





Abstract
Chromatin immunoprecipitation (ChIP) has been used to detect binding of DNA-binding proteins to sites in nuclear and mitochondrial genomes. Here, we describe a method for detecting protein-binding sites on chloroplast DNA, using modifications to the nuclear ChIP procedures. The method was developed using the lac operator ( lacO )/ lac repressor (LacI) system from Escherichia coli . The lacO sequences were integrated into a single site between the rbcL and accD genes in tobacco plastid DNA and homoplasmic transplastomic plants were crossed with transgenic tobacco plants expressing a nuclear-encoded plastid-targeted GFP-LacI fusion protein. In the progeny, the GFP-LacI fusion protein could be visualized in living tissues using confocal microscopy, and was found to co-localize with plastid nucleoids. Isolated chloroplasts from the lacO /GFP-LacI plants were lysed, treated with micrococcal nuclease to digest the DNA to fragments of ∼600 bp and incubated with antibodies to GFP and protein A-Sepharose. PCR analysis on DNA extracted from the immunoprecipitate demonstrated IPTG (isopropylthiogalactoside)-sensitive binding of GFP-LacI to lacO . Binding of GFP-LacI to endogenous sites in plastid DNA showing sequence similarity to lacO was also detected, but required reversible cross-linking with formaldehyde. This may provide a general method for the detection of binding sites on plastid DNA for specific proteins.
INTRODUCTION
Chromatin immunoprecipitation (ChIP) has been widely used as a means of identifying regions of DNA interacting with specific proteins in nuclei of many organisms ( 1–5 ). ChIP has also been used to examine interactions of proteins with mitochondrial DNA in yeast and in a human cell line ( 6–8 ). So far there has been only one report of the use of ChIP to investigate protein–DNA interactions in chloroplasts ( 9 ), but the WHY1 protein under investigation bound to all regions of maize chloroplast DNA and sequence-specific protein-binding sites were not detected ( 9 ). Several specific protein–DNA interactions in chloroplasts have been described ( 10–17 ), but these interactions have either been based on experiments in vitro , using footprinting ( 10 , 11 ), electrophoretic mobility shift assays ( 10 , 11 , 13 ) or South–western blotting ( 12 ) or on functional effects of gene knockouts or silencing the expression of genes encoding DNA-binding proteins in vivo ( 15–17 ).
We have attempted to develop a generic method for investigating protein–DNA interactions in chloroplasts, based on the protocols developed for ChIP with nuclear and mitochondrial genes. Unfortunately, none of the specific DNA–protein interaction systems so far described for chloroplasts seemed sufficiently robust to form the basis for developing a ChIP protocol, because of insufficient knowledge of the affinity of the protein for the binding site. It was decided to introduce a very well-characterized DNA–protein interaction system, the lac operator ( lacO )/ lac repressor (LacI) system from Escherichia coli ( 18 ), into tobacco chloroplasts to provide plant material for the development of a ChIP protocol. The lacO /LacI system has previously been shown to provide IPTG-inducible regulation of gene expression in tobacco chloroplasts ( 19 ).
An advantage of the lacO /LacI system is the ability of GFP-tagged fusion proteins of LacI to bind to lacO sites in a variety of organisms in vivo . In Chinese hamster ovary and yeast cells, a GFP-LacI fusion protein has been employed to detect tandem lacO sequences inserted at various places around the genome thereby enabling observations of operator-tagged chromosomal DNA in vivo ( 20 , 21 ). In E. coli , the technique has allowed investigations of DNA segregation ( 22 ). In higher plants, Kato and Lam ( 23 ) used lacO /GFP-LacI to tag chromosomes in transgenic Arabidopsis thaliana and thus enable visualization of genomic loci, nucleolar locations and chromatin organization. Similarly Matzke et al. ( 24 ) reported the successful tagging of transgenic loci in A. thaliana with lacO and a GFP-LacI fusion protein and subsequent observation of fluorescent spots in interphase nuclei of various cell types. The use of GFP-tagged LacI in chloroplasts should provide additional visual evidence for its interaction with plastid DNA in vivo .
Plastid DNA appears as tightly coiled particulate structures, known as nucleoids ( 25 ), which are composed of DNA-binding proteins, plastid DNA and uncharacterized RNA ( 26 ). Nucleoids can be visualized by fluorescence microscopy after staining with a DNA-specific fluorochrome, such as 4′,6-diamidino-2-phenylindole (DAPI; 27). Plastid nucleoids have also been visualized by fusing GFP to the plastid envelope DNA-binding (PEND) protein ( 28 ), providing a major advantage over DAPI in that plastid DNA can be located in living, intact tissues, including non-chlorophyll-containing tissues such as roots and flowers. This article describes the development of a ChIP protocol for the detection of GFP-LacI binding to lacO and related sequences in tobacco chloroplasts. It may provide a generic method for examining protein–DNA interactions in chloroplasts.
MATERIALS AND METHODS
Plant material
Plants of Nicotiana tabacum cv ‘Petite Havana’ and experimental lines used for DNA immunoprecipitation were grown axenically as described in Birch-Machin et al. ( 29 ). Plants were grown under 16-h light/8-h dark at 23°C with light intensity of 50 µmol m −2 s −1 . Young leaves ∼2.5-cm long from 8–12-week-old plants of Petite Havana were used for particle bombardment while similar-sized leaves from lacO , GFP-LacI, lacO/ GFP-LacI and P rrn::gfp lines were used for DNA immunoprecipitation. Fully expanded 5-cm leaves of Petite Havana from axenically grown plants maintained by monthly subculture were used for nuclear transformation. Experimental lines used for crosses and seed production were grown to maturity in a greenhouse with supplementary lighting from 400-W high-pressure sodium lamps (TRS400L Grolux 40; http://www.sylvania.com ) to give a 16-h day length at a temperature of 23°C.
Gene constructs
Constructs for plastid transformation containing lacO repeats were produced in pZS-JH1, a derivative of pZS197 ( 30 ) containing a polylinker in the unique Bsu36I site ( 31 ). A plasmid containing the chimeric gene construct P psbA-gfp in pZS-JH1 ( 31 ) was digested with HindIII and SmaI to remove P psbA-gfp. lacO repeat sequences of 1 or ∼2.5 kb were excised from plasmids pAFS52 or pAFS59 ( 21 ) by restriction digestion with HindIII and SmaI, and ligated into the corresponding restriction sites of digested pZS-JH1.
Constructs encoding chloroplast-targeted protein fusions of LacI and GFP were assembled in pUC18 and transferred to pBIN19 ( 32 ) for nuclear transformation. For the gfp-lacI construct, the lacI coding sequence ( 33 ) was PCR-amplified from E. coli XL1-Blue (Stratagene, http://www.stratagene.com ) using forward (5′-ACG GGATCC GTGAAACCAGTAACGTTATACGATGTC) and reverse (5′-TTATAA GAGCTC TCACTGCCCGCTTTCCAGTCGGG) primers, introducing a stop codon and inserting BamHI and SacI restriction sites (underlined) at the 5′- and 3′-ends, respectively. The gfp coding region was excised from pUC Rbc STP- gfp containing the CaMV 35S promoter, a region encoding the pea Rubisco small subunit transit peptide ( Rbc STP), gfp and nos terminator, by digestion with BamHI and SacI. The amplified fragment containing the lacI coding region was then inserted between the BamHI and SacI sites in digested pUC Rbc STP- gfp to produce pUC Rbc STP- lacI . The gfp coding region was amplified from pUC Rbc STP- gfp using forward (5′-TGA TCTAGA CCTAAAGGAGAAGAACTTTTC) and reverse (5′-ATAT GGATCC TTTGTATAGTTCATCCATGCC) primers resulting in the removal of the stop codon and introduction of XbaI and BamHI restriction sites (underlined) at the 5′- and 3′-ends, respectively. This fragment was inserted into XbaI and BamHI-digested pUC Rbc STP- lacI directly upstream of lacI to produce pUC Rbc STP- gfp-lacI .
