correspondences; and the distributions are used as ‘gold standard’ for other queries.
The tails of the length-regularized score distributions were found to be well-described by the extremal Gumbel statistics which was accordingly used to compute the statistical significance of a specific alignment. The latter is quantified by means of a P-value or, equivalently, through a  -score. The former is the probability that an alignment of unrelated proteins returns a score as high as the observed one, while the second measures by how many standard deviations the observed score exceeds the one expected for random alignments. Statistically significant matches are therefore associated to small P-values and large
-score. The former is the probability that an alignment of unrelated proteins returns a score as high as the observed one, while the second measures by how many standard deviations the observed score exceeds the one expected for random alignments. Statistically significant matches are therefore associated to small P-values and large  -scores.
-scores.
Integration of non-aligned degrees of freedom and RMSIP
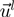 and
and 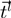 , the
, the 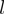 -th effective modes of the proteins, one defines
-th effective modes of the proteins, one defines 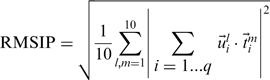
 labels the
labels the  corresponding amino acids. The RMSIP takes on the value 1 in case of perfect correspondence of the spaces, and 0 in case of their complete orthogonality. When the number of compared amino acids,
corresponding amino acids. The RMSIP takes on the value 1 in case of perfect correspondence of the spaces, and 0 in case of their complete orthogonality. When the number of compared amino acids,  , is of the order of 100 amino acids, RMSIP values equal to 0.7 or higher are typically deemed as statistically significant (30).
, is of the order of 100 amino acids, RMSIP values equal to 0.7 or higher are typically deemed as statistically significant (30).Finally, in addition to the RMSIP value, the root mean square distance, RMSD, of the matching amino acid pairs is also used to convey the quality of the alignment.
Web server: input and output
In the input form, users are asked to provide the two proteins to align: this can be done either by uploading PDB coordinate files or by entering their PDBids (and, optionally, the chain identifier). The algorithm's running time scales approximately proportionally to the product of the lengths of the input proteins. In fact, the time required for the alignment of two proteins of about 250 amino acids is typically <1 min on the modern multicore server that hosts ALADYN, while two proteins of about 500 amino acids are completed in ∼4 min. Clearly, the run time can vary depending on the number of jobs submitted at the same time.
Upon successful completion, users are finally directed to an interactive graphical representation of the superposed proteins, based on the Jmol (31) applet, which is complemented by a summary of the salient properties of the alignment, number of aligned amino acids, RMSIP, RMSD and the statistical significance conveyed by the  -score and P-value.
-score and P-value.
The applet controls can be used to visualize the matching regions and/or the matching modes ranked for decreasing mutual similarity. This ranking, which entails a redefinition of the basis of the low energy modes (and, as such, does not affect the alignment score nor the RMSIP), is carried out with the linear optimization procedure introduced in ref. (16).
The links provided at the bottom of the results page allow users to download data files containing all details of the alignment output, namely: the coordinate files of the optimally aligned structures, the list of corresponding amino acids, the corresponding ten lowest energy modes of the aligned amino acids and a VMD (32) state file for a convenient off-line visualization of the results.
RESULTS AND DISCUSSION
We discuss here two test cases in order to illustrate the performance of the ALADYN alignment tool, namely human β-secretase (BACE) versus HIV-1 protease and exonuclease III versus human adenovirus proteinase.
HIV-1 PR and β-secretase
The additional insight offered by the dynamics-based alignment with respect to ‘static’ alignment approaches is aptly illustrated by the comparison of HIV-1 PR (PDBid: 1aid) and human β-secretase (PDBid: 3hvgA). The two enzymes, which are both aspartic proteases, present major structural differences. In fact, HIV-1 PR is a 198-amino acid long homo dimer, and is almost entirely composed of β sheets. On the contrary, β-secretase is a monomeric enzyme consisting of 379 amino acids and rich in α helices. Despite the differences in symmetry, oligomeric state, length and secondary structure content, the two enzymes share several segments of the primary sequence and are hence believed to be evolutionarily related (33). In fact, they admit a partial, but significant, structural superposition: their DALIlite alignment (34) returns 94 corresponding residues with an associated RMSD of 3.4 �, while the MISTRAL alignment returns 128 equivalent amino acids at 2.4 � RMSD. In addition to the partial structural correspondence previous studies, based on atomistic molecular dynamics (MD) simulations, had highlighted the similarity of the low-energy modes of the two molecules (35,36).
The dynamics-based alignment returned by ALADYN is statistically significant, as the associated P-value is appreciably smaller than the conventional threshold of 0.05. and is fully consistent with the above-mentioned findings. The alignment consists of more than 140 amino acid pairs at an RMSD <4 �. The good correspondence of the modes is highlighted by the large RMSIP value of the matching modes, which is ∼0.8.
