





Abstract
Mutations in the Werner gene promote the segmental progeroid Werner syndrome (WS) with increased genomic instability and cancer. The Werner gene encodes a DNA helicase (WRN) that can engage in direct protein–protein interactions with DHX9, also known as RNA helicase A or nuclear DNA helicase II, which represents an essential enzyme involved in transcription and DNA repair. By using several synthetic nucleic acid substrates we demonstrate that WRN preferably unwinds RNA-containing Okazaki fragment-like substrates suggesting a role in lagging strand maturation of DNA replication. In contrast, DHX9 preferably unwinds RNA–RNA and RNA–DNA substrates, but fails to unwind Okazaki fragment-like hybrids. We further show that the preferential unwinding of RNA-containing substrates by WRN is stimulated by DHX9 in vitro , both on Okazaki fragment-like hybrids and on RNA-containing ‘chicken-foot’ structures. Collectively, our results suggest that WRN and DHX9 may also cooperate in vivo , e.g. at ongoing and stalled replication forks. In the latter case, the cooperation between both helicases may serve to form and to dissolve Holliday junction-like intermediates of regressed replication forks.
INTRODUCTION
The progeroid human disease Werner’s syndrome (WS) is a widely used model for studying molecular aspects of human aging [for recent reviews see e.g. ( 1 , 2 )]. WS represents a monogenic disease with mutations in the WRN gene, which encodes a member of the superfamily 2 (SF2) subgroup of RecQ helicases with 3′-5′ unwinding and a unique 3′-5′-exonuclease activity [see e.g. ( 3 )]. Initially, the recQ gene product was identified in bacteria as an enzyme presumed to be involved in recombination repair and that, among others, clears replication blockades from stalled or collapsed replication forks ( 4 , 5 ). Similarly, mutants in the SGS1 gene, the only RecQ helicase in Saccharomyces cerevisiae , display replisome instability and replication fork collapses ( 6 ). In addition, Sgs1 helicase is necessary to maintain the DNA polymerases α and ε at stalled replication forks ( 7 ). Cells from WS patients show a retarded progression through the S phase of the cell cycle ( 8 , 9 ), and in normally proliferating primary human fibroblast, about 60% of replication foci contain WRN ( 10 , 11 ). Together, this indicates an important but non-essential role in unperturbed DNA replication. Yet, whenever a replication fork comes to a halt, WRN is preferentially attracted to these sites ( 12 ). In line with this, WRN syndrome cells are hypersensitive to drugs that block ongoing replication forks, such as 4-nitroquinoline-N-oxide (4-NQO) or camptothecin ( 13 , 14 ). Apparently, WS cells display defects in the resolution of homologous recombination intermediates, particularly Holliday junctions (HJs) and ‘chicken-foot’ structures that result from collapsed replication forks ( 15 , 16 ).
Recently we have demonstrated that WRN functionally cooperates with DHX9 ( 17 ) [unofficial HUGO nomenclature used; other names are nuclear DNA helicase II (NDH II) or RNA helicase A (RHA)], another member of the SF2 family helicases with a 3′-5′ directionality ( 18 ). DHX9 coprecipitates with WRN and stimulates its 3′-5′ exonuclease activity, whereas DNA unwinding is inhibited ( 17 ). Moreover, a fraction of WRN and DHX9 colocalizes at centrosomes during interphase ( 19 ), and both enzymes interact with the mediator of homologous recombination BRCA1 ( 20 , 21 ). In addition, WRN and DHX9 interact with DNA-dependent protein kinase ( 22 , 23 ) and Ku70/Ku80 ( 24 , 25 ) that together form a checkpoint kinase involved in DNA damage signaling. Both helicases also bind to the sliding clamp for replicative DNA polymerases PCNA ( 26 , 27 ), while until now only WRN has been shown to interact with DNA polymerase δ ( 28 ), flap endonuclease I (FEN-1) ( 29 ), and the single-strand DNA-binding protein RPA ( 30 ), all of which are components of the Okazaki fragment maturation apparatus [see e.g. ( 31 )]. Collectively, these findings suggest a joint role for both enzymes in lagging strand maturation as well as in the rescue of stalled replication forks. Since every Okazaki fragment starts with a short piece of RNA, typically 8–12 nt in length ( 32 ), we tried to mimic this situation by designing appropriate template-primers and exploring the influence of 5′-RNAs on the unwinding kinetics of WRN. Using this approach we could show that WRN preferentially unwinds Okazaki fragment-like hybrids in a reaction that was stimulated by DHX9. Although WRN has been well characterized enzymologically, this new function further elucidates the role of WRN in DNA replication and lagging strand maturation. Based on our data we also suggest a model for the formation and dissolution of chicken-foot structures by the joint action of WRN and DHX9 that may facilitate a recombinational bypass of DNA damages.
MATERIALS AND METHODS
Expression and purification of recombinant proteins
Recombinant human DHX9 and WRN proteins were produced and purified from baculovirus-infected High-Five™ cells as described previously ( 17 ).