For the lacI::gfp construct, the lacI coding sequence was PCR amplified from E. coli XL1-Blue using forward (5′-AG TCTAGA GTGAAACCAGTAACGTTATACGATGTC) and reverse (5′-TTATTT AGGCCT CTGCCCGCTTTCCAGTCGGGAAAC) primers, omitting the lacI stop codon and introducing XbaI and StuI sites (underlined) at the 5′- and 3′-ends, respectively. The amplified fragment was inserted into XbaI and StuI sites located directly upstream of gfp in pUC RbcS TP- gfp , to produce pUC RbcS TP- lacI-gfp .
The two chimeric gene-fusion constructs in pUC18, consisting of the CaMV 35S promoter, regions encoding the RbcS transit peptide and the GFP fusion protein, and the nos terminator, were removed by digestion with HindIII and EcoRI and ligated into the HindIII and EcoRI sites of pBIN19 ( 32 ). The constructs in pBIN19 were then introduced into Agrobacterium tumefaciens strain LBA4404 ( 34 ) by electroporation ( 35 ).
Plant transformation
Plastid transformation of tobacco with plasmids containing 1 or ∼2.5 kb of lacO sequence was carried out by particle bombardment as described by Birch-Machin et al. ( 29 ). Nuclear transformation of tobacco with A. tumefaciens strain LBA4404 containing either gfp-lacI or lacI-gfp was accomplished by the leaf disk method, as described by Horsch et al. ( 36 ), using kanamycin for selection. Regenerated lines were assessed for GFP expression by confocal microscopy. Plastid transformants containing lacO were used as female parents in crosses with gfp-lacI nuclear transformants in order to combine lacO and gfp-lacI ; expression of GFP was confirmed by confocal microscopy.
Microscopy
Chloroplasts were viewed by laser scanning confocal microscopy (Leica TCS-NT, DMRXA light microscope stand, Leica Microsystems Wetzlar GmbH, http://www.leica.com ). GFP and chlorophyll fluorescence, following excitation at 488 nm, were collected through TRITC and FITC filters, respectively. Leaf tissue stained with DAPI (1 µg/ml) was observed with a Nikon Optiphot-2 microscope adapted for epifluorescence, using filters XF06 and XF76 (Omega Optical Inc., http://www.omegafilters.com ) for DAPI and GFP fluorescence, respectively.
Southern blot and PCR analysis of lacO transplastomic plants
Total DNA was isolated from young leaves of wild-type and transplastomic tobacco plants using a ChargeSwitch gDNA Plant Kit (Invitrogen; http://www.invitrogen.com ) and analysed by Southern blotting as described previously ( 29 ). Total leaf DNA (0.3 µg) was cut with EcoRV and SacII and fractionated by electrophoresis in a 1% agarose gel; DNA was blotted onto GeneScreen Plus nylon membrane ( http://www.perkinelmer.com ) and probed with a 32 P-labelled 1.24 kb BamHI fragment from rbcL , as described previously ( 29 ). Total leaf DNA of wild-type and transplastomic lacO plants was analysed by PCR in a 25 µl reaction volume containing 0.5-µl DNA template, 0.5 u KOD Hot Start DNA polymerase (Novagen, http://www.emdbiosciences.com ), 2.5 µl of 10× reaction buffer supplied with the enzyme, 1 mM MgSO 4 , 200 µM each of dATP, dCTP, dGTP, dTTP and 0.3 µM of each primer. The PCR programme was as follows: 94°C (2 min), 30 cycles of 94°C (30 s), 55°C (1 min), 72°C (2 min) and 72°C (10 min). PCR fragments were resolved by electrophoresis on a 1% agarose gel containing 0.2 µg ethidium bromide ml −1 and visualized with an AlphaImager 1200 (AlphaInnotech Corp., http://www.alphainnotech.com ). Primers used to confirm the presence of lacO included a forward primer at the 3′-terminal end of rbcL (5′-TTGACTAAGTATATACTTACCTAG) and a reverse primer in the accD flanking region (5′-TTCAGGGAAACTACAAACTACAGG).
Chloroplast DNA immunoprecipitation
Chloroplast DNA–protein complexes were isolated from young leaves of tobacco as follows. Leaf tissue (2.5 g) was homogenized in 50 ml ice-cold sucrose isolation medium [SIM; 0.35 M sucrose, 25 mM HEPES-NaOH pH 7.6, 2 mM EDTA, 2 mM L-ascorbic acid, 1 mM phenylmethylsulphonyl fluoride (PMSF), 1 mM benzamidine, 5 mM 6-aminocaproic acid, 0.1% BSA], using a Polytron PT3100 fitted with homogenizer PT-DA 3020/2MEC/KL (Kinematica AG, http://www.kinematica.ch ) for 30 s at speed 8. The resulting suspension was filtered through eight layers of muslin into a 50 ml centrifuge tube and centrifuged at 1600 g for 5 min at 4°C. The pellet was resuspended in 20 ml SIM and recentrifuged. The washed pellet was resuspended in 1 ml suspension buffer (1 ml SIM to which was added 2.5 mM DTT, 0.5 mM PMSF, 0.5 µg antipain ml −1 , 0.5 µg leupeptin ml −1 ). If a formaldehyde cross-linking step was included, cross-linking of proteins to DNA was carried out by adding 28 µl of 37% formaldehyde to 1 ml chloroplast suspension followed by incubation at 22°C for 10 min. Glycine (150 µl of 1M) was added to remove excess formaldehyde and the suspension incubated for a further 5 min. Chloroplasts were pelleted at 5900 g in a microcentrifuge for 5 min at 4°C and the pellet resuspended in 0.5 ml lysis buffer (2% Nonidet P-40, 50 mM Tris–HCl pH 7.6, 1.2 mM spermidine, 1 mM PMSF, 0.1 µg antipain ml −1 , 0.1 µg leupeptin ml −1 ). At this stage, the DNA was sheared either by sonication or by micrococcal DNase digestion. In the early stages of development of the method, the suspension was subjected to sonication with an MSE Soniprep 150 (Sanyo-Gallenkamp, http://www.sanyogallenkamp.com ), incubating the suspension on ice and keeping the sonication times to short bursts of 10 s at 18–20 µm amplitude and allowing the suspension to rest on ice for 5 min between each sonication. For micrococcal DNase digestion, 125 µl of 50 mM Tris–HCl pH 7.6, 5 mM MgCl 2 and 125 µl of 6× nuclease digestion buffer (90 mM NaCl, 24 mM CaCl 2 ) were added to 500 µl of lysed chloroplasts. Micrococcal DNase (0.03 units) (micrococcal nuclease from Staphylococcus aureus ; Sigma-Aldrich, http://www.sigmaaldrich.com ) was added to the tube and the suspension was incubated at 37°C. After 10 min, 62 µl of 0.5 M EGTA was added to stop nuclease activity and the tube was incubated 5 min on ice. The sample was centrifuged at 5900 g for 10 min at 4°C and 0.8 ml supernatant collected and mixed with 4.2 ml immunoprecipitation buffer (IPB; 1% Triton X-100, 10 mM EDTA, 20 mM Tris–HCl pH 7.6, 150 mM NaCl, 0.17 mM PMSF, 0.17 µg antipain ml −1 , 0.17 µg leupeptin ml −1 ). To reduce non-specific background in subsequent PCR analyses, the lysed chloroplast suspension was cleared by adding 100 µl protein A-Sepharose beads (50% slurry in 10 mM Tris–HCl pH 7.6, 1 mM EDTA, 0.1% BSA) and 10 µl sonicated bacteriophage λ DNA (0.66 µg ml −1 ). The mixture was incubated on a rotation wheel for 1 h at 4°C. Beads were removed by centrifugation at 2200 g for 5 min and the supernatant aliquotted into 1 ml portions. To one tube was added 2 µl rabbit polyclonal antibody to A. thaliana TRANSPARENT TESTA GLABRA 1 (TTG1; 37) as a negative control; to a second, 2 µl rabbit polyclonal anti-GFP (ab6556, Abcam plc, http://www.abcam.com ); a third received no antibody. These three tubes were incubated on a rotation wheel overnight at 4°C. A fourth tube was stored at 4°C for extraction of total DNA later. All of these steps from extraction onwards were performed on the same day to avoid storage and possible degradation of DNA–protein complexes. Following overnight incubation, 40 µl of protein A-Sepharose beads (50% slurry in 10 mM Tris–HCl pH 7.6, 1 mM EDTA, 0.1% BSA) and 1 µl sonicated λ DNA (0.66 µg ml −1 ) were added to each of the first three tubes which were then incubated on a rotation wheel for 2 h at room temperature. The beads were pelleted at 4500 g in a microcentrifuge for 15 s and the supernatant removed and discarded. The protein A-Sepharose beads were washed twice with 400 µl wash buffer 1 (0.1% SDS, 1% Triton-X 100, 2 mM EDTA, 20 mM Tris–HCl pH 7.6, 150 mM NaCl), once with 400 µl wash buffer 2 (0.25 M LiCl, 1% Nonidet P-40, 1 mM EDTA, 10 mM Tris–HCl pH 7.6, 24 mM sodium deoxycholate) and twice with 400 µl TE, with spins of 4500 g for 15 s between each wash. Protein–DNA complexes were eluted from the beads by washing the beads three times with 160 µl elution buffer (1% SDS, 0.1 M NaHCO 3 ) with a low-speed (4500 g ) spin between each wash. To 480 µl of eluate was added 20 µl 5 M NaCl; 60 µl 5 M NaCl was also added to the 1 ml portion of chloroplast lysate stored at 4°C. Where a formaldehyde cross-linking step had been included in the immunoprecipitation process, all tubes were incubated in a waterbath for 5–6 h at 65°C to reverse the cross-linking. Two volumes of ethanol were added to each tube and samples were incubated overnight at −20°C to precipitate the protein–DNA complexes. Precipitates were collected by centrifugation, dissolved in 100 µl TE to which 2 µl proteinase-K (18.6 µg ml −1 ) was added and incubated for 2 h at 42°C. After addition of 300 µl TE to each tube, the DNA was extracted with 400 µl phenol:chloroform:iso-amyl alcohol (25:24:1 by weight) followed by 400 µl chloroform; care was taken at the final step to collect the same volume of upper layer from each tube to facilitate DNA characterization. Purified DNA was precipitated with two volumes of ethanol in the presence of 5 µg glycogen overnight at −20°C. The precipitate was collected by centrifugation, washed with 70% cold ethanol and dissolved in 20 µl TE for analysis by PCR.
Protein immunoblotting
Protein–DNA complexes eluted from washed protein A-Sepharose beads with 1% SDS, 0.1 M NaHCO 3 as described above were used as the source of protein for immunoblotting. Protein–DNA complexes were precipitated with two volumes of ethanol overnight, collected by centrifugation, washed twice in 70% ethanol, air dried for 10 min and then dissolved in 40 µl 10mM Tris–HCl pH 7.6. Protein sample buffer (13 µl of 4× concentration: 65 mM Tris–HCl pH 6.8, 10% glycerol, 2% SDS, 5% 2-mercaptoethanol, 0.05% bromophenol blue) was added to each of the samples, which were then boiled for 5 min before being cooled on ice. Total lysate protein (25 µl) and 35 µl each of anti-GFP, anti-TTG and no antibody treatments were subjected to SDS–PAGE with a 10% resolving gel and a 5% stacking gel ( 38 ). Biotinylated protein markers (#7727, Cell Signalling, http://www.cellsignal.com ) and 50 ng recombinant GFP (8360-1, Clontech Laboratories Inc., http://www.clontech.com ) were separated on the same gel. Proteins were transferred to nitrocellulose (Protran BA83, Schleicher & Schuell, http://www.sigmaaldrich.com ) using a semi-dry blotting apparatus. The membrane was incubated with rabbit polyclonal antibody to GFP (ab290, Abcam plc, http://www.abcam.com ) at a 1:5000 dilution, followed by biotinylated anti-rabbit Ig antibody (RPN1004, GEHealthcare UK Limited, http://www.gehealthcare.com ) and streptavidin-biotinylated horseradish peroxidase complex (RPN1051V, GE Healthcare UK Limited, http://www.gehealthcare.com ) and detected with enhanced chemiluminescence (Western Lightning, Perkin Elmer, http://www.perkinelmer.com ).
PCR of immunoprecipitated chloroplast DNA
Immunoprecipitated chloroplast DNA was analysed by PCR in a 25 µl reaction volume containing 1 µl DNA template, 0.5 u DNA polymerase (BIOTAQ; Bioline UK, http://www.bioline.com ), 2.5 µl of 10× reaction buffer supplied with the enzyme, 2 mM MgCl 2 , 200 µM each of dATP, dCTP, dGTP, dTTP and 0.3 µM of each primer. The PCR programme was as follows: 94°C (2 min), 26–28 cycles of 94°C (30 s), 50–60°C (30 s), 72°C (30 s) and 72°C (10 min). PCR fragments were resolved by electrophoresis on a 1.2% agarose gel containing 0.2 µg ethidium bromide ml −1 and visualized with an AlphaImager 1200. Primers used to confirm the presence of lacO in immunoprecipitated DNA included a forward primer in the psbA 3′-terminal end of aadA (5′-GAAATAAGAAAGAAGAGCTATATTCG) and a reverse primer in the accD flanking region (5′-TTCAGGGAAACTACAAACTACAGG). Primers to investigate binding in the atpBE region were forward (5′-TCAGCATATCGATTTATGCCTAGC) and reverse (5′-TCTCACAACAACAAGGTCTACTCG) to amplify a fragment of 396 bp. Primers to amplify a 233-bp fragment of 23S rDNA ( rrn23 ) were forward (5′-TATCGTGCCCACGGTAAACGCTGG) and reverse (5′-CGTAATGATAAACGGCTCGTCTCG). Amplification of a 167-bp fragment from psbT was carried out using forward (5′-AAACCACGATCGAATCTATGG) and reverse (5′-GAGGCTCAGTACTTCAACTG) primers. Amplification of a 3.8 kb fragment from accD to atpBE , accomplished using the same forward primer for atpBE and the reverse accD primer as described above, encompassing the lacO insertion site, was carried out as described above but with a modified PCR programme incorporating 28 cycles of 94°C (1 min), 55°C (2 min) and 72°C (3 min).
RESULTS
Creation of tobacco lines with plastid-located lacO sequences
Chloroplast transformation by biolistics was used to introduce multiple copies of lacO into a single site between the rbcL and accD genes in tobacco chloroplast DNA. Detached leaves of the tobacco cultivar Petite Havana were bombarded with lacO plasmids containing either 1 (32 copies) or ∼2.5 kb of lacO sequences (68 copies) ( Figure 1 a). Spectinomycin-resistant transplastomic plants regenerated following bombardment with either construct were found to contain a shortened length of lacO repeat sequence compared with the transforming DNA. PCR analyses of several regenerated lines using primers which flanked the lacO insertion site indicated that ∼200 bp could be attributed to the inserted lacO DNA regardless of the transforming construct ( Figure 1 b). Transplastomic lines were also subjected to Southern blot analysis. Probing of total leaf DNA digested with EcoRV and SacII with a 1.24 kb BamHI fragment from the plastid rbcL gene adjacent to the construct insertion site was expected to give a 2.8 kb band in untransformed plants, and a 5.3 kb band if 2.5 kb of lacO DNA had been transferred. The Southern blot detected a band of ∼4.4 kb in four of the lines ( Figure 1 c) confirming the PCR results and indicating that <300 bp of lacO DNA was located between the aadA selectable marker gene and the accD recombination region. Weak hybridization to a band at 3.7 kb was also obtained; this has been observed previously with pZS197-based vectors ( 29 , 31 ) and is probably due to recombination between introduced chloroplast DNA sequences and identical endogenous sequences ( 39 ). Recombination frequencies between nearby repeated sequences in chloroplasts are known to be high ( 40 ), and recombination between the repeated lacO sequences probably accounts for the substantial reduction in numbers of lacO elements in all independent lines tested. This appears to have occurred within 11 weeks of bombardment of the leaves and no further shortening of the lacO sequences was observed. Sequencing of PCR products from the transplastomic lines confirmed the presence of three lacO repeat sequences of 32 bp each, separated by HaeIII restriction sites and flanked by Eco RI sites ( Figure 2 a), an arrangement similar to the original pAFS plasmid where repeats of eight lacO sequences, each separated by a HaeIII restriction site, were themselves separated by EcoRI restriction sites ( 21 ).
![Constructs for plastid and nuclear transformations. ( a ) Construct for plastid transformation. lacO DNA (1 or ∼2.5 kb; dashed lines) was inserted between the HindIII and SmaI restriction sites into a polylinker in the unique Bsu36I site ( 31 ) of pZS197 ( 30 ). P rrn , plastid rrn promoter driving the aadA gene conferring resistance to spectinomycin; T psbA , terminator region from plastid psbA . Plastid rbcL and accD sequences are used for recombination into the plastid genome; their direction of transcription is shown by the arrowheads. The EcoRV restriction site was destroyed in the production of pZS197 but is restored in transplastomic plants. 1: position of 1.24 kb probe used in Southern DNA analysis. 2: position of the rbcL primer used to determine the size of the lacO insert in (b); 3: position of the forward and reverse primers used to analyse immunoprecipitated DNA for the presence of lacO . ( b ) PCR analysis of leaf DNA from transplastomic lacO lines. Lanes 1–4 and 5–8, putative transplastomic lines containing lacO constructs with 1 or ∼2.5 kb lacO DNA, respectively. Lane 9, no-template negative control. Lane 10, positive control, PCR on plasmid containing 1 kb lacO. Marker sizes are shown on the left. Primers were used to amplify a region extending from rbcL to accD and were expected to give band sizes of 2.6 or ∼4.0 kb with 1 kb or ∼2.5 kb of lacO , respectively. ( c ) Southern blot analysis of transplastomic tobacco lines, regenerated following bombardment with a plasmid containing ∼2.5 kb lacO repeat sequence. Total leaf DNA (0.3 µg) was cut with EcoRV and SacII and probed with a 32 P-labelled 1.24 kb BamHI fragment from rbcL [see (a) above]. Lanes 1–6, putative transplastomic lines. WT, ‘Petite Havana’ wild-type. Band sizes are shown on the left. ( d , e ) Constructs for nuclear transformation containing the CaMV 35S promoter and nos terminator in pBIN19 ( 32 ). (d) gfp-lacI DNA was inserted between the coding region for the RbcS transit peptide and the nos terminator using XbaI and SacI restriction sites. (e) lacI-gfp has been inserted between the coding region for the RbcS transit peptide and the nos terminator using XbaI and StuI restriction sites.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/38/14/10.1093_nar_gkq413/2/m_gkq413f1.jpeg?Expires=1750246233&Signature=F4qwTFzg~T3WMnFA~4u0l3~Czll4owR0JLMgcE8Ay4a9yXvCuGDH0gg2y0b7wF0jtyRrLIAVP3JAUbDmHNfZIIi2A4NnpU7myXarDMM1HfqBNqoUzcTvDCxjfffVVMqCgLfzO-97DyLGVhvSwvO-h8GpudFv1PqKIDN8D7~b1hN~NKHBDJOp1Sk3D1YeMMy~DcYWCC57hMTvXTX0tpm4Sy6V-F-rZTU7aFjpPkUQF0V9eFDheYEjyJ~c~rWCkIk8YGV4KRCNGoa6sYRmoIdkDylM1euZWKmHgSZy5JkKrlDG-6I2fs-BYkQ3bgYMylMbAgwJi-XZkYE4EfXdgHW0lQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Constructs for plastid and nuclear transformations. ( a ) Construct for plastid transformation. lacO DNA (1 or ∼2.5 kb; dashed lines) was inserted between the HindIII and SmaI restriction sites into a polylinker in the unique Bsu36I site ( 31 ) of pZS197 ( 30 ). P rrn , plastid rrn promoter driving the aadA gene conferring resistance to spectinomycin; T psbA , terminator region from plastid psbA . Plastid rbcL and accD sequences are used for recombination into the plastid genome; their direction of transcription is shown by the arrowheads. The EcoRV restriction site was destroyed in the production of pZS197 but is restored in transplastomic plants. 1: position of 1.24 kb probe used in Southern DNA analysis. 2: position of the rbcL primer used to determine the size of the lacO insert in (b); 3: position of the forward and reverse primers used to analyse immunoprecipitated DNA for the presence of lacO . ( b ) PCR analysis of leaf DNA from transplastomic lacO lines. Lanes 1–4 and 5–8, putative transplastomic lines containing lacO constructs with 1 or ∼2.5 kb lacO DNA, respectively. Lane 9, no-template negative control. Lane 10, positive control, PCR on plasmid containing 1 kb lacO. Marker sizes are shown on the left. Primers were used to amplify a region extending from rbcL to accD and were expected to give band sizes of 2.6 or ∼4.0 kb with 1 kb or ∼2.5 kb of lacO , respectively. ( c ) Southern blot analysis of transplastomic tobacco lines, regenerated following bombardment with a plasmid containing ∼2.5 kb lacO repeat sequence. Total leaf DNA (0.3 µg) was cut with EcoRV and SacII and probed with a 32 P-labelled 1.24 kb BamHI fragment from rbcL [see (a) above]. Lanes 1–6, putative transplastomic lines. WT, ‘Petite Havana’ wild-type. Band sizes are shown on the left. ( d , e ) Constructs for nuclear transformation containing the CaMV 35S promoter and nos terminator in pBIN19 ( 32 ). (d) gfp-lacI DNA was inserted between the coding region for the RbcS transit peptide and the nos terminator using XbaI and SacI restriction sites. (e) lacI-gfp has been inserted between the coding region for the RbcS transit peptide and the nos terminator using XbaI and StuI restriction sites.

LacI binding sites in E. coli and chloroplasts. ( a ) Arrangement of lacO repeats in transplastomic lacO lines. lacO sequences are separated by HaeIII restriction sites, with an EcoRI restriction site at each end. The sequence of the 118-bp region is given below the diagram. The gap indicates the centre of the dyad symmetry. ( b ) Core regions of three lacO sequences from E. coli . Bases identical to lacO1 are in bold, non-identical bases are lower case. ( c ) Chloroplast DNA sequences showing similarity to lacO . Bases identical to lacO1 are in bold, non-identical bases are lower case. The locations of the sequences in the tobacco genome (Genbank Z00044) are: rrn23 , 109075-109095 and 133535-133555; ndhB , 98558-98578; ndhJ , 51302-51322; rpl14 , 83504-83524; atpBE , 57377-57397; psaA , 42101-42121; rrn16 , 103808-103828 and 138802-138822.