The functional relevance of the alignment returned by ALADYN is underscored by the following facts. First, the returned alignment superposes the catalytic dyads of the two enzymes. This is a non-trivial aspect in consideration that no information about the chemical composition (such as the primary sequence) was used. The second observation regards the consensus movements in the two proteins, which entail the modulation of the region accommodating the peptide chain to be cleaved. It is known that in order for the proteolytic reaction to occur, both BACE and HIV-1 PR must ‘stretch’ the substrate in a β-extended conformation (35,36), and the consensus motion captured by ALADYN (see Figure 1) is consistent with the required deformation (37).
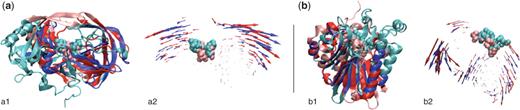
Examples of alignments returned by the ALADYN web server. The structural correspondences and the consistency of the fluctuation dynamics of the aligned regions are shown side-by-side for each of the test cases discussed in the Results section. (a) The alignment of HIV-1 protease (pink/red) and β-secretase (cyan/blue) are shown in subpanels a1 and a2. (b) The alignment of human adenovirus proteinase (pink/red) and exonuclease III (cyan/blue) are shown in subpanels b1 and b2. Aligned regions are shown with saturated colors (i.e. red and blue), while the active sites are highlighted using a Van der Waals representation.
The dynamics-based alignment therefore vividly illustrates the existence of a fundamental similarity underlying the internal dynamics of these enzymes, which is instrumental to produce analogous, functionally oriented deformation patterns in spite of the overall structural differences.
Exonuclease III and human adenovirus proteinase
Exonuclease III (PDB: 1ako) and the human adenovirus proteinase (PDB: 1avp) are not evolutionarily related and are structurally dissimilar at the CATH architecture level. Their structural alignment has a P-value >0.1 according to MISTRAL and, similarly, it is ruled out as ‘not significant’ by DALIlite.
Despite these differences, the enzymes process chemically-similar substrates. In fact, both exonuclease III and human adenovirus proteinase bind DNA (in double- and single-stranded forms, respectively). In the study of Zen et al. (21) the dynamics-based alignment of the enzymes was found to have a good statistical significance. As for the case of BACE and HIV-1 PR, the functional relevance of the dynamical correspondence was underscored by the fact that the known active sites of the proteins (38) were spatially superposed by the alignment and by the fact that the consensus motion was compatible with the expected functionally oriented structural changes (39,40).
All the above established results are reproduced by the ALADYN alignment that employs a more general search scheme than the method of Zen et al. (21) (on which is conceptually-based). As visible in Figure 1, the two proteins align over more than 90 amino acids, at an RMSD <4 �. The consistency of the dynamics of the aligned regions is high (RMSIP value ∼0.7). It is readily noticed that the alignment yields a good spatial overlap of the active sites of the two enzymes. In accordance with the previous findings (21), the latter are located in a region at the interface between two oppositely moving ‘domains’. As suggested for other enzymes (41), this characteristic ought to preserve the catalytic geometry at the active site, while facilitating the accommodation/processing of the substrate.
CONCLUSION
We have presented the ALADYN server that can be used to establish significant pairwise correspondences in proteins based on similarities of their large-scale internal dynamics, which expectedly assists or accompanies their biological functionality. The server is conceptually related to the dynamics-based alignment first introduced in (21). With respect to the original method, the ALADYN alignment scheme is both more general (being non-sequential) and faster and hence lends naturally to be interactively used through a web server.
The input required from the user is kept at a minimum and merely consists of the PDBid's of the proteins to be compared or, alternatively, of their structural coordinates (in PDB format). Alignments of proteins of up to 250 amino acids are typically completed in <1 min. The results are returned through a graphical interface based on the Jmol applet which allows users to interactively visualize the aligned regions and the associated large-scale motion (computed via an elastic network model). The graphical summary is accompanied by quantitative details about the quality and significance of the alignment. Further quantitative data, such as the list of corresponding amino acids and the deformations entailed by the low-energy modes of the matching regions, are provided as downloadable data files.
FUNDING
Democritos - Consiglio Nazionale delle Ricerche - Istituto Officina dei Materiali. Funding for open access charge: CUBENET, an HPC project sponsored by Friuli Venezia Giulia Region.
ACKNOWLEDGEMENTS
We are indebted to Vincenzo Carnevale, Henri Orland and Andrea Zen for valuable discussions.
Conflict of interest statement. None declared.

{kind=link}
Comments