Radioactive labeling and annealing
5′-end labeling was achieved by T4 polynucleotide kinase from Fermentas (St. Leon-Rot, Germany) and γ- 33 P-ATP (5000 Ci/mmol) (Hartmann Analytical, Braunschweig, Germany) in ‘forward’ reaction buffer (80 mM Tris–HCl, pH 7.4, 10 mM MgCl 2 and 5 mM DTT) from Fermentas. Unincorporated material was removed from the labeled oligonucleotide using Microspin TM-25 columns from GE (Amhearst, UK), and, if necessary, by a successive run through an RNase-free Micro Bio-Spin P-30 column (Bio-Rad, Munich, Germany), equilibrated with 10 mM Tris-buffer, pH 8.0.
Helicase substrates
Highly purified oligonucleotides (listed in Table 1 ) were purchased from Purimex (Grebenstein, Germany). Oligonucleotides were synthesized in a 1000 nmol scale and subsequently purified either by polyacrylamide electrophoresis (RNA) or by HPLC (DNA). Oligonucleotide primers were designed to form a series of DNA structures as follows: Simple hybrids: 1–10 pmol/µl M1, M2, R1, R2, R1D, R4D and R8D ( Table 1 ) were 5′-labeled with [γ- 33 P]ATP and annealed to oligonucleotides containing a complementary region of 17 nt and a 3′-overhang. HJs were prepared by mixing the labeled oligonucleotide with a 2-fold molar excess of the complementary unlabeled strands. Samples were heated to 100°C for 5 min followed by incubation at 25°C for 3–4 h or cooling down overnight to room temperature.
Oligonucleotides used in this study
| Name of the oligonucleotide | Length | Sequence |
|---|---|---|
| DNA M1 | 17 | 5′CCTGCAGGCATGCAAGC3′ |
| DNA M2 | 34 | 5′GCTTGCATGCCTGCAGGCCAGCCTCAATCTCATC3′ |
| DNA M3 | 34 | 5′CTACTCTAACTCCGACCGCTTGCATGCCTGCAGG3′ |
| DNA M4 | 34 | 5′ATCCTCTCTAGAGTCGACCTGCAGGCATGCAAGC3′ |
| DNA-S1 | 34 | 5′GATGAGATTGAGGCTGGGAAAAGTTACTGTAGCC3′ |
| DNA-S2 | 34 | 5′GGCTACAGTAACTTTTCTCGACTCTAGAGAGGAT3′ |
| RNA R1 | 17 | 5′CCUGCAGGCAUGCAAGC3′ |
| RNA R2 | 34 | 5′GCUUGCAUGCCUGCAGGCCAGCCUCAAUCACAUC3′ |
| RNA–DNA R8D | 17 | 5′rCrCrUrGrCrArGrGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R4D | 17 | 5′rCrCrUrGrdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R1D | 17 | 5′rCdCdUdGdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R8M4 | 34 | 5′rArUrCrCrUrCrUrCdTdAdGdAdGdTdCdGdAdC dCdTdGdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
| Name of the oligonucleotide | Length | Sequence |
|---|---|---|
| DNA M1 | 17 | 5′CCTGCAGGCATGCAAGC3′ |
| DNA M2 | 34 | 5′GCTTGCATGCCTGCAGGCCAGCCTCAATCTCATC3′ |
| DNA M3 | 34 | 5′CTACTCTAACTCCGACCGCTTGCATGCCTGCAGG3′ |
| DNA M4 | 34 | 5′ATCCTCTCTAGAGTCGACCTGCAGGCATGCAAGC3′ |
| DNA-S1 | 34 | 5′GATGAGATTGAGGCTGGGAAAAGTTACTGTAGCC3′ |
| DNA-S2 | 34 | 5′GGCTACAGTAACTTTTCTCGACTCTAGAGAGGAT3′ |
| RNA R1 | 17 | 5′CCUGCAGGCAUGCAAGC3′ |
| RNA R2 | 34 | 5′GCUUGCAUGCCUGCAGGCCAGCCUCAAUCACAUC3′ |
| RNA–DNA R8D | 17 | 5′rCrCrUrGrCrArGrGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R4D | 17 | 5′rCrCrUrGrdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R1D | 17 | 5′rCdCdUdGdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R8M4 | 34 | 5′rArUrCrCrUrCrUrCdTdAdGdAdGdTdCdGdAdC dCdTdGdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
Oligonucleotides used in this study
| Name of the oligonucleotide | Length | Sequence |
|---|---|---|
| DNA M1 | 17 | 5′CCTGCAGGCATGCAAGC3′ |
| DNA M2 | 34 | 5′GCTTGCATGCCTGCAGGCCAGCCTCAATCTCATC3′ |
| DNA M3 | 34 | 5′CTACTCTAACTCCGACCGCTTGCATGCCTGCAGG3′ |
| DNA M4 | 34 | 5′ATCCTCTCTAGAGTCGACCTGCAGGCATGCAAGC3′ |
| DNA-S1 | 34 | 5′GATGAGATTGAGGCTGGGAAAAGTTACTGTAGCC3′ |
| DNA-S2 | 34 | 5′GGCTACAGTAACTTTTCTCGACTCTAGAGAGGAT3′ |
| RNA R1 | 17 | 5′CCUGCAGGCAUGCAAGC3′ |
| RNA R2 | 34 | 5′GCUUGCAUGCCUGCAGGCCAGCCUCAAUCACAUC3′ |
| RNA–DNA R8D | 17 | 5′rCrCrUrGrCrArGrGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R4D | 17 | 5′rCrCrUrGrdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R1D | 17 | 5′rCdCdUdGdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R8M4 | 34 | 5′rArUrCrCrUrCrUrCdTdAdGdAdGdTdCdGdAdC dCdTdGdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
| Name of the oligonucleotide | Length | Sequence |
|---|---|---|
| DNA M1 | 17 | 5′CCTGCAGGCATGCAAGC3′ |
| DNA M2 | 34 | 5′GCTTGCATGCCTGCAGGCCAGCCTCAATCTCATC3′ |
| DNA M3 | 34 | 5′CTACTCTAACTCCGACCGCTTGCATGCCTGCAGG3′ |
| DNA M4 | 34 | 5′ATCCTCTCTAGAGTCGACCTGCAGGCATGCAAGC3′ |
| DNA-S1 | 34 | 5′GATGAGATTGAGGCTGGGAAAAGTTACTGTAGCC3′ |
| DNA-S2 | 34 | 5′GGCTACAGTAACTTTTCTCGACTCTAGAGAGGAT3′ |
| RNA R1 | 17 | 5′CCUGCAGGCAUGCAAGC3′ |
| RNA R2 | 34 | 5′GCUUGCAUGCCUGCAGGCCAGCCUCAAUCACAUC3′ |
| RNA–DNA R8D | 17 | 5′rCrCrUrGrCrArGrGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R4D | 17 | 5′rCrCrUrGrdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R1D | 17 | 5′rCdCdUdGdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
| RNA–DNA R8M4 | 34 | 5′rArUrCrCrUrCrUrCdTdAdGdAdGdTdCdGdAdC dCdTdGdCdAdGdGdCdAdTdGdCdAdAdGdC3′ |
Helicase assays
The helicase assays contained the 33 P-labeled substrate in 10 µl of 20 mM Tris–HCl, pH 7.5, 3.5 mM MgCl 2 , 3.5mM ATP, 0.1 mg/ml BSA, 5mM dithiothreitol and 10% glycerol. Reactions were initiated by the addition of enzyme; incubation was between 0 and 30 min at 37°C. DNA unwinding was terminated by rapid cooling on ice and by the addition of 3.5 µl stop buffer (0.2% SDS, 50 mM EDTA, 0.1% bromophenol blue, 0.1% xylene cyanol, 40% glycerol) along with the addition of 10-fold excess of unlabeled oligonucleotide to prevent reannealing of the unwound strand. The products were separated by electrophoresis through 10–12 % non-denaturing polyacrylamide gels at 4°C, and visualized using a PhosphorImager and quantified using the ImageQuant software (GE Healthcare). WRN and DHX9 helicase were inactivated using 50 mM N-ethylmaleimide that, by forming irreversible cysteine bonds, knocked-down helicase activities ( 33 ). The rate of unwinding was calculated as (ssDNA/total DNA) × nM substrate/µM enzyme/time. The percentage of unwound substrate was determined as [(% unwinding) = 100 × (P/(S + P)], where P is the product and S is the residual substrate as obtained from the PhosphorImager ( 34 , 35 ). The values of P and S were corrected after subtracting background values as obtained from controls with no enzyme. Unwinding rates were determined using a linear regression through the initial linear part of the time course.
Electrophoretic mobility gel shift assay
DNA-binding reactions were performed in 10 µl 20 mM triethanolamine–HCl, pH 7.5, 2 mM MgCl 2 , with or without 1 mM ATPγS as indicated, 0.1 µg/ml BSA, 1 mM dithiothreitol, DNA and protein concentrations as given. Following incubation at room temperature for 30 min, the reaction was stopped with loading buffer (6% Ficoll, 0.1% bromophenol blue, 0.1% xylene cyanol, 40% glycerol) and the products were separated by electrophoresis through 4% nondenaturing polyacrylamide gels at 4°C for 2 h. Gels were dried, scanned and quantified as described above. The percentage of binding was calculated as the ratio between bound and unbound radiolabeled substrate. The Kd values were estimated from Scatchard analyses.
RESULTS
Both WRN and DHX9 preferably unwind RNA-containing heteroduplexes
We constructed several homo- and heteroduplexes with DNA and RNA ( Table 1 ) and determined the activities of WRN and DHX9 in parallel. To avoid different GC contents and a potential formation of stable secondary structures within one strand we always used the same sequence for DNA and RNA substrates in direct comparisons. Also, a 10-fold molar excess of the unlabeled oligonucleotide was used to mask out a potential reannealing of the unwound strand. Both enzymes unwound DNA:DNA duplexes with unwinding rates of about 0.24 and 0.08 nM (17-mer) min −1 µM −1 enzyme for WRN and DHX9, respectively ( Figure 1 A and B). WRN unwound an RNA:RNA 17-mer duplex with a 17-mer 3′ single-strand extension extremely poorly ( Figure 1 C), whereas DHX9 melted RNA:RNA strands with a rate of 0.72 nM min −1 µM −1 enzyme ( Figure 1 D), which confirms and extends earlier findings ( 36 , 37 ). DHX9 unwound the double-stranded (ds) RNA substrate R2:R1 within 5 min to about 80% and the DNA:RNA hybrid, M2:R1, to 72%. If one of the two strands consisted of DNA, DHX9 was slightly slower than WRN yielding unwinding rates of 0.74 and 0.86 nM (17-mer) min −1 µM −1 , respectively ( Figure 1 E and F). Quantifications of duplex unwinding by WRN and DHX9 are depicted in Figure 1 G and H, respectively. In summary, both enzymes unwound RNA-containing heteroduplexes considerably faster than dsDNA.
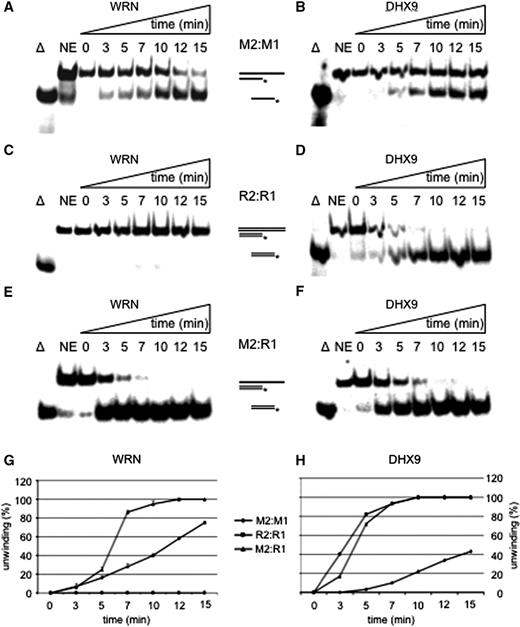
Unwinding of short fully hybridized strands. Time course for the unwinding of M2:M1 by WRN ( A ) or DHX9 ( B ), unwinding of R2:R1 by WRN ( C ) and DHX9 ( D ), and melting of M2:R1 by WRN ( E ) or DHX9 ( F ) 28 nM of WRN or of DHX9 and 1 nM of substrates were used for each experiment. Triangle represents heat-denatured DNA substrate control, NE no enzyme. The positions of the substrates and reaction products are indicated; double lines depict RNA containing strands. The asterisk represents the 5′-labeled nucleotide. Quantifications of duplex unwinding by WRN ( G ) and DHX9 ( H ) are also depicted. Error bars indicate SD as derived from three independent experiments.
WRN helicase preferably unwinds Okazaki fragment-like structures
Since WRN acted best on RNA–DNA hybrids, we next looked at Okazaki fragment-like substrates, consisting of a mixed 17-mer with an 8-mer of RNA at the 5′-end followed by a 9-mer of DNA that was hybridized to a 34-mer DNA template (R8D:M2, Table 1 ). Unexpectedly, DHX9 unwound R8D:M2 poorly ( Figure 2 A, lanes 8–12), even at a lower rate than the corresponding DNA:DNA substrate ( Figure 1 B), and about 10-fold slower than the corresponding RNA:DNA hybrid ( Figure 1 D). In striking contrast, 28 nM WRN dissociated half of the R8D:M2 construct in 1.5 min ( Figure 2 A, lanes 3–7), yielding an unwinding rate of ∼1.5 nM (17-mer) min −1 µM −1 enzyme. Hence, WRN displaced Okazaki fragment-like primers 6.2- and still 1.8-fold faster than the comparable DNA:DNA or RNA:DNA constructs (see also Supplementary Figure S1A ).
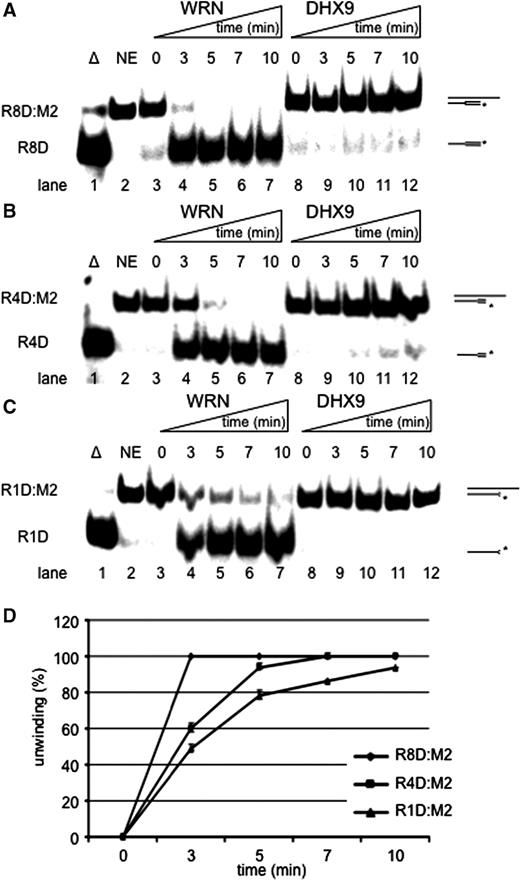
Unwinding of Okazaki fragment-like structures. ( A ) Time course of unwinding of R8D:M2. ( B ) Time course of unwinding of R4D:M2. ( C ) Time course of unwinding of R1D:M2. Unwinding of either construct by 28 nM WRN (left) or DHX9 (right) and 1 nM of substrate is shown in a 10-min time scale. Triangle represents, heat-denatured control; NE, no enzyme. 8R, 4R and 1R indicate the numbers of ribonucleotides at the 5′ ends. ( D ) Quantification of unwinding of R8D (diamonds), R4D (squares) and R1D (triangles) by WRN. Error bars indicate SD as derived from three independent experiments.
Next we examined the size of RNA that stimulated unwinding by WRN. To this end we designed 17-mer oligonucleotides with 5′-ends consisting of four (R4D) and only one (R1D) ribonucleotide and measured the efficiency of unwinding. Compared to R8D:M2, DHX9 unwound R4D:M2 and R1D:M2 even slower ( Figure 2 B and C, lanes 8–12). On the other hand, WRN displaced R4D and R1D from DNA in 3 min ( Figure 2 B and C, lanes 3–7), which was 2-fold slower than the unwinding of R8D but similar to the unwinding of a 17-mer RNA strand. 28 nM WRN melted R8D:M2 completely within 3 min while R4D:M2 and R1D:M2 became unwound to 60 and 49%, respectively ( Figure 2 D).
Therefore, apparently only one ribonucleotide at the 5′-end sufficed to mark the corresponding oligonucleotide for a rapid unwinding by WRN. On the first glance this may indicate that WRN recognizes the transition from RNA to DNA on one of the two strands. Alternatively, 5′-RNA–DNA hybrids or their assumed A-conformation may better fit into the active site of WRN and thereby facilitate unwinding. This scenario would also explain why full hybrids were better unwound than DNA double-strands.
DHX9 stimulates WRN-catalyzed unwinding of Okazaki fragment-like structures
Since DHX9 and WRN copurified and influenced each other’s activities ( 17 ) we looked at possible cooperative effects on Okazaki fragment-like substrates. Unwinding of R8D:M2 by WRN ( Figure 3 A, lanes 3 and 6) was stimulated by DHX9 ( Figure 3 A, lanes 5 and 8), in a time-dependent manner, while 14 nM DHX9 alone did not unwind the same template-primer ( Figure 3 A, lanes 4 and 7). Interestingly, DHX9 did not stimulate WRN on simple DNA:RNA ( Figure 3 B, lanes 3–8) or DNA:DNA substrates ( Figure 3 C, lanes 3–8) neither did it with RNA:RNA hybrids (data not shown). Taken together, DHX9 stimulated WRN on Okazaki fragment-like substrates about 2-fold ( Figure 3 D). Since this had not been observed with the RNA containing substrate M2:R1 ( Figure 3 B), we conclude that the stimulation was due to protein–protein interactions rather than to the trapping of melted pieces of RNA by DHX9. To decide whether this enhancement was due to a DHX9-mediated stimulation of WRN or, alternatively, due to the stimulation of DHX9 by WRN, we treated both helicases with 50 mM N -ethylmaleimide for 10 min. This procedure inactivated the unwinding activity of both enzymes ( Figure 3 E, lanes 3–5). However, NEM-inactivation could be rescued by supplementing the inactivation buffer with 100 mM DTT ( Figure 3 E, lanes 6–8). An amount of 14 nM each of NEM-treated WRN (w) and untreated DHX9 (D) did not unwind R8D:M2 ( Figure 3 F, lane 6). Instead, addition of 14 nM of poisoned DHX9 (d) stimulated WRN (W) as efficiently as untreated DHX9 did (cf. Figure 3 E, lane 8 with Figure 3 F, lane 7). This indicates that DHX9 exerts a stimulatory effect onto WRN independent of its helicase activity. Because of its high affinity for dsRNA, DHX9 should be well suited to resolve any potential RNA secondary structures at the 5′-end of an Okazaki fragment, which otherwise may prevent cleavage by FEN1 and/or unwinding by WRN ( 38 ).
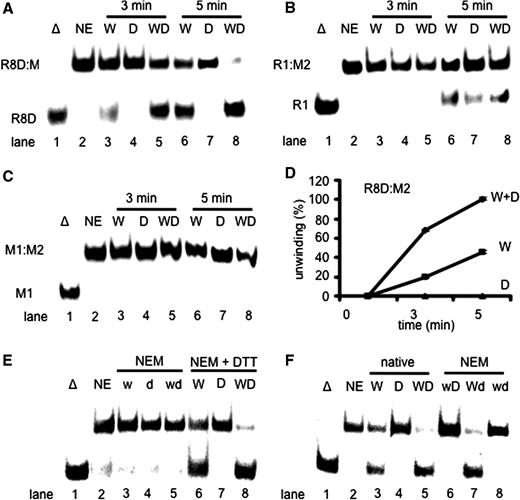
DHX9 stimulates the unwinding of Okazaki fragment-like structures by WRN. ( A ) Unwinding of 1 nM R8D:M2 catalyzed by 14 nM WRN (lanes 3 and 6), 14 nM DHX9 (lanes 4 and 7) or 14 nM each of WRN and DHX9 (lanes 5 and 8) after 3 min (lanes 3–5) or 5 min (lanes 6–8) incubation at 37°C. ( B ) The same experiment as shown in panel A except that 1 nM R1:M2 was used as substrate. ( C ) The same experiment as shown in panel A except that 1 nM M1:M2 was used. ( D ) Quantification of the unwinding of R8D:M2 by 14 nM WRN in the presence of 14 nM DHX9. ( E ) Unwinding of 1 nM R8D:M2 catalyzed by 21 nM poisoned WRN (w, lane 3), 21 nM poisoned DHX9 (d, lane 4), and 21 nM each of NEM-poisoned WRN and DHX9 (wd, lane 5). Poisoning could be rescued by adding 0.1 M DTT together with NEM (lanes 6–8). ( F ) Unwinding of 1 nM R8D:M2 catalyzed by 21 nM WRN (lane 3), DHX9 (lane 4), WRN plus DHX9 (lane 5), NEM-poisoned WRN plus untreated DHX9 (w, lane 6), NEM-poisoned DHX9 plus untreated WRN (Wd, lane 7), and poisoned WRN plus poisoned DHX9 (wd, lane 8). Error bars indicate SD as derived from three independent experiments.
WRN prefers binding to Okazaki fragments and other DNA–RNA substrates
To further explore the mode of binding of WRN to various substrates we performed gel shift assays. In the absence of ATP, WRN bound DNA–DNA (M2:M1) better than DNA–RNA (M2:R1) with Kd values of about 1 × 10 −7 M and 5 × 10 −7 M, respectively ( Figure 4 A). Surprisingly, there was little binding to the Okazaki fragment-like substrate R8D:M2 that only became detectable at the highest amount of the enzyme giving rise to a Kd of about 4 × 10 −6 M ( Figure 4 A). But in the presence of the poorly hydrolysable nucleotide ATPγS the affinity of WRN for the DNA–DNA substrate decreased significantly, yielding a Kd of about 3 × 10 −6 M (compare Figure 4 A and B), whereas binding to the DNA–RNA substrate appeared unaffected (5 × 10 −7 M) ( Figure 4 A and B). Interestingly, in the presence of ATPγS, WRN bound the Okazaki fragment-like oligonucleotide R8D:M2 much better ( Kd = 2 × 10 −7 M) than without a nucleotide cofactor (cf. lanes 3 and 4 in Figure 4 A and B). This behavior is not without precedence. The 5′-3′ helicase Pif1, which is also thought to be involved in Okazaki fragment unwinding, bound forked DNA–DNA and DNA–RNA substrates equally well in the absence of any nucleoside triphosphate ( 39 ). In the presence of ATPγS however, i.e. the condition that comes closest to DNA unwinding, DNA–DNA forks were bound much tighter than DNA–RNA forks ( 39 ). Despite this, Pif1 unwound RNA–DNA forks much better than the corresponding DNA–DNA forks, whereas WRN unwound the best-bound substrate R8D:M2 with the largest rate. Our data suggest that ATP stimulates binding of WRN to R8D:M2 and, as a consequence, its unwinding. Therefore, ATP may facilitate initial loading of the enzyme to this substrate and/or stabilize the enzyme substrate complex after loading to stimulate any unwinding.
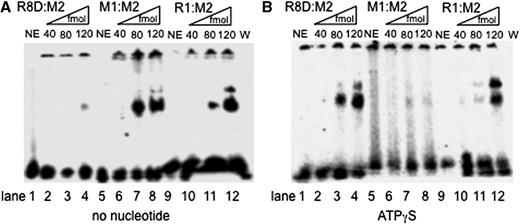
WRN binds to R8D:M2, M2:R1, and M2:M1 in an ATPγS-dependent manner. ( A ) Substrate (50 fmol) was incubated without (NE, lane 1) and with increasing amounts of WRN (as indicated) in the absence of any nucleotide. ( B ) The same experiment as shown in panel A, but this time in the presence of 1 mM ATPγS. The radiolabeled DNA species were analyzed on a native 4% PAGE and visualized by PhosphorImaging. The bands in the slots on top of the gel were also visible in many no enzyme controls and probably represent aggregated substrate.
DHX9 stimulated the unwinding of HJs by WRN helicase when one strand starts with an RNA piece
All our data strongly suggest that WRN plays a role in Okazaki fragment maturation. However, WRN is not an essential enzyme and other enzymes, such as the 5′-helicases Pif1 and Dna2, have been suggested to perform lagging strand maturation after strand displacement synthesis by DNA polymerase delta ( 32 ). Also, in normally proliferating primary human fibroblast, only 60% of replication foci contain WRN ( 10 , 11 ), while WRN clearly becomes attracted to sites where replication forks come to a halt, most likely because of an underlying DNA damage ( 12 ). Stalled replication forks may regress and form HJs and chicken-foot structures ( 40 , 41 ) that, because of an inevitable involvement of an Okazaki fragment, must begin with a short piece of RNA on one of the four strands. To account for this particular situation, we designed a HJ that started with an 8-nt long piece of RNA on one strand followed by 24 nt of DNA ( Table 1 , R8M4:M2:S1:S2). WRN unwound both the RNA and DNA containing HJ equally well ( Figure 5 A, lane 3, and Supplementary Figure S1 ), whereas DHX9 was not able to unwind either construct ( Figure 5 A, lane 4, and Supplementary Figure S2 ). In agreement with the observed stimulation of unwinding of RNA-containing pieces by WRN, DHX9 stimulated the unwinding of an RNA-primer containing HJ by WRN at about stoichiometric amounts ( Figure 5 A, lane 5), but apparently inhibited it when the junction consisted only of DNA. On this substrate an about 2-fold molar excess of WRN was needed to overcome the inhibitory action of DHX9 ( Supplementary Figure S2B ). Therefore in the presence of DHX9, WRN could only resolve HJ when these contained primer RNA on one of the four arms.
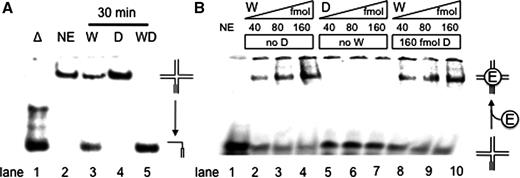
DHX9 stimulates the WRN-catalyzed branch migration of an RNA-primer containing HJ. ( A ) WRN-catalyzed (28 nM) unwinding of 1 nM of the synthetic RNA-primer HJ substrate R8M4:M2*:S1:S2. WRN unwound about half of the substrate within 30 min at 37°C (lane 3) while DHX9 failed to do so (lane 4). With 28 nM each of DHX9 and WRN complete unwinding was achieved (lane 5). The reaction products were resolved on a native 10% polyacrylamide gel and visualized by PhosphorImaging. ( B ) 60 fmol radiolabeled HJ were incubated with the indicated amounts of WRN and/or DHX9. After 20 min preincubation at 37°C in the presence of 1 mM ATPγS, the products were separated by electrophoresis through a 4% non-denaturing polyacrylamide gel at 4°C and visualized using a PhosphorImager. The bands in the slots on top of the gel were also visible in the no enzyme control (lane 1) and probably represent aggregated substrate.
The binding of WRN to an RNA-primer containing HJ was directly determined by gel mobility shift assays ( Figure 5 B). An amount of 40–160 fmol WRN produced a significant amount of shifted HJ products ( Figure 5 B, lanes 2–4), whereas the same amounts of DHX9 did not bind to the substrate ( Figure 5 B, lanes 5–7). Approximately 80% of the RNA-HJ was bound by 160 fmol of WRN ( Figure 5 B, lane 4), with a Kd value of about 5 × 10 −8 M. Substrate binding of WRN was not affected by the presence of 160nM DHX9 ( Figure 5 B, cf. lanes 2–4 and lanes 8–10). This indicates that DHX9 exerts its stimulatory effect on the branch migration activity of WRN by enhancing its catalytic constant ( kcat ) rather than by increasing its substrate affinity ( Ka ).
DISCUSSION
WRN and DHX9 represent a pair of helicases that copurify, coimmunoprecipitate, and cooperate at a functional level. Earlier, the interaction sites had been mapped to the N-terminal exonuclease domain of WRN as well as to the double-strand RNA-binding domain II (dsRBD II) and the arginine/glycine-rich RGG box of DHX9 that are situated N- and C-terminally, respectively. In this study, we observed that both helicases unwound fully hybridized DNA-strands, but with rather slow rates, whereas DNA:RNA heteroduplexes were melted considerably faster than DNA:DNA strands. This is remarkable since only a few of the currently reported helicases are known to unwind DNA–RNA hybrids efficiently. The effective unwinding of double-stranded hybrids initially indicated to us that RNA primers, which mark the beginning of Okazaki fragments, might be ideal substrates for both enzymes. This turned out to be true for WRN, since this helicase unwound 5′-R 8 D 9 -3′ oligonucleotides much faster than the corresponding DNA:DNA and even RNA:DNA strands. A successive shortening of the RNA piece from eight over four to only one nucleotide revealed that a single ribonucleotide at the 5′-end was sufficient to stimulate the unwinding activity of WRN, although unwinding became slower when there were fewer RNA nucleotides at the 5′-end. Nevertheless it is noteworthy that a DNA:DNA construct with a single 5′-ribonucleotide at a 17-mer strand became unwound as fast as the corresponding RNA:DNA hybrid. A single 5′-ribonucleotide remains whenever one of the two (potential) mammalian primer removing enzymes RNase H1 or 2 cleave off the RNA primer from an Okazaki fragment [for a recent review see ( 42 )]. Thus, even one remaining 5′-ribonucleotide sufficed to direct WRN helicase to Okazaki fragment unwinding. Here WRN may catalyze melting of the RNase H-cleaved fragment as a prerequisite for the subsequent cleavage by the FEN-1 endonuclease, which in turn is necessary to remove the error-prone DNA primer inserted by DNA polymerase α ( 43 ). Surprisingly, in vitro the WRN-catalyzed unwinding of Okazaki fragment-like mixed-hybrids was stimulated by DHX9 indicating that the latter enzyme may also become loaded into these sites in vivo . Because DHX9 preferentially removes RNA duplexes ( Figure 1 D), it is particularly suited to unwind RNA containing hairpin structures that in turn may obstruct cleavage by FEN-1 ( 41 ) or digestion by RNase H ( 42 ). In agreement with this view, both DHX9 and WRN interact with PCNA ( 26 , 27 ) and WRN has been shown to stimulate both the lagging strand DNA polymerase δ ( 28 ) and the primer-removing endonuclease FEN-1 ( 29 ). Therefore, WRN may play a role in Okazaki fragment unwinding as suggested earlier [see e.g. ( 44 )], perhaps together with the also discussed 5′-3′ helicases Dna2 and Pif1 ( 45 , 46 ).
In addition to the discussed role of DHX9 in Okazaki fragment unwinding this enzyme seems to aid WRN in restarting halted replication forks. The melting of the 5′-end of the ultimate Okazaki fragment is a prerequisite for the formation of a chicken-foot structure. At stalled replication forks DNA polymerase δ cannot accomplish strand displacement synthesis that in term is required for the loading of the 5′-3′-helicases Dna2 or Pif1. Thus a 3′–5′-helicase like WRN is necessary to displace the 5′-end of the Okazaki fragment next to the stalled fork. According to our results DHX9 would stimulate this reaction and contemporarily prevent secondary structure formation of the outmost 5′ RNA portion ( Figure 6 , top). The leading strand must also be unwound, perhaps by Dna2 or Pif1, or alternatively by the 5′–3′ helicase BACH1 (BRCA1 associated C-terminal helicase, also known as BRIP1 and FancJ), which may become attracted via the WRN- and DHX9-binding protein BRCA1 ( 47 ), and whose activity is required for the timely progression through S phase of the cell cycle ( 48 ). BACH1 senses and unwinds damages in one of the duplex strands ( 49 ) and therefore might act as the prime candidate 5′ helicase for chicken-foot formation on damaged DNA. Also, fork regression either requires positive supercoiling ahead of the stalled fork and/or the action of BLM helicase, which interestingly is part of the BRAFT super complex ( 50 ), comprising the BLM complex and the Franconia anemia complex including BACH1/FancJ ( 51 ). Since the strand annealing activity of DHX9 is stronger than that of WRN (unpublished observation), DHX9 may also help to form the RNA-containing HJ ( Figure 6 , center). Furthermore, once the regressed fork becomes resuscitated, e.g. after the underlying DNA damage has been repaired, DHX9 can stimulate dissolution of the chicken-foot by WRN ( Figure 6 , bottom). Hence, similar as and together with WRN, DHX9 may play a role in the removal of replication blockades.
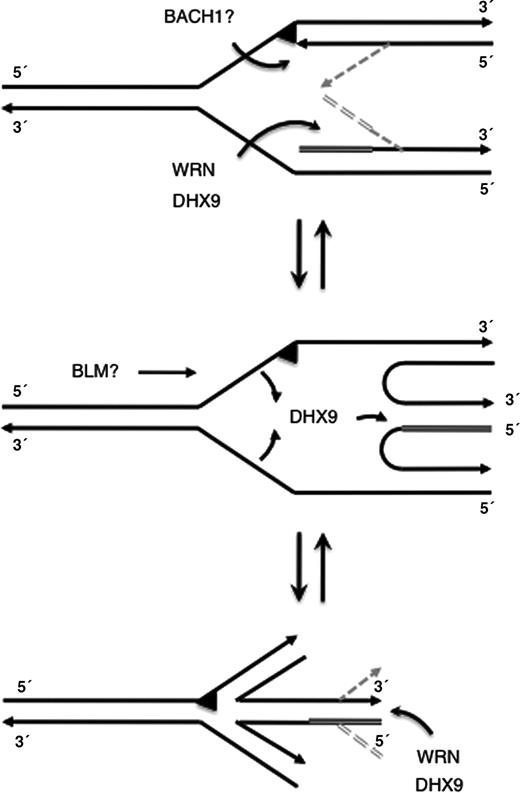
Model for a cooperation of WRN and DHX9 at stalled replication forks. The arrowheads indicate 3′-ends, the triangle indicates a damaged site, the double line shows primer RNA, and the gray lines display proposed intermediate structures. See text for more details.
Collectively, our findings strongly suggest cooperative functions for WRN and DHX9 at both migrating and stalled replication forks, where they seem to be involved in the removal of primer RNA-containing Okazaki fragments and the removal of replication hindrances by stimulating the dissolution and, as suggested here, also the formation of chicken-foot structures. Therefore, like WRN, DHX9 may significantly contribute to the maintenance of genomic stability. However, further investigations are warranted for a better understanding of the mechanism(s) involved in processing chicken-foot structures and intermediates of stalled replication forks.
FUNDING
Deutsche Forschungsgemeinschaft (grant number Gr 895/17-1); Fritz Lipmann Institute by State of Thuringia and the Federal Republic of Germany. Funding for open access charge: Fritz Lipmann Institute.
Conflict of interest statement . None declared.
ACKNOWLEDGEMENTS
The authors are indebted to Dr Helmut Pospiech for helpful suggestions and critically reading of the manuscript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments