





ABSTRACT
Structural information on interacting proteins is important for understanding life processes at the molecular level. Genome-wide docking database is an integrated resource for structural studies of protein–protein interactions on the genome scale, which combines the available experimental data with models obtained by docking techniques. Current database version (August 2009) contains 25 559 experimental and modeled 3D structures for 771 organisms spanned over the entire universe of life from viruses to humans. Data are organized in a relational database with user-friendly search interface allowing exploration of the database content by a number of parameters. Search results can be interactively previewed and downloaded as PDB-formatted files, along with the information relevant to the specified interactions. The resource is freely available at http://gwidd.bioinformatics.ku.edu.
INTRODUCTION
Function of proteins in the living cell is determined by their ability to interact with other biologically relevant molecules (other proteins, DNA, RNA, small ligand, etc.). Thus understanding mechanisms of these interactions is critically important for studying life processes at the molecular level. Genome sequencing provided vast amount of information on proteins, spanning the entire universe of life from viruses to the highest eukaryotic organisms. In the post-genomic era, the efforts focus on the function assignment of the sequenced proteins based on their three-dimensional (3D) structures and/or participation in interactions. Because of the limitations of the experimental techniques for structural characterization, computational methods play a vital role (1).
Success in recreating maps of interactions for specific organisms and/or specific biochemical pathways emphasize the need for large-scale modeling efforts to deliver 3D structures of the protein complexes. Computational methods for structural modeling of the protein–protein interactions (PPIs) historically started with ab initio (or template free) methods based on shape complementarity and were later supplemented by the constraints derived from statistical analysis of properties of known protein complexes or from the experimentally acquired additional biochemical/biophysical knowledge (2). Most of the existing docking servers (3) employ constrained-based template-free approach. Despite the significant progress in development of the template-free algorithms, their accuracy in the high-throughput applications is limited.
Accumulation of experimental data in the last decade have caused paradigm shift in 3D modeling of individual proteins from ab initio to template-based techniques. A similar trend is underway in structural modeling of protein complexes (protein docking). Recently, several groups assessed quality of the models produced by the homology/threading docking techniques where a protein complex is modeled based on similarity to another protein complex with the known structure (4–8). It was demonstrated that the majority of the homology-docking models are of acceptable and medium quality, according to the CAPRI criteria (3). It was estimated that the homology docking can account for a significant part (15–20%) of known PPI (7). Structural alignment techniques were also benchmarked on various sets of protein complexes (9,10).
Success in developing the high-throughput modeling techniques makes it feasible to create a long-needed comprehensive resource, which would reflect large-scale efforts in structural modeling of known protein complexes. Genome-wide docking database (GWIDD) provides annotated collection of experimental and modeled 3D structures of protein–protein complexes from the entire universe of life spanning from viruses to humans. The database provides user-friendly interface for searching and browsing database content and downloading experimental and modeled structures of protein complexes.
DATABASE CONTENT AND DESCRIPTION
Source of PP1s data
PPIs are imported to GWIDD from external sources specialized in collecting and curating PPI. Currently they include BIND (http://www.bind.ca) (11) and DIP (http://dip.doe-mbi.ucla.edu) (12,13) databases. These databases were chosen because their content is not restricted to a single genome or group of genomes like in many other PPI databases [e.g. different flavors of MINT (14) or MIPS (15)]. They are also regularly updated providing up-to-date pool of the initial data. The interactions are obtained through either high-throughput discovery methods or small-scale experiments and thus are of diverse reliability. However, at the current stage, evaluation of credibility of the PPI data is outside the scope of GWIDD.
Current content of the database
The ultimate goal of the GWIDD resource is to provide the 3D structures for all known PPI. At the current stage, the following steps are taken toward this goal. First, if an interaction is found in protein data bank (PDB), this structure is used and no modeling is performed. Otherwise, a search for a pair of homologous sequences from a complex with known structure is performed and the model is build by homology docking (6,7). We used earlier-described criteria for statistical significance of the sequence alignments (7) with an additional requirement that both alignments contain at least 80% of the target sequences. In the future, for the interactions not covered by these two steps, we will use other docking methods (e.g. structural alignment, template free docking), which will be incorporated in the upcoming GWIDD releases. However, even the current limited modeling approach provides structures for 14 635 PPI, which together with the available non-redundant X-ray structures (10 924) constitutes >20% of the currently known PPI. Summary of the GWIDD content is provided in Table 1. As of August 2009, GWIDD contained 126 897 binary interactions, involving 43 976 proteins from 771 different organisms spanning the entire universe of life (Table 1). Among those, 6079 entries are either cross-organism interactions or do not have organism annotation in the source data. Thus they are not present in Table 1, although will appear in search results. The distribution of available structures (X-ray and modeled) is shown in Figure 1 for 10 organisms with the largest numbers of structured GWIDD entries. The database is automatically updated every half year.
Distribution of GWIDD entries for various categories of living organismsa
| Living organisms | Number of speciesb | Number of interactionsc | Number of model structuresd | Number of experimental structures |
| Archaea | 41 | 1128 | 369 | 723 |
| Bacteria | 288 | 13 871 | 3183 | 5488 |
| Lower eukaryotae | 80 | 29 289 | 2058 | 811 |
| Plants | 79 | 2055 | 365 | 399 |
| Animals | 136 | 72 395 | 7858 | 2746 |
| Viruses | 147 | 2080 | 802 | 757 |
| Total | 771 | 120 818 | 14 635 | 10 924 |
| Living organisms | Number of speciesb | Number of interactionsc | Number of model structuresd | Number of experimental structures |
| Archaea | 41 | 1128 | 369 | 723 |
| Bacteria | 288 | 13 871 | 3183 | 5488 |
| Lower eukaryotae | 80 | 29 289 | 2058 | 811 |
| Plants | 79 | 2055 | 365 | 399 |
| Animals | 136 | 72 395 | 7858 | 2746 |
| Viruses | 147 | 2080 | 802 | 757 |
| Total | 771 | 120 818 | 14 635 | 10 924 |
The data is for protein–protein interactions where both partners are from the same organisms, except for the viruses where interactions are between a protein from the virus and a protein from the host organism.
Number of species for which at least one protein–protein interaction is present in DIP and BIND databases.
As in DIP and BIND, including interactions with no modeled structures.
Modeled by homology docking.
Includes primitive organisms and fungi.
Distribution of GWIDD entries for various categories of living organismsa
| Living organisms | Number of speciesb | Number of interactionsc | Number of model structuresd | Number of experimental structures |
| Archaea | 41 | 1128 | 369 | 723 |
| Bacteria | 288 | 13 871 | 3183 | 5488 |
| Lower eukaryotae | 80 | 29 289 | 2058 | 811 |
| Plants | 79 | 2055 | 365 | 399 |
| Animals | 136 | 72 395 | 7858 | 2746 |
| Viruses | 147 | 2080 | 802 | 757 |
| Total | 771 | 120 818 | 14 635 | 10 924 |
| Living organisms | Number of speciesb | Number of interactionsc | Number of model structuresd | Number of experimental structures |
| Archaea | 41 | 1128 | 369 | 723 |
| Bacteria | 288 | 13 871 | 3183 | 5488 |
| Lower eukaryotae | 80 | 29 289 | 2058 | 811 |
| Plants | 79 | 2055 | 365 | 399 |
| Animals | 136 | 72 395 | 7858 | 2746 |
| Viruses | 147 | 2080 | 802 | 757 |
| Total | 771 | 120 818 | 14 635 | 10 924 |
The data is for protein–protein interactions where both partners are from the same organisms, except for the viruses where interactions are between a protein from the virus and a protein from the host organism.
Number of species for which at least one protein–protein interaction is present in DIP and BIND databases.
As in DIP and BIND, including interactions with no modeled structures.
Modeled by homology docking.
Includes primitive organisms and fungi.
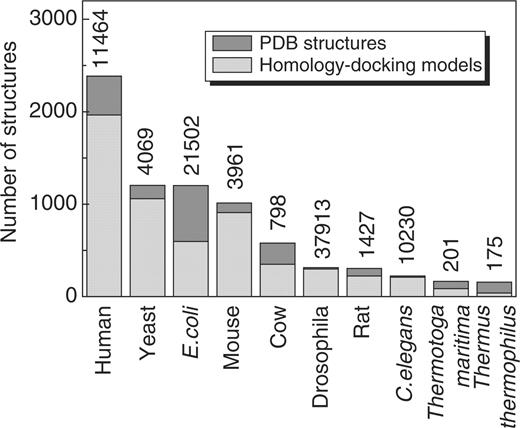
Number of experimental structures (dark gray bars) and structures modeled by homology docking (light gray bars) for 10 organisms with the largest structural coverage in GWIDD. Numbers at the bars indicate the total amount of non-identical interactions, including those with no structure, in DIP and BIND databases.
Implementation of the database and its user interface
The data from the external source databases have different formats and different levels of details. Thus such data are unified into a single dataset of PPI, removing redundancy and retaining common data fields for all the sources. Due to the large amount of data and complex data dependency as well as complex query requirement, all interaction data are stored in a relational database, except for large files, such as PDB ones, which are stored directly in the file system and are linked from the relational database. Implementation of the web interface is based on LAPP (Linux-Apache-PostgreSQL-PHP) software stack. Web user interface is built using PHP and jQuery library, where PHP is for web presentation and logic as well as back-end database access. jQuery is responsible for AJAX and other JavaScript-based dynamic features. Visualization of protein structures is implemented utilizing Jmol (http://www.jmol.org) technology. Homology docking was performed by NEST (16), BLAST (17) and in-house profile-to-profile alignment program used previously for the benchmarking of homology docking (7). The above parts are joined by a set of Python scripts.
User interface description
The database can be freely accessed at http://gwidd.bioinformatics.ku.edu. The default option offered to users is search of the database by keywords related to a single interaction partner (‘Protein A’ part in Figure 2A). Other search options are available by clicking tabs ‘Sequence’ (explicit input or upload of sequence in the FASTA format) or ‘Structure’ (upload of a PDB-format file). When searching by keywords, user can either enter any keyword in the protein description (name of organism, cellular location, biological function, etc.) or choose from the series of drop-down menus containing lists of all organisms currently in GWIDD. By repeating the selection with the box ‘Add another organism to the list’ checked, user can choose several organisms. When the box is unchecked, the search will clear the list of previously selected organisms. Also, in each submenu, user can select all listed organisms by a single click on the top ‘Select All’ position. An option to search by standard taxonomy ID with link to taxonomy database http://www.uniprot.org is also provided for convenience. Search results for the ‘Keyword’ tab can be, for example, all PPI related to a certain pathway (defined by the keyword) or all interactions within certain organism or group of organisms. Search results for the ‘Sequence’ and ‘Structure’ tabs contain all interactions with the input sequence as one of the interaction partners (in the case of input PDB file the sequence extracted from the SEQRES tags or, if the SEQRES part is not available, from ATOM tags for the Cα atoms). The amino acid sequences from different sources can differ in length even for the same protein (e.g. due to unresolved residues in the X-ray structure). Thus advanced options are provided in the sequence search parts. An example of search by organism and its results is shown in Figure 2.
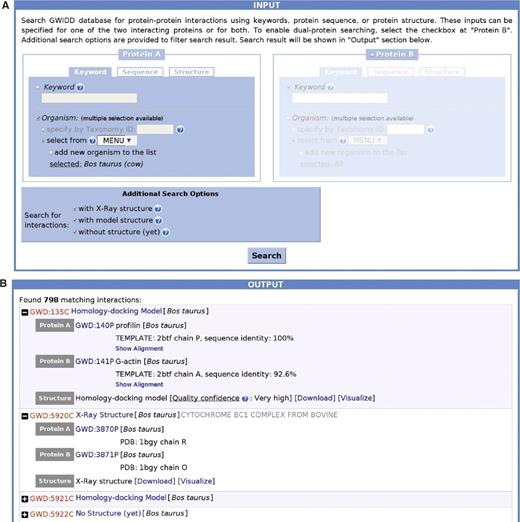
Example of a search by organism (A) and the results of this search (B).
If information related to the other interaction partner is also known, user can enable the second part of the search interface (‘Protein B,’ see Figure 2A) by checking the corresponding box and input the information similarly to ‘Protein A’. In addition, search results can be filtered by the availability of different types of GWIDD entries (experimental structures, modeled structures or interactions with no structures). Online help is provided in pop-up windows (question marks inside blue circles, see Figure 2). Search results screen (Figure 2B) displays all interactions in the database satisfying the input search criteria in the form of expandable list of GWIDD interaction IDs with minimum additional information. The expanded item in the list contains the name and the GWIDD IDs of the interacting partners along with information on the type of 3D structure available for this interaction (if applicable). For the homology-docking models, the alignments used to build the model are provided and the model quality is assessed by the sequence identity criteria (5). For the available structures, links are provided to download the PDB-format file along with the text file containing relevant information, as well as to the visualization screen where the structure is displayed in colored-by-chain space-filled interactive representation.
COMPARISON TO OTHER EXISTING RESOURCES
There are several resources available that are similar in spirit (genome-wide approach to PPI) to the GWIDD resource. Michigan molecular interactions (MiMIs), database (http://mimi.ncibi.org) (18) provides one cohesive view of molecules found in several popular interaction databases, including BIND, HPRD, IntAct, GRID and others, with complementary or conflicting data among the sites highlighted. POINT (http://point.bioinformatics.tw) (19) is a functional database for prediction of the human protein–protein interactome based on available orthologous interactome datasets with the emphasis on extraction of mouse, fruit fly, worm and yeast PPI datasets from DIP, followed by their conversion to predicted human interactome. 3D-GENOMICS (http://www.sbg.bio.ic.ac.uk/3dgenomics) (20) provides structural annotations for proteins from sequenced genomes and in August 2003 included data for 93 proteomes. NCBI Inferred Biomolecular Interactions Server (IBIS, http://www.ncbi.nlm.nih.gov/Structure/ibis/ibis.cgi) reports physical interactions observed in experimentally determined structures for sequences homologous to the input amino acid sequence, thus inferring interacting partners and binding sites. However, none of the above resources provide single integrated and searchable pool of experimental and modeled 3D structures for all genomes for which at least one PPI is annotated. Recently developed ProtInfo PPC server (http://protinfo.compbio.washington.edu/ppc) (21) provides model structures for user's supplied sequences, but lacks the annotated database of 3D structures.
FUTURE DEVELOPMENT
The major direction in the future development of GWIDD is expanding the pool of available structures modeled by other modeling techniques, such as docking by structural alignment (to be submitted) and template-free docking by GRAMM methodology (22–24). To assess the applicability of these methods to the high-throughput, genome-wide modeling, large-scale benchmarking is currently underway. New sources of PPI will be incorporated as they become available.
ACKNOWLEDGEMENTS
Andrey Tovchigrechko and Tatiana Baronova made important contributions to GWIDD project at the earlier stages of development.
FUNDING
Funding for open access charge: National Institutes of Health (R01 GM074255).
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
Comments