Creation of tobacco lines expressing nuclear-encoded plastid-targeted GFP-LacI and LacI-GFP
Two chimeric genes encoding plastid-targeted N- or C-terminal fusions of LacI to GFP were produced in the binary vector pBIN19 ( 32 ), under control of the CaMV 35S promoter ( Figure 1 d and e) for Agrobacterium -mediated transformation of tobacco. GFP-LacI and LacI-GFP were targeted to chloroplasts with the pea RbcS transit peptide. Eleven kanamycin-resistant transgenic lines with the GFP-LacI construct were examined for GFP fluorescence by confocal microscopy. All lines showed a punctate pattern of GFP fluorescence within the plastids ( Figure 3 a), unlike the uniform GFP fluorescence throughout the stroma found in transplastomic lines expressing soluble GFP (31; Figure 3 b). In the highest expressing lines, there was a slight background of GFP fluorescence throughout the chloroplasts, in addition to the punctate pattern. This punctate pattern was similar to the pattern of nucleoids shown in other studies ( 28 ). Staining of leaf tissue with the DNA-specific fluorochrome DAPI and examination by epifluorescence microscopy established the co-localization of DAPI and GFP fluorescence ( Figure 3 c and d), indicating that GFP-LacI was located in plastid DNA-containing nucleoids. A similar punctate pattern of GFP fluorescence within the plastids was observed in lines containing LacI-GFP ( Figure 3 e). Modification of the C-terminus of the LacI by deleting the last 3–5 amino acid residues has been shown to result in dimeric proteins rather than the wild-type tetramers ( 41 ). Attaching GFP to the C-terminus of LacI was therefore likely to interfere with tetramer formation and produce dimeric LacI-GFP fusion protein, which might affect the visualization of GFP binding to lacO . However, no differences in the patterns of GFP fluorescence from GFP-LacI and LacI-GFP were observed. Several of the most strongly expressing LacI-GFP lines showed yellowing of the interveinal regions of the leaves, but this phenotype was not observed with the strongly expressing GFP-LacI lines.
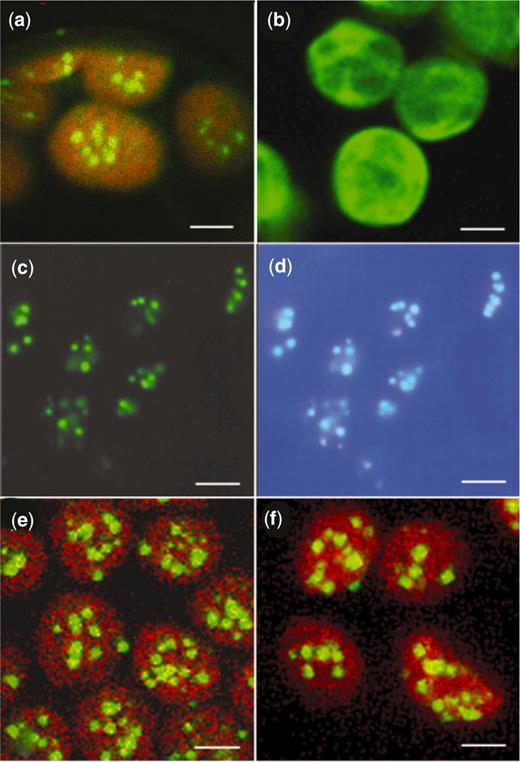
Chloroplasts from tobacco leaf mesophyll cells in water-mounted tissue visualized by confocal or epifluorescence microscopy. (a,b,e,f) Chloroplasts viewed by laser scanning confocal microscopy (TCS-NT, DMRXA light microscope stand, Leica Microsystems Wetzlar GmbH). Images of GFP and chlorophyll fluorescence, using an excitation wavelength of 488 nm, were collected through TRITC and FITC filters, respectively. Images (a), (e) and (f) result from merging chlorophyll and GFP fluorescence. Image (b) shows GFP fluorescence only. ( a ) GFP-LacI line showing the appearance of plastid DNA as fluorescent nucleoids (bar = 2.8 µm). ( b ) P rrn::gfp line showing GFP fluorescence distributed throughout the chloroplasts (bar = 2.8 µm). (c,d) Leaf tissue from a lacO /GFP-LacI line stained with DAPI and viewed by epifluorescence microscopy. ( c ) GFP fluorescence (bar = 2 µm). ( d ) DAPI fluorescence of the same cells, merged with GFP fluorescence, showing co-localization of GFP and DAPI-stained DNA (bar = 2 µm). ( e ) LacI-GFP line showing the appearance of plastid DNA as fluorescent nucleoids (bar = 2.8 µm). ( f ) lacO /GFP-LacI line showing fluorescent nucleoids (bar = 2.8 µm).
Pollen from a representative homozygous transgenic GFP-LacI line was used to pollinate transplastomic lacO lines to combine plastid-located lacO and plastid-targeted GFP-LacI in the same plant. The presence of three lacO repeats in progeny of the crosses was confirmed by PCR (data not shown) and by DNA sequencing. Examination of the leaves by confocal microscopy indicated a punctate pattern of GFP fluorescence within the chloroplasts ( Figure 3 f). Progeny from one of these crosses was used for the development of a chloroplast ChIP protocol.
Development of a chloroplast ChIP protocol
In order to develop a chloroplast ChIP protocol, chloroplasts were isolated from leaves of the lacO /GFP-LacI plants described above and from three control lines: transplastomic lacO plants without GFP-LacI, transgenic GFP-LacI plants without lacO and transplastomic P rrn::gfp plants which contain high levels of soluble GFP distributed more or less uniformly throughout the stroma with no sign of localization to nucleoids ( 31 ). Leaves from 8 to12 week-old tissue culture-grown plants were routinely used as they contained low levels of starch, which did not interfere with chloroplast extraction, in comparison with plants grown in a growth chamber. A sucrose-based isolation medium was used routinely to aid in stabilization of nucleoid structure within the chloroplasts ( 26 ). Fractionation of chloroplasts in a 40/80% Percoll step gradient after extraction resulted in an approximate ratio of 2:1 non-intact:intact chloroplasts. However, under an epifluorescence microscope chloroplasts from both fractions showed bright distinct nucleoids, and therefore unfractionated chloroplast preparations were used in order to maximize recovery of immunoprecipitated material.
Sonication is usually the method of choice to fragment chromatin to protein–DNA complexes containing 300–600 bp DNA in nuclear ChIP methods ( 1 , 3 ). However, sonication of chloroplasts lysed by treatment with 2% Nonidet P-40 was unsuccessful in shearing chloroplast DNA in nucleoids of lacO/ GFP-LacI plants to small enough fragments to reduce the likelihood of immunoprecipitating long regions that would lower the sensitivity of subsequent PCR analyses. Sonication of the suspension produced by lysing chloroplasts in 2% Nonidet P-40 detergent for 120 s in 30-s bursts reduced the bulk of the DNA to fragments of ∼1–3 kb, as estimated by agarose gel electrophoresis. However, it was still possible to amplify regions of 3.8 kb by PCR using a forward primer in the atpB/E region 2 kb upstream from the construct insertion site and a reverse primer in the accD region adjacent to the 3′-terminus of the inserted DNA, indicating that long fragments of DNA remained. Compact fluorescent nucleoids were also still visible with epifluorescence microscopy following extended sonication of lysed chloroplasts. Digestion with AluI was used by Prikryl et al. ( 9 ) to fragment maize chloroplast DNA for their ChIP experiments, but digestion with a specific restriction enzyme was thought not to be advisable for generating random fragments of chloroplast DNA. Micrococcal nuclease was therefore tested under various conditions for its ability to digest plastid DNA to smaller fragments ( 42 ). A 10-min incubation of lysed chloroplasts with micrococcal nuclease (0.06 u ml −1 ) at 37°C in Tris–HCl pH 7.6 buffer containing 0.8 mM MgCl 2, 15 mM NaCl and 4 mM CaCl 2 , followed by addition of 38 mM EGTA to stop nuclease activity, produced DNA fragments of ∼600–1200 bp and it was not possible subsequently to PCR-amplify fragments of 3.8 kb.
The ability of anti-GFP antibodies to immunoprecipitate GFP-LacI complexes from chloroplast extracts was examined by protein immunoblot analysis. Following micrococcal nuclease treatment, chloroplast extracts were incubated overnight at 4°C with protein A-Sepharose beads and antibodies to GFP, antibodies to TTG1 (a non-chloroplast-located protein) or no antibodies. Proteins eluted from the beads with 1% SDS, 0.1 M NaHCO 3 were separated by SDS-PAGE, blotted onto nitrocellulose and GFP detected with antibodies to GFP and enhanced chemiluminescence. A band of 66 kDa corresponding to the GFP-LacI fusion protein is present in the total chloroplast lysate ( Figure 4 , lane 4) and in the immunoprecipitate with antibody to GFP ( Figure 4 lane 2) but is not present in the immunoprecipitate with antibody to TTG1 or in the absence of antibodies ( Figure 4 lanes 1 and 3). The strongly reactive band of ∼55 kDa in Figure 4 , lanes 1 and 2, is due to the heavy chain of rabbit IgG from the immunoprecipitation incubation cross-reacting with the biotinylated anti-rabbit IgG antibody used in the detection process. It can be concluded that GFP-LacI fusion protein was successfully immunoprecipitated using the antibodies to GFP and protein A-Sepharose.
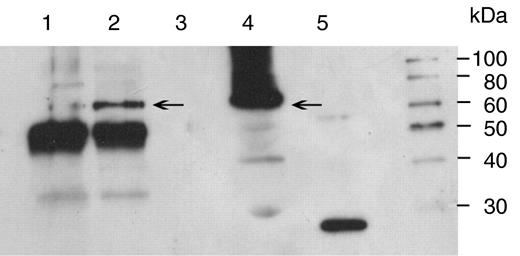
Immunoblot of immunoprecipitated protein obtained by incubation of lysed chloroplast extracts with or without antibody prior to addition of protein-A Sepharose, elution of the protein–DNA complexes from the protein-A Sepharose and subsequent precipitation in ethanol. The precipitates were solubilized in 10 mM Tris–HCl pH 7.6 and 25–35 µl fractionated by electrophoresis on a SDS-10% polyacrylamide gel, blotted onto nitrocellulose and incubated with rabbit polyclonal anti-GFP. Immunoreactive bands were detected by enhanced chemiluminescence. Lane 1, rabbit anti- A. thaliana TTG1; lane 2, rabbit anti-GFP; lane 3, no antibody; lane 4, total soluble protein; lane 5, 50 ng recombinant GFP. Arrows mark the position of the GFP-LacI monomer at 66 kDa. A marker lane is shown to the right.
Formaldehyde is usually used to produce DNA–protein cross-links in nuclear chromatin both in vivo and in vitro ( 1 , 43 , 44 ) and, in comparison to other cross-linking reagents, cross-linking can be reversed relatively easily ( 45 ). However, formaldehyde cross-linking is not necessary for stabilizing interactions between purified LacI and lacO -containing DNA in vitro ( 46 ). Therefore, the formaldehyde cross-linking step was assessed for its effect on immunoprecipitation of chloroplast DNA–protein complexes. Isolated chloroplasts from lacO /GFP-LacI plants were incubated with 1% formaldehyde for 10 min at 22°C, followed by addition of glycine to remove excess formaldehyde and lysis in a buffer containing 2% Nonidet P-40. Following shearing of chloroplast DNA by micrococcal nuclease, complexes containing GFP-LacI were immunoprecipitated with rabbit antibodies to GFP and protein A-Sepharose, eluted from the protein A-Sepharose with SDS and incubated at 65°C for 5–6 h to reverse the cross-links. The extracted immunoprecipitated chloroplast DNA was analysed for the presence of lacO by PCR using primers that flanked the lacO insertion site ( Figure 1 a) to produce a 409-bp fragment. The lacO region was successfully amplified from chloroplast DNA immunoprecipitated by anti-GFP ( Figure 5 a) but not by anti-TTG1 or no antibody, providing a clear indication that GFP-tagged LacI had bound to the lacO sequence in plastid DNA. No amplification of the lacO fragment was obtained from the immunoprecipitates of chloroplasts of the three control lines lacO , GFP-LacI and Prrn::gfp ( Figure 5 a). Immunoprecipitation of the lacO sequence was obtained irrespective of the use of the formaldehyde cross-linking treatment, corroborating the finding that cross-linking was not necessary to stabilize the binding of LacI to lacO ( 46 ).
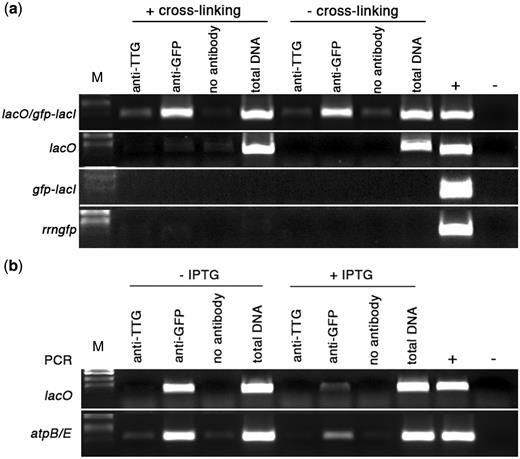
Binding of GFP-LacI to chloroplast-located lacO sequences. ( a ) PCR analysis of immunoprecipitated chloroplast DNA. Immunoprecipitation was carried out with the following treatments: antibody to A. thaliana TTG1, antibody to GFP or no antibody. Total DNA was extracted from a fraction of the chloroplast lysate that did not undergo immunoprecipitation. Control reactions for PCR included genomic DNA isolated from a transplastomic lacO line (+) and water (−). M, DNA size markers. (a) Binding of GFP-LacI to lacO. Primers were used to amplify a 409-bp sequence, which includes plastid-localized lacO (marked as position 3 in Figure 1(a). Experimental lines are listed on the left. Treatments shown in the left half of the panel were carried out following formaldehyde cross-linking, those on the right were not subjected to formaldehyde cross-linking. ( b ) Effect of IPTG on binding of GFP-LacI to lacO. PCR analysis of immunoprecipitated chloroplast DNA from lacO /GFP-LacI lines, using primers to amplify a 409-bp fragment encompassing lacO and a 396-bp fragment from atpBE (shown on left side). Treatments shown in the left half of the panel were carried out in the absence of IPTG, those in the right half were carried out in the presence of 20 mM IPTG.
The affinity of LacI for lacO is reduced 1000-fold in the presence of isopropyl β- D -1-thiogalactopyranoside (IPTG) ( 47 ) and, although IPTG can remove LacI from lacO binding sites, it does not affect non-specific binding. The addition of IPTG during chloroplast extraction was tested to ascertain whether it would affect binding of LacI to lacO . Addition of 20 mM IPTG to chloroplasts prior to cross-linking and inclusion in the lysis buffer, immunoprecipitation buffer and at all subsequent stages, led to a pronounced reduction in the amplified product generated by PCR when primers specific for lacO were used ( Figure 5 b). Even in the presence of excessive amounts of LacI, de-repression by IPTG may not always be complete ( 48 ) and, since a high level of GFP-LacI was being expressed from the CaMV 35S promoter ( Figure 4 and ‘Discussion’ section), this may be a possible explanation for the small amount of binding of LacI to lacO that still seems to be taking place. However, the GFP-LacI fusion protein clearly showed an affinity for lacO that could be disrupted by the addition of IPTG, indicating binding of a specific nature.
The appearance of fluorescent GFP foci that co-localized with DNA in plastids of plants transformed with nuclear-encoded GFP-LacI ( Figure 3 a) suggested that LacI was binding to plastid DNA even in the absence of lacO . This might be due to non-specific binding of GFP-LacI to all regions of plastid DNA or to specific binding to endogenous plastid DNA sequences showing similarity to lacO . Comparison of the three naturally occurring lacO sequences from E. coli ( Figure 2 b) with tobacco plastid DNA identified several locations around the plastome with some sequence similarity to lacO ( Figure 2 c) . The lacO sequences show pseudo-dyad symmetry with the central base pairs stabilizing the LacI– lacO complex ( 49 , 50 ). We chose to examine binding of GFP-LacI to two regions of plastid DNA ( atpBE and rrn23 ) containing sequences showing similarity to lacO , and to a control region encompassing psbT , which was located at least 2 kb away from any region showing similarity to lacO . The lacO similarity in the atpBE promoter region (nucleotides 57372–57392) includes eight consecutive base pairs in the central region identical to lacO3 ( 49 ) suggesting high probability as a binding site for GFP-LacI. The atpBE region is located ∼3.4 kb from the lacO insertion site in the transplastomic lacO plants and PCR using atpBE and accD primers confirmed that the micrococcal nuclease treatment of plastid nucleoids did not produce long fragments of plastid DNA containing both sites. To examine GFP-LacI binding to the atpBE region, PCR was performed on immunoprecipitated DNA using primers to amplify a sequence of 396 bp from the atpB-rbcL intergenic region. The PCR results ( Figure 6 a) demonstrate that atpBE DNA had been immunoprecipitated from formaldehyde cross-linked treatments of chloroplasts from GFP-LacI lines with or without lacO , suggesting that GFP-LacI had bound to plastid DNA in this region. In the absence of formaldehyde, however, there was no amplified DNA band, implying that cross-linking was necessary to stabilize the linkage of weaker protein–DNA complexes during the course of immunoprecipitation. PCR carried out with primers for the atpBE region also resulted in a reduced amount of amplified DNA in samples that had been treated with IPTG ( Figure 5 b), suggesting specific recognition of the atpBE site by GFP-LacI.
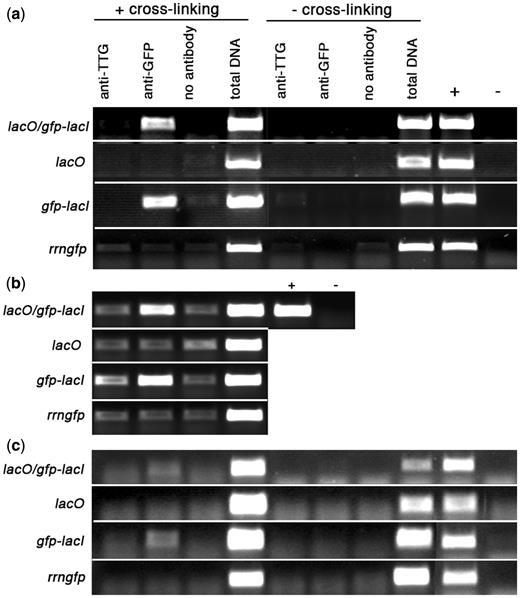
Binding of GFP-LacI to chloroplast sequences. Experimental lines are listed on the left; treatments shown in the left half of the panels were carried out following a formaldehyde cross-linking step, those on the right were not subjected to cross-linking. ( a ) PCR analysis of immunoprecipitated chloroplast DNA using primers to amplify a 396-bp fragment from plastid atpB/E . ( b ) PCR analysis of formaldehyde cross-linked, immunoprecipitated chloroplast DNA using primers to amplify a 233-bp fragment from plastid 23S rDNA. ( c ) PCR analysis of immunoprecipitated chloroplast DNA using primers to amplify a 167-bp fragment from plastid psbT . Total DNA was extracted from a fraction of the lysate that did not undergo immunoprecipitation. Control reactions for PCR included genomic DNA isolated from a transplastomic lacO line (+) and water (−).
The other potential GFP-LacI binding site examined occurs within the 23S rDNA sequence where there are 11 consecutive base pairs in the central region identical to lacO1 ( Figure 2 c). The PCR results clearly indicate that GFP-LacI also bound to a 233-bp region containing this site. PCR on the anti-GFP-immunoprecipitated DNA gave a positive band in lacO /GFP-LacI and GFP-LacI plants, but not in lacO or P rrn::gfp lines ( Figure 6 b). The positive results for binding to lacO -related sequences in atpBE and rrn23 are in contrast to the results obtained for binding to chloroplast psbT region. This region did not appear to contain a lacO -related sequence, and the distance to the nearest potential binding site appeared to be ∼2 kb. It was not possible to amplify a PCR product of the correct size from any of the immunoprecipitated material ( Figure 6 c) suggesting that there was little or no binding of GFP-LacI in this region of the genome. This indicates that the location of GFP-LacI in plastid nucleoids is not due to non-specific binding over the complete length of plastid DNA and is likely to be due to specific binding to lacO -related sequences. These endogenous binding sites for GFP-LacI appear to be predictable based upon DNA sequence similarity to lacO.
DISCUSSION
The binding of a GFP-LacI fusion protein to lacO sequences integrated into the chloroplast genome has been demonstrated by chromatin immunoprecipitation. The binding of GFP-LacI to lacO was repressed in the presence of IPTG and its detection by ChIP did not require treatment with formaldehyde, which is commonly used as a reagent to cross-link proteins to DNA. Both these results support the concept that GFP-LacI was binding specifically to lacO , as IPTG specifically reduces binding of LacI to lacO ( 47 ) and formaldehyde cross-linking is not necessary to stabilize lacO/ LacI binding ( 46 ). Binding of GFP-LacI to other sites on the chloroplast genome, as evidenced by the presence of fluorescent nucleoids in the absence of lacO ( Figure 3 ), could be detected by ChIP only following cross-linking in the presence of formaldehyde. Two, at least, of these endogenous binding sites appeared to be predictable based on their sequence similarity to naturally occurring lacO sequences. The requirement for formaldehyde to stabilize the linkages probably indicates a weaker affinity of the fusion protein for the endogenous sites. However, the affinity of LacI for these endogenous sites would appear to be greater than that of LacI for non-specific binding to DNA. The association constant Ka for LacI binding to lacO is ∼10 12 M −1 ( 51 ), whereas non-specific binding of LacI to DNA has a Ka of ∼10 5 M −1 ( 52 ). The absence of any detectable binding of GFP-LacI to the psbT region of chloroplast DNA ( Figure 6 ) implies that the ChIP protocol, even in the presence of formaldehyde, does not detect such low-affinity non-specific binding.
The binding of LacI to endogenous binding sites might possibly affect the use of the lacO /LacI system to provide an inducible expression system for transgenes in the chloroplast genome. Mühlbauer and Koop ( 19 ) demonstrated 20-fold induction of GFP expression from a chimeric rrn promoter containing lacO sequences on spraying tobacco leaves with IPTG. Binding of LacI to endogenous sites may affect the expression of endogenous chloroplast genes, potentially affecting the growth and development of the plant. No such growth effects were reported ( 19 ), suggesting that LacI binding to chloroplast sequences did not have major effects on chloroplast gene expression. In our experiments, yellowing of the interveinal areas of leaves of the most highly expressing LacI-GFP lines was detected, possibly indicating an effect on chloroplast gene expression. However, this was not observed with the most highly expressing GFP-LacI lines, suggesting that the presence of LacI in chloroplasts was not normally detrimental to growth and development.
The amounts of the LacI fusion proteins in chloroplasts are considerably higher than the amount of LacI normally found in E. coli , which has been estimated as 5–10 molecules of LacI per cell ( 48 ). From western blots of chloroplast extracts with antibodies to GFP, as in Figure 4 , it is possible to estimate the ratio of GFP to Rubisco in chloroplasts, assuming that Rubisco accounts for 50% of the chloroplast protein ( 53 ). This produces a molar ratio of Rubisco to GFP of 200:1. The number of molecules of Rubisco per chloroplast can be obtained from estimates of mean chloroplast volume and Rubisco concentration. A mean chloroplast volume of 25 µm 3 can be calculated from the face area of the chloroplasts in Figure 3 , assuming a chloroplast thickness of 1 µm ( 54 ). Assuming the concentration of Rubisco in the stroma of tobacco chloroplasts is ∼200 mg/ml, based on the protein concentration of crystals of tobacco Rubisco ( 55 ), the number of Rubisco molecules per chloroplast is 6 million. This number is similar to the value of 4.2 million that can be calculated from the Rubisco content (3.6 pg Rubisco per chloroplast) of chloroplasts in the middle region of a developing wheat leaf ( 56 ). The estimate of 6 million Rubisco molecules per chloroplast leads to an estimate of 30 000 GFP-LacI molecules per chloroplast. However, these chloroplasts will contain ∼100 chloroplast DNA molecules, so the ratio of LacI to DNA in chloroplasts is ∼300:1, considerably greater than the 5–10:1 ratio in E. coli . This large excess of GFP-LacI is probably responsible for the binding to lower-affinity sites on chloroplast DNA. However, the punctate pattern of GFP fluorescence was still observed in the absence of lacO in lower expressing GFP-LacI lines, estimated on the basis of western blotting to have 10-fold less GFP (data not shown), suggesting that such binding to chloroplast DNA can occur at LacI:DNA ratios not much greater than found in E. coli .
The ChIP protocol developed used micrococcal nuclease to fragment the chloroplast genome into suitable small DNA fragments. We decided against the use of a specific restriction enzyme, such as AluI ( 9 ), because it would not produce random, overlapping fragments of chloroplast DNA that we believed should maximize the probability of detecting specific protein binding at any site on the chloroplast genome. We failed to identify suitable conditions for the use of sonication, which is the method of choice for fragmenting nuclear and mitochondrial DNA ( 1 , 4 , 7 ). Neither mitochondria nor chloroplasts contain histones and therefore lack the nucleosome structure of nuclear chromatin. The difference in the susceptibility of the chloroplast and mitochondrial genomes to fragmentation by sonication may therefore be due to differences in the higher order organization of DNA in the two organelles. Sonication produced ∼500-bp fragments of DNA from yeast mitochondria ( 7 ), but with chloroplasts it was possible to detect DNA fragments longer than 3 kb following sonication. We therefore used nuclease digestion to ensure that the chloroplast DNA was sheared into small enough fragments to eliminate potentially misleading results of proteins binding at remote sites on long DNA fragments. The micrococcal nuclease digestion protocol was developed to ensure the separation of the atpBE region showing similarity to lacO and the introduced lacO sequences. These sites are ∼3.4 kb apart and following digestion it was not possible to amplify long fragments containing both sites. The ChIP experiment should then be able to detect binding at each of the sites independently. By slight modifications of the nuclease digestion protocol it should be possible to detect binding at two even-closer proximal sites by ensuring consistent nuclease cleavage between the two sites.
We believe the ChIP method developed using the introduced lacO /LacI system should provide a generic means of identifying specific endogenous protein-binding sites in chloroplast DNA, provided specific antibodies to the DNA-binding protein of interest are available. These antibodies could be raised against the purified protein or synthetic peptides, or against added epitopes, such as HA or MYC tags, or fusion proteins, such as GFP. The use of antibodies against epitope tags or fusion proteins requires the availability of transgenic plants expressing the epitope-tagged or fused DNA-binding protein and this increases the potential risk of aberrant binding to non-physiological target sites by the over-expressed protein. However, the expression of the tagged or fusion protein from its native promoter in a gene-knockout background should minimize this risk. The identification of all binding sites for a particular DNA-binding protein might be achieved by combining the ChIP protocol developed here with a DNA microarray ( 57 ). A similar method was used by Prikryl et al. ( 9 ) to identify fragments of maize chloroplast DNA bound by the WHY1 protein. Although they detected binding to all fragments of AluI-digested maize stromal DNA, the method should be able to detect binding to individual specific regions of the chloroplast genome.
It may be possible to use the chloroplast ChIP protocol to investigate the binding sites of the PEND protein in organello . Previous studies have identified potential binding sites in vitro by South-western blotting of pea chloroplast fractions ( 9 ) or by mobility-shift assay and binding-site selection using the expressed DNA-binding region of PEND ( 58 ). The soluble bZIP region of PEND was shown to have a Ka of ∼4 × 10 6 M −1 for a double-stranded oligonucleotide containing two copies of the identified binding-site sequence TAAGAAGT ( 58 ). It is difficult to judge if the binding affinity of membrane-associated PEND protein is likely to be similar to that of the soluble bZIP domain, but a higher affinity (higher Ka ) is likely to be required to assure success with the ChIP assay. Similar problems may be encountered in trying to use the ChIP assay to detect sigma factor binding at specific sites. Sigma factors provide the specificity for holo-RNA polymerase binding to −35 and −10 promoter elements, but do not bind DNA in the absence of RNA polymerase ( 59 ). The association constant of E. coli RNA polymerase holoenzyme containing the σ 70 sigma factor for canonical promoters is ∼10 9 M −1 ( 60 ), four orders of magnitude greater than values of ∼10 5 M −1 for non-specific binding to DNA ( 61 , 62 ). If the affinity of the chloroplast RNA polymerase holoenzyme containing a specific sigma factor for chloroplast promoter elements is similar to the values of the E. coli holoenzyme, the ChIP protocol may be able to detect specific binding, provided the sigma factors are accessible to antibodies and can be immunoprecipitated.
The introduction of the GFP-LacI/ lacO system into tobacco has made it possible to locate plastid nucleoids throughout plant tissue, in the same way as that demonstrated with the PEND-GFP fusion protein system reported by Terasawa and Sato ( 28 ). Plastid nucleoids can be easily visualized in living tissues throughout the plant, including those tissues with non-green plastids that are more difficult to locate using light microscopy. The GFP-LacI/ lacO plants provide an alternative to the PEND-GFP system ( 28 ) that does not require over-expression of an endogenous protein for investigating plastid nucleoid behaviour in various plant organs.
FUNDING
European Commission as part of FP6 projects PLASTOMICS (grant number LSHG-CT-2003-503238); PHARMA-PLANTA (grant number LSHB-CT-2003-503565). Funding for open access charge: European Commission grant LSHB-CT-2003-503565.
Conflict of interest statement . None declared.
ACKNOWLEDGEMENTS
We would like to thank A. Straight for the gift of plasmids pAFS52 and pAFS59, and T. Kavanagh and J. Jouhet for many helpful discussions during the course of this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments