





Abstract
DNA transcription depends on multimeric RNA polymerases that are exceptionally conserved in all cellular organisms, with an active site region of >500 amino acids mainly harboured by their Rpb1 and Rpb2 subunits. Together with the distantly related eukaryotic RNA-dependent polymerases involved in gene silencing, they form a monophyletic family of ribonucleotide polymerases with a similarly organized active site region based on two double-Ψ barrels. Recent viral and phage genome sequencing have added a surprising variety of putative nucleotide polymerases to this protein family. These proteins have highly divergent subunit composition and amino acid sequences, but always contain eight invariant amino acids forming a universally conserved catalytic site shared by all members of the two-barrel protein family. Moreover, the highly conserved ‘funnel’ and ‘switch 2’ components of the active site region are shared by all putative DNA-dependent RNA polymerases and may thus determine their capacity to transcribe double-stranded DNA templates.
INTRODUCTION
Transcription is catalysed by two distinct DNA-dependent RNA polymerases (RNAPs). Monomeric RNAPs, involved in the transcription of mitochondrial, chloroplastic and phage genomes, belong to a super-family of ‘right-handed’ DNA and RNA polymerases ( 1 ). However, all other genomes are transcribed by a highly conserved family of RNA polymerases, that was discovered 50 years ago in animal cells and has since then been found to operate in all cellular organisms. These enzymes have a remarkably conserved multi-subunit structure which has been investigated in great details in the bacterial, archaeal and yeast context, and also operate in nucleo-cytoplasmic DNA viruses ( 2–12 ). Together with the single-subunit RNA-dependent RNAPs involved in eukaryotic gene silencing, they form a monophyletic family of ‘two-barrel’ RNAPs ( 13 , 14 ), with a common organization based on two double-Ψ barrels structure with an invariant DxDxD Mg 2+ -binding signature.
These cellular and viral DNA-dependent RNAPs share a surprisingly large active site region of ∼500 amino acids, which are present in all bacterial, archaeal and eukaryotic lineages investigated so far and must therefore have already characterized the transcription enzyme of their last common unknown ancestor (LUCA). On the other hand, there is mounting evidence that a variety of extra-chromosomal genomes from bacteria and eukaryotes encode proteins with a far more distant kinship to canonical RNAPs, representing less than one-tenth of the active site mentioned above. These proteins have remarkably divergent amino acid sequences, but share a small core of eight amino acids with the transcription and gene-silencing enzymes mentioned above. Moreover, two additional motifs, corresponding to the switch 2 and funnel domains in the yeast and bacterial RNAPs, are present in all putative DNA-dependent RNAPs and in the proven but atypical transcription enzymes of insect Baculoviruses ( 15 ). The present survey will discuss these highly conserved elements and the structural and functional diversity of the new members of the ‘two-barrel’ protein family.
A COMPLEX ACTIVE SITE SHARED BY ALL CELLULAR DNA-DEPENDENT RNAPs
Following early studies on the bacterial ( Thermus aquaticus ) and yeast (RNAP II of Saccharomyces cerevisiae ) transcription enzymes ( 3 , 7 ), the complete crystal structure of several bacterial, archaeal and yeast DNA-dependent RNAPs has now been determined at an atomic level of resolution ( 4 , 5 , 9 , 11 , 16–18 ). Their strong conservation extends to five subunits (Rpb1, Rpb2, Rpb3, Rpb6 and Rpb11 in the eukaryotic RNAP II nomenclature adopted here) present in all cellular organisms and forming the entire core structure of bacterial RNAPs ( Table 1 ). In addition, the eukaryotic RNAP I, II and III have seven other core subunits (Rpb4, Rpb5, Rpb7, Rpb8, Rpb9, Rpb10 and Rpb12 in the case of RNAP II) which are also orthologous to archaeal subunits ( 2 , 4 , 5 , 19 , 20 ). Finally, several lineages of nucleo-cytoplasmic DNA viruses encode multi-subunit RNAPs which presumably derive from the eukaryotic RNAP II form ( 12 , 21 ). Their multi-subunit structure systematically includes the two largest subunits, Rpb1 and Rpb2, as well as the Rpb5 subunit which plays a critical role in the organization of the active site region ( 22 ). Subunits akin to Rpb3, Rpb6, Rpb8 and Rpb10 also exist in some viral lineages ( Table 1 ).
Subunit composition of DNA-dependent RNA polymerases
| Organisms | Subunit homology | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Eukaryotes | Pol II ( S. cerevisiae ) | Rpb1 | Rpb2 | Rpb3 | Rpb4 | Rpb5 | Rpb6 | Rpb7 | Rpb8 | Rpb9 | Rpb10 | Rpb11 | Rpb12 | |
| Pol I ( S. cerevisiae ) | Rpa190 | Rpa135 | Rpc40 | Rpa14 | Rpb5 | Rpb6 | Rpa43 | Rpb8 | Rpa12 | Rpb10 | Rpc19 | Rpb12 | ||
| Pol III ( S. cerevisiae ) | Rpc160 | Rpc128 | Rpc40 | Rpc17 | Rpb5 | Rpb6 | Rpc25 | Rpb8 | Rpc11 | Rpb10 | Rpc19 | Rpb12 | ||
| Archaea | M. jannaschii | A′ + A″ | B′ + B″ | D | F | H | K | E | M/TFS | N | L | P | ||
| S. solfataricus | A′ + A″ | B | D | F | H | K | E | G | M/TFS | N | L | P | ||
| Viruses | Ap MV | + | + | + | + | + | ||||||||
| ASFV | + | + | + | + | + | + | ||||||||
| Eh V | + | + | + | + | + | + | ||||||||
| Iridoviruses | + | + | + | |||||||||||
| Poxiviruses | Rpo1 | Rpo2 | + | Rpo22 | Rpo30 | Rpo7 | ||||||||
| Bacteria | Bacteria (e.g. E. coli ) | β′ | β | α | ω | α | ||||||||
| Cyanobacteria | γ + β′ | β | α | ω | α | |||||||||
| Helicobacteria | β::β′ | α | ω | α | ||||||||||
| Yeast plasmids | pGKL2 ( K. lactis ) | Rpb2::Rpb1 (Orf6) | ||||||||||||
| Organisms | Subunit homology | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Eukaryotes | Pol II ( S. cerevisiae ) | Rpb1 | Rpb2 | Rpb3 | Rpb4 | Rpb5 | Rpb6 | Rpb7 | Rpb8 | Rpb9 | Rpb10 | Rpb11 | Rpb12 | |
| Pol I ( S. cerevisiae ) | Rpa190 | Rpa135 | Rpc40 | Rpa14 | Rpb5 | Rpb6 | Rpa43 | Rpb8 | Rpa12 | Rpb10 | Rpc19 | Rpb12 | ||
| Pol III ( S. cerevisiae ) | Rpc160 | Rpc128 | Rpc40 | Rpc17 | Rpb5 | Rpb6 | Rpc25 | Rpb8 | Rpc11 | Rpb10 | Rpc19 | Rpb12 | ||
| Archaea | M. jannaschii | A′ + A″ | B′ + B″ | D | F | H | K | E | M/TFS | N | L | P | ||
| S. solfataricus | A′ + A″ | B | D | F | H | K | E | G | M/TFS | N | L | P | ||
| Viruses | Ap MV | + | + | + | + | + | ||||||||
| ASFV | + | + | + | + | + | + | ||||||||
| Eh V | + | + | + | + | + | + | ||||||||
| Iridoviruses | + | + | + | |||||||||||
| Poxiviruses | Rpo1 | Rpo2 | + | Rpo22 | Rpo30 | Rpo7 | ||||||||
| Bacteria | Bacteria (e.g. E. coli ) | β′ | β | α | ω | α | ||||||||
| Cyanobacteria | γ + β′ | β | α | ω | α | |||||||||
| Helicobacteria | β::β′ | α | ω | α | ||||||||||
| Yeast plasmids | pGKL2 ( K. lactis ) | Rpb2::Rpb1 (Orf6) | ||||||||||||
Subunit symbols are as in refs ( 2 , 3 , 7 , 19 ). Note that M. jannaschii and the other euryarchaea have no G subunit ( 20 ). The (+) symbol corresponds to predicted viral subunits, except for poxiviruses, where a specific nomenclature exists for vaccinia ( 12 ). The abbreviated species names correspond to Saccharomyces cerevisiae , Methanocaldococcus jannaschii , Sulfolobus solfataricus, Escherichia coli and Kluyveromyces lactis. Ap MV, ASFV and Eh V stand for the Acanthamoeba polyphaga mimivirus, African swine fever virus and Emiliana huxleyi virus, respectively.
Subunit composition of DNA-dependent RNA polymerases
| Organisms | Subunit homology | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Eukaryotes | Pol II ( S. cerevisiae ) | Rpb1 | Rpb2 | Rpb3 | Rpb4 | Rpb5 | Rpb6 | Rpb7 | Rpb8 | Rpb9 | Rpb10 | Rpb11 | Rpb12 | |
| Pol I ( S. cerevisiae ) | Rpa190 | Rpa135 | Rpc40 | Rpa14 | Rpb5 | Rpb6 | Rpa43 | Rpb8 | Rpa12 | Rpb10 | Rpc19 | Rpb12 | ||
| Pol III ( S. cerevisiae ) | Rpc160 | Rpc128 | Rpc40 | Rpc17 | Rpb5 | Rpb6 | Rpc25 | Rpb8 | Rpc11 | Rpb10 | Rpc19 | Rpb12 | ||
| Archaea | M. jannaschii | A′ + A″ | B′ + B″ | D | F | H | K | E | M/TFS | N | L | P | ||
| S. solfataricus | A′ + A″ | B | D | F | H | K | E | G | M/TFS | N | L | P | ||
| Viruses | Ap MV | + | + | + | + | + | ||||||||
| ASFV | + | + | + | + | + | + | ||||||||
| Eh V | + | + | + | + | + | + | ||||||||
| Iridoviruses | + | + | + | |||||||||||
| Poxiviruses | Rpo1 | Rpo2 | + | Rpo22 | Rpo30 | Rpo7 | ||||||||
| Bacteria | Bacteria (e.g. E. coli ) | β′ | β | α | ω | α | ||||||||
| Cyanobacteria | γ + β′ | β | α | ω | α | |||||||||
| Helicobacteria | β::β′ | α | ω | α | ||||||||||
| Yeast plasmids | pGKL2 ( K. lactis ) | Rpb2::Rpb1 (Orf6) | ||||||||||||
| Organisms | Subunit homology | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Eukaryotes | Pol II ( S. cerevisiae ) | Rpb1 | Rpb2 | Rpb3 | Rpb4 | Rpb5 | Rpb6 | Rpb7 | Rpb8 | Rpb9 | Rpb10 | Rpb11 | Rpb12 | |
| Pol I ( S. cerevisiae ) | Rpa190 | Rpa135 | Rpc40 | Rpa14 | Rpb5 | Rpb6 | Rpa43 | Rpb8 | Rpa12 | Rpb10 | Rpc19 | Rpb12 | ||
| Pol III ( S. cerevisiae ) | Rpc160 | Rpc128 | Rpc40 | Rpc17 | Rpb5 | Rpb6 | Rpc25 | Rpb8 | Rpc11 | Rpb10 | Rpc19 | Rpb12 | ||
| Archaea | M. jannaschii | A′ + A″ | B′ + B″ | D | F | H | K | E | M/TFS | N | L | P | ||
| S. solfataricus | A′ + A″ | B | D | F | H | K | E | G | M/TFS | N | L | P | ||
| Viruses | Ap MV | + | + | + | + | + | ||||||||
| ASFV | + | + | + | + | + | + | ||||||||
| Eh V | + | + | + | + | + | + | ||||||||
| Iridoviruses | + | + | + | |||||||||||
| Poxiviruses | Rpo1 | Rpo2 | + | Rpo22 | Rpo30 | Rpo7 | ||||||||
| Bacteria | Bacteria (e.g. E. coli ) | β′ | β | α | ω | α | ||||||||
| Cyanobacteria | γ + β′ | β | α | ω | α | |||||||||
| Helicobacteria | β::β′ | α | ω | α | ||||||||||
| Yeast plasmids | pGKL2 ( K. lactis ) | Rpb2::Rpb1 (Orf6) | ||||||||||||
Subunit symbols are as in refs ( 2 , 3 , 7 , 19 ). Note that M. jannaschii and the other euryarchaea have no G subunit ( 20 ). The (+) symbol corresponds to predicted viral subunits, except for poxiviruses, where a specific nomenclature exists for vaccinia ( 12 ). The abbreviated species names correspond to Saccharomyces cerevisiae , Methanocaldococcus jannaschii , Sulfolobus solfataricus, Escherichia coli and Kluyveromyces lactis. Ap MV, ASFV and Eh V stand for the Acanthamoeba polyphaga mimivirus, African swine fever virus and Emiliana huxleyi virus, respectively.
Sequence alignments based on current genome databases ( http://blast.ncbi.nlm.nih.gov/Blast.cgi ) reveal ∼1000 amino acid positions conserved in all archaeal and eukaryotic nuclear RNAPs. Thus, complex RNAPs essentially similar to those of present-day eukaryotes and archaea existed before the ancestral divergence of the archaeal and eukaryotic kingdoms. The spatial distribution of these amino acids is illustrated in Figure 1 A for yeast RNAP II. About half of them are also present in bacterial RNAPs, as predicted by inspecting the genome sequences of all bacterial lineages ( Supplementary Data S1 ). They correspond to some 25 discrete domains of the two large subunits, Rpb1 and Rpb2, with some minor contribution of Rpb3, Rpb6 and Rpb11 ( Figure 1 B and C), and collectively form a very complex active site organized around two catalytic Mg 2+ ions, called MgA and MgB. Each Mg 2+ is harboured by one of the double-Ψ barrels mentioned above (hereafter called barrel A and B), which are connected by a large ‘bridge’ helix and are associated with a complex network of conserved domains ( 6–9 , 17 ).
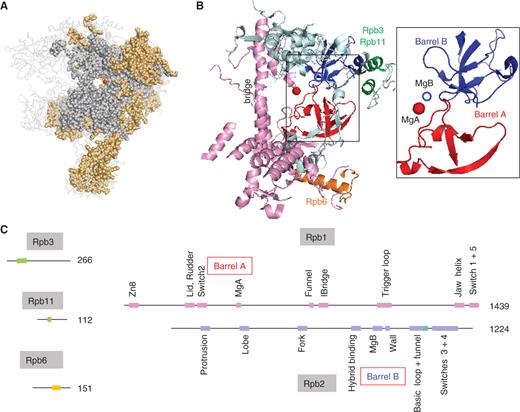
Conserved motifs shared by the bacterial, archaeal and eukaryotic RNAPs. ( A ) Conserved amino acids are shown as grey (all cellular organisms) or orange spheres (archaeal RNAPs and eukaryotic RNAP I, II and III). The red dot denotes the catalytic MgA. This figure corresponds to the sequence alignments provided as Supplementary Data S1 and is based on the 1WCM PDB coordinates showing the complete twelve-subunit structure of yeast RNAP II ( 11 ), using the Pymol software ( http://www.pymol.org ). ( B ) Spatial organization of the amino acid positions common to all bacterial, archaeal and eukaryotic RNAPs (I, II and III). Experimental data are as in Figure 1 A above. Rpb1, Rpb2, Rpb3/11 and Rpb6 domains are in pink, blue, green and orange, respectively. The box illustrates the Barrel A (red) and B (blue) motifs. The corresponding yeast RNAP II positions are 346–365 and 444–487 (Rpb1) and 822–844, 974–1011 and 1074–1089 (Rpb2). ( C ) Distribution of the corresponding motifs on the Rpb1, Rpb2, Rpb3, Rpb6 and Rpb11 subunits. Colour symbols are as in (B) above. Individual domains are named according to ref. ( 7 ), expect for part of the Rpb2 hybrid binding domain (positions 964–1028) which is referred to as the basic loop and Rpb2 funnel domains (see text for explanations).
YEAST PLASMIDS ENCODE A PUTATIVE CYTOPLASMIC RPB2::RPB1 FUSION
Several budding yeast species are able to direct killer toxins again other yeast species and to resist their own toxin production. Early studies have shown that, in the dairy yeast Kluyveromyces lactis , these properties are governed by two small cytoplasmic DNA genomes, pGKl1 and pGKl2 ( 23 ). The evolutionary origin of these cytoplasmic plasmids is quite puzzling. They are apparently restricted to a small number of closely related yeasts ( K. lactis, Lachancea kluyveri, Pichia acacia and P. etschelli ) and are so far unknown in other eukaryotic lineages, including well-characterized yeasts such as S. cerevisiae or Schizosaccharomyces pombe . In K. lactis , pGKl1 ensures the production of the yeast toxin and the corresponding toxin resistance mechanism, whereas pGKl2 provides cytoplasmic replication and transcription enzymes which are extremely simplified version of their nuclear counterparts ( 24 , 25 ).
One of the pGKl2 gene products is a 982 amino acid protein, Orf6, which has not been biochemically characterized so far but clearly corresponds to an Rpb2::Rpb1-like fusion and thus belongs to the cytoplasmic transcription system ( 24 , 25 ). Orf6 presumably derives from some ancestral yeast Rpb1 and Rpb2 (or their RNAP I and RNAP III paralogues), which was followed by a strongly divergent evolution that fused these two genes, deleted about half of their coding sequence and inserted Orf6-specific domains downstream of the N-terminal Rpb2 ‘protrusion’ and at the junction of the Rpb2 and Rpb1 halves, respectively. It was initially thought that the homology of Orf6 to Rpb2 and Rpb1 was restricted to a few amino acids ( 24 , 25 ). However, updated sequence alignments now extend this homology to ∼250 amino acids. They correspond to 11 motifs forming about half of the active site shared by all bacterial, archaeal and eukaryotic RNAPs ( Figure 2 A, Supplementary Data S1 ).
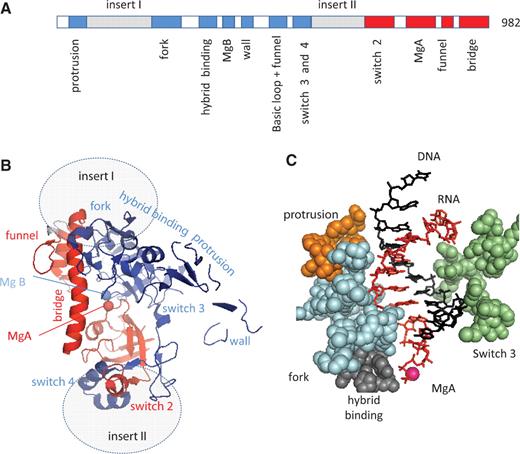
Conserved motifs of the Orf6 protein. ( A ) Distribution of the Orf6-specific (inserts I and II) or Rpb1 (red) and Rpb2 (blue) conserved domains on the Orf6 amino acid chain, based on the sequence alignments provided as Supplementary Data S1 . ( B ) Spatial organization in the yeast RNAP II crystal structure (upper view). This figure is based on the 1R9S PDB coordinates showing the elongating RNAP II ( 8 ) using the same colour code and same orientation as in Figure 1 B above. Grey ovals denote the hypothetical insertion sites of the Orf6-specific domains. ( C ) Spatial organization of the conserved protrusion, fork, hybrid binding and switch 3 domains of Rpb2 on each side the RNA/DNA hybrid structure (side view). The orange, blue, grey and green spheres highlight positions 199–210 (protrusion), 466–512 (fork), 763–778 (hybrid binding) and 1106–1131 (switch 3) of yeast Rpb2, respectively. The first nine complementary bases of the RNA and template DNA strand are shown in red and blue, respectively.
Figure 2 B shows the spatial distribution of these conserved domains, as projected on the yeast RNAP II crystal structure. It contains the two double-Ψ barrels mentioned above, connected by the bridge helix and combined with most of the conserved Rpb2 domains also present in the bacterial, archaeal and yeast RNAP II structure. Among them, the protrusion, fork, hybrid binding and switch 3 domains are close to barrel B and surround the RNA/DNA hybrid ( Figure 2 C). On the other hand, Orf6 has no detectable homology to the highly conserved trigger, switch 1 and switch 5 domains of Rpb1, and also lack the Rpb1 rudder and lid, which form a somewhat less conserved protruding loop domain ( 6–9 , 17 ), suggesting that these domains may not be strictly crucial for transcription, at least in terms of amino acid sequences.
INSECT DNA VIRUSES ENCODE ATYPICAL RNAPs AND TFIIS FACTORS
Viral multi-subunit RNAPs are typically encoded by nucleo-cytoplasmic large DNA viruses with genomes ranging between ∼120 kb (poxviruses) and the 1.2 Mb of the Amoeba polyphaga mimivirus ( 12 , 21 , 26 ). These RNAPs operate during the cytoplasmic phase of their infectious cycle, when the viral DNA is not accessible to the host nuclear transcription system. Other viruses such as the algal Phycodnaviruses have genomes ranging between 185 and 325 kb, but do not reside in their host cytoplasm and are transcribed by their host nuclear transcription system ( 27 ). Presumably, this also holds for other large DNA viruses such as the shrimp white spot virus (305 kb) and hypertrophic salivary gland virus of Glossinia pallidipes (190 kb) which have no RNAP subunit genes ( 28 , 29 ).
The well-characterized insect Baculoviruses, which have DNA genomes of up to 160 kb, do not reside in the cytoplasm of their host, but have an interesting pattern of transcription since a relatively small number of viral genes, specifically expressed during their late infectious cycle, depends on a virus-specific multi-subunit RNAP ( 15 ). These enzymes are made of four subunits (called p47, LEF-4, LEF-8 and LEF-9, where LEF stands for late expression factor). They are unrelated to canonical RNAPs, except for a limited similarity of its LEF-9 and LEF-8 subunits to Rpb1 and Rpb2, respectively. Nudiviruses, a sister-group to Baculoviruses, also contain open reading frames weakly but significantly related to the four baculoviral RNAP subunits ( 30 ). Moreover, cDNAs encoding the LEF-5 and LEF-8 proteins occur in a library prepared from polydnaviral particles of braconid wasps, which presumably originate from a nudiviral ancestor ( 31 ).
The LEF-8 and LEF-9 proteins are therefore restricted to insect viruses, and are themselves rather poorly related between the nudi- and baculoviral lineages. This is best explained by assuming that they originate from eukaryotic Rpb1 and Rpb2 ancestor genes (or of the equivalent subunits of the other two RNAPs) which underwent a fast and highly divergent evolution obliterating anything that was not strictly needed for DNA transcription. Indeed, a combination of Psi-blast search and visual alignment indicates that their overall similarity to Rpb1 and Rpb2 is limited to ∼40 amino acids, which may correspond to a minimal active site region strictly required for the transcription of DNA templates ( Figure 3 A and Supplementary Data S1 ). These amino acids belong to the MgA and MgB barrels mentioned above, to the Rpb1 switch 2 domain (positions 329–367 in S. cerevisiae ) and to a bipartite ‘funnel’ formed by the conserved Rpb1 (746–758) and Rpb2 (1004–1028) motifs near the NTP entry site of yeast RNAP II ( 8 ).
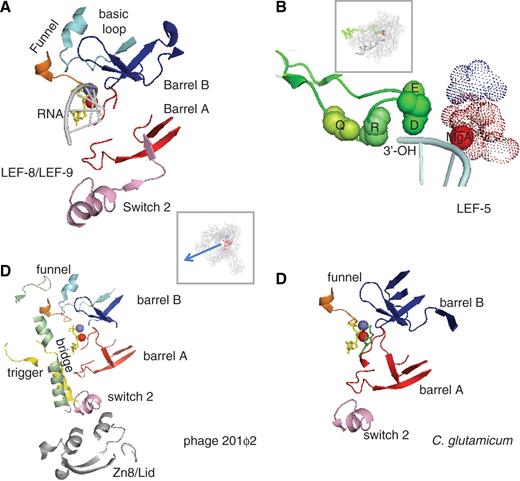
Conserved active centre in non-canonical RNAPs. ( A ) Conserved amino acids in LEF-9/Rpb1 and LEF-8/Rpb2. This figure is based on the 1R9S PDB coordinates which corresponds to the ‘entry’ position incoming NTP ( 8 ). Conserved amino acids were as defined in Supplementary Data S1 . The Rpb1 and Rpb2 components of the funnel domain are in orange and light blue, respectively. The box shows the entire RNAP II structure in the same spatial orientation. ( B ) Conserved Lef5/TFIIS C-terminal domain. Four invariant amino acids (Q285, R287, D290 and E291 in the yeast TFIIS) are space-filled (green spheres). The dotted spheres correspond to the invariant carboxylic acids of LEF-9/Rpb1 (D479, D481, D483 of Rpb1, in red) and LEF-8/Rpb2 (D837 of Rpb2, shown in blue). The spatial organization of the corresponding TFIIS, Rpb1 and Rpb2 motifs was taken from the PDB 3GTM2 coordinates which represent RNAP II in its back-tracking conformation ( 36 ). The box shows the 10-subunit RNAP II structure in the same spatial orientation. ( C ) Conserved amino acids in gp275, gp139 and gp273/274 (phage 201ϕ2). Conserved amino acids were as defined in Supplementary Data S1 . This figure is based on the 1R9S PDB coordinates ( 8 ), with the same colour code. The bridge, trigger and Zn8/lid domains, which are additionally conserved in gp275, gp130 and gp139 (see Supplementary Data S1 ) are shown in green, yellow and grey, respectively. Same spatial organization as in (A). ( D ) Conserved amino acids in NCgl 1702 ( C. glutamicum ). Same spatial organization as in (A).
Baculoviral transcription also depends on LEF-5, another highly conserved late expression factor which functionally interacts with the viral RNAP ( 32 ). LEF-5 has homology with the eukaryotic TFIIS factor associated with RNAP II, RNAP III and perhaps RNAP I ( 33 ), with the archaeal TFS, which is akin the eukaryotic subunits Rpa12 (RNAP I) and Rpc11 (RNAP III) and may itself be regarded as a dissociable RNAP subunit ( 10 , 34 ). TFIIS-like factors exist in all lineages of nucleo-cytoplasmic large DNA viruses investigates so far ( Table 1 ) and are also encoded by Phycodnaviruses ( 27 ) and by the G. pallidipes insect virus ( 29 ) which, has mentioned above, lack RNAP subunit genes. Thus, a highly conserved family of TFIIS-like proteins exists in all archaeal, eukaryotic and viral transcription systems, including the atypical late expression transcription of baculo- and nudiviruses. All these proteins share a C-terminal Zn loop with an invariant QxRxxDE motif ( Supplementary Data S1 ), where D and E are immediately close to the catalytic MgA and MgB in a yeast TFIIS/RNAP II binary complex ( Figure 3 B). This triggers the latent transcript cleavage activity of the RNAP, which removes the backtracked part of the RNA in stalled RNAP molecules and allow them to resume RNA synthesis ( 35 , 36 ).
TFIIS, Rpc11 and the archaeal TFS primarily act as elongation factors, whereas LEF-5 factor appears to be a transcription initiation factor ( 32 ), but recent data suggest that yeast TFIIS may be especially critical in a very early phase of the transcription cycle ( 10 ). Hence, these factors appear to have a universally conserved role based on the activation of a latent transcript cleavage activity of their cognate RNAP. Strikingly, the transcript cleavage activity of bacterial RNAPs depends on conserved GreA/B factors present in all bacterial lineages except cyanobacteria. They are unrelated to TFIIS but also contain an invariant GDLxEN signature with two Mg-binding carboxylic acids ( 37 ). Intriguingly, they are encoded by the chromosomes of a few Eukaryotes such as Trichomonas vaginalis ( 38 ), where they presumably result from horizontal gene transfer ( Supplementary Data S1 ).
ATYPICAL RNAPs MAY ENSURE THE TRANSCRIPTION OF LARGE DNA PHAGES
The transcription of DNA phages usually depends on the host transcription system or on phage-encoded RNAPs similar to the monomeric enzyme of phage T7. However, the recent sequencing of three closely related lytic phages isolated from Pseudomonas aeruginosa (ϕEL, ϕKZ180) and P. chlororaphis (201ϕ2) has revealed two sets of proteins distantly related to the β′ (Rpb1) and β (Rpb2) subunits of canonical RNAPs ( 39 , 40 ). Mass spectrometry data show that three of them (gp275, gp139 and gp273/724) are present in the 201ϕ2 virion. The gp275 and gp139 proteins correspond to β′-like N- and C-halves, respectively, whereas gp273/274 is homologous to β for its C-terminal gp274 part, which separated from gp273 separated by a mobile intron. In 201ϕ2, gp273/274 forms a single band in SDS/polyacrylamide gel electrophoresis, but its two domains may correspond to distinct open reading frames in ϕEL. A second pair of β′- and β-related proteins corresponds to the gp129 (β-like) and to gp107 (β′-like N-half) and gp130 (β′-like C-half) gene products of 201ϕ2. They are not detected in the virion and are thus probably expressed upon infection of the bacterial host ( 39 ).
These data strongly suggest the existence of two atypical and functionally distinct phage RNAPs ( 39 ). Given the lack of α- or ω-like gene products, these transcription enzymes may possibly have a dimeric structure limited to two large subunits. Their amino acid sequences have a limited homology to the canonical β′ (Rpb1) and β (Rpb2), and part of this homology corresponds to regions that are specific of the bacterial β′ or β subunits (see the sequence alignments shown as Supplementary Data S1 ). As in the case of the insect baculo- and nudiviruses mentioned above, this is best explained by assuming that these phage proteins derive from bacterial β′- and β-encoding genes which underwent a fast and highly divergent evolution obliterating anything that was not strictly needed for DNA transcription.
In summary, there are now four well-documented examples of proven (Baculoviruses) or putative DNA dependent RNAPs which, as far we can tell, originated from the multi-subunit RNAPs of some some bacterial ( Pseudomonas phages) or eukaryotic ancestors (insect viruses) and independently evolved towards highly atypical transcription enzymes. Strikingly, the phage enzymes share a common set of ∼40 conserved amino acids, that are closely associated in the spatial structure of multi-subunit RNAPs and correspond to the conserved two-barrel, switch 2 and funnel domains also characterizing the LEF-9 and LEF-8 RNAP subunits of the baculo- and nudiviruses ( Figure 3 C and Supplementary Data S1 ). As noted below, the latter two domains are absent from RNA-dependent RNA polymerases, thus strongly suggesting that they are strictly critical for the transcription of DNA templates.
ϕEL and ϕKZ180 have genomes of 211 and 280 kb, respectively, and 201ϕ2 (317 kb) is actually the largest phage genome sequenced so far ( 39 ). This raises the possibility that other long-genome phages, which are notably under-represented in current genome sequences, may have undetected atypical RNAPs. Two pieces of evidence suggest that this is indeed the case. First, the Bacillus subtilis phage PBS2 has a poorly characterized RNAP with four polypeptides of 48, 58, 76 and 80 kDa ( 41 ), which happen to match the calculated size of the 201ϕ2 RNAP subunits mentioned above. Second, the Corynebacterium glutamicum genome encodes an unusually large (2169 amino acids) NCg1 1702 protein which is distantly related to a β::β′ fusion ( 13 ). A close inspection of its amino acid sequence reveals ∼40 conserved amino acids, which again correspond to the conserved two-barrel, switch 2 and funnel (Rpb1) domains common to the baculoviral and putative phage RNAPs mentioned above ( Figure 3 D, Supplementary Data S1 ). The remaining 98% of NCg11702 bear no obvious relation to any other protein, even in closely related corynebacterial genomes. NCg11702 is therefore an unusually a fast-evolving protein, which may have been recruited to the C. glutamicum from an unidentified phage or mobile element.
PHAGE AND BACTERIAL YONO-LIKE PROTEINS
Single-subunit RNA-dependent RNAPs participate in various forms of RNA-mediated gene silencing in a large range of eukaryotes. Within the same organism, they often coexist under structurally and functionally diverse forms such as the three distinct enzymes of Neurospora crassa : Sad1 (involved in meiotic silencing), the poorly characterized Rrp3 protein and Qde1, distantly related to the first two and participating in post-transcriptional gene silencing ( 14 , 42 ). Their homology is chiefly restricted to a bipartite domain of ∼60 amino acids, which corresponds to positions 691–747 and 951–1014 of Qde1 and is present in all eukaryotic RNA-dependent RNAPs identified so far ( Figure 4 A and Supplementary Data S2 ). As shown in Figure 4 B, this bipartite domains forms the most conserved part of the Qde1 two-barrel structure ( 13 ).
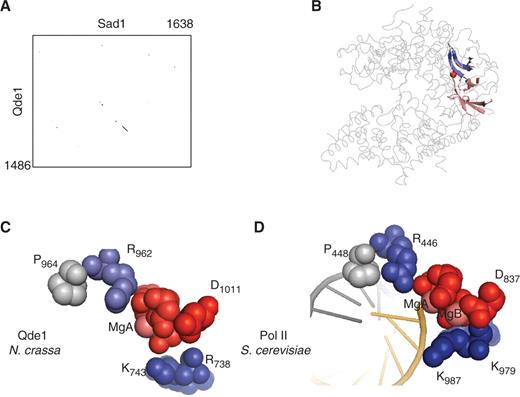
A minimal active site shared by all two-barrel RNAPs. ( A ) Homology of the Neurospora crassa RNA-dependent RNAPs (Qde1 and Sad1). The two sequences were aligned by a Blosum 6 matrix at a stringency of 6/23, using the Strider 1.4f6 software ( 61 ). ( B ) Spatial organization of the conserved domain in eukaryotic RNA-dependent RNAPs. This figure is based on the 2J7N PDB coordinates ( 13 ). Positions 691–747 (blue) and 955–1014 (red) correspond to the conserved domains shown as in Supplementary Data S2 . The less conserved part of the two barrels is in dark grey. The rest of the Qde1 structure, which is poorly conserved, is represented by light grey lines. The red sphere denotes MgA. ( C ) Blow-up of the eight invariant amino acids shared by all two barrel RNAPs in Qde1 ( N. crassa ). Spheres correspond to MgA, MgB and eight invariant amino acid positions shown in red (D 709 , D 1007 , D 1009 and D 1011 ), blue (R 738 , K 743 , R 962 ) and grey (P 964 ), based on the PDB 2J7N coordinates ( 14 ). ( D ) Blow-up of the eight invariant amino acids shared by all two barrel RNAPs in RNAP II ( S. cerevisiae ). The corresponding invariant positions are Rpb1 D 479 , D 481 , D 483 and Rpb2-D 837 (red), Rpb1-R 446 , Rpb2-K 979 and Rpb2-K 987 (blue) and Rpb1-P 448 (grey), as taken from PDB 1R9T ( 8 ).
Iyer et al. ( 13 ) noted a distant similarity of this bipartite domain to the YonO protein of B. subtilis ( 43 ), originating from an inserted prophage (SPβc2), and a similar open reading frame occurs in the 219 kb DNA genome of the ϕ8–36 phage of Bacillus thuringensis ( 44 ). Our updated Psi-blast search now reveals Yono-like proteins in five different firmicute phages. Moreover, some 50 related proteins are sporadically encountered in the main genomes of firmicutes, cyanobacteria and in two members of the CFB group ( Bacteroides capillosus and Bacteroides pectinophilus ). All these proteins have a distant kinship to eukaryotic RNA-dependent RNAPs, but only at the level of the bipartite domain mentioned above, with a common set of eight invariant amino acids that are close to the catalytic MgA in the crystal structure of DNA- and RNA-dependent RNAPs ( Figure 4 C and D). This strongly argues that these proteins are bona fide nucleotide polymerases. On the other hand, their very heterogeneous amino acid sequences (see Supplementary Data S2 ) indicate a particularly rapid evolution, perhaps associated with phage-mediated lateral gene transfers.
Based on their limited sequence similarity, one may tentatively allocate these proteins to five distantly related families corresponding to the proteins of phages sβC2, ϕ8-36 and 1706, from B. subtilis, B. thuringensis and Lactobacillus lactis , respectively ( 43–45 ), to the cyanobacterial gene products mentioned above and to a single protein encoded by three closely related strains of B. thuringensis . Their distant homology to the eukaryotic RNA-dependent RNAPs suggests a common ancestry, which may correspond to a very early branch of the two-barrel protein family, that diverged from DNA-dependent RNAPs before the emergence of LUCA and was subsequently lost in Archaea ( 13 ). Alternatively, these proteins could have initially originated from DNA-dependent RNAP precursor(s), followed by an extremely divergent evolution associated with the gain of new functions such as RNA amplification or some other form of nucleotide polymerization. This tentatively suggests a role in some RNA-mediated regulation of the corresponding bacterial or (pro)phage genomes. However, such RNA-mediated controls are currently unknown in bacteria, and the biological role of these proteins therefore remains an enigma.
A HIGHLY CONSERVED CATALYTIC SITE SHARED BY ALL TWO-BARREL RNAPs
All the proteins reviewed in this survey are characterized by eight invariant amino acids located in the immediate vicinity of their catalytic MgA and (when available) MgB cations ( Figure 4 C and D). This includes the well-characterized Asp (Rpb1 479D F D G D483 and Rpb2- D837 in yeast RNAP II), coordinating the two catalytic Mg 2+ , as well as three basic amino acids (Rpb1-R 446 , Rpb2-K 979 , Rpb2-K 987 in yeast RNAP II), combined with Rpb1-P 448 which presumably defines the precise position of Rpb1-R 446 . These amino acids have an almost identical spatial distribution in the crystal structure of RNA and DNA-dependent RNAPs. Moreover, site-directed mutagenesis in yeast RNAP II or III have shown that six of these positions are strictly critical in vivo ( 46–48 ). It should be noted, however, that there is no Rpb1-P 448 mutation available so far, and that the rpb1-R446A replacement of yeast RNAP II appears to have no growth defect despite its strict invariance ( 49 ).
In keeping with the generic two-metal ions mechanism which probably operates in all nucleotide polymerases ( 50 ), studies on yeast and bacterial RNAPs have established that MgA stably resides in the catalytic site, where it is chelated by the Asps of the D.D.D loop and co-ordinates the NTP α-phosphate and the RNA 3′-OH ( 5 , 7 , 16 ). MgB, on the other hand, enters with or is stabilized by the incoming NTP. It is co-ordinated by its γ-phosphate and by Rpb1-D 481 , Rpb1-D 483 and Rpb2-D 837 in the yeast RNAP II ( 8 ), but occupies a somewhat different position in the bacterial structure of Thermus thermophilus ( 6 , 17 ), where it directly interacts with β′-D 739 (equivalent to Rpb1-D 479 ) and forms water interactions with β-D 686 (corresponding to Rpb2-D 837 ). The Qde1 crystal has no MgB, but the almost identical distribution of its 1007D Y D G D1011 and D709 positions leaves little doubt that the eukaryotic RNA-dependent RNAPs and bacterial Yono-like proteins are all characterized by two catalytic Mg 2+ . However, they lack a fifth carboxylic residue (Rpb2-E 836 ) present in nearly all other two-barrel RNAPs ( Supplementary Data S1 ) and holding MgB by an additional water-mediated bond ( 6 , 17 ).
Recent X-Rays studies on yeast RNAP II indicate that NTPs are loaded through a pore leading to the active centre ( 8 ). It has been suggested that the incoming NTP first binds to an entry (E) site and then rotates around MgB to reach the final nucleotide addition (A) site ( 8 ). This provides a rationale for the extreme conservation of three positively charged residues (Rpb1-R 446 , Rpb2-K 979 and Rpb2-K 987 ) in the catalytic site. Rpb1-R 446 and the Rpb2-K 979 /Rpb2-K 987 pair occupy the end of this NTP entry pore, on each side of the two Mg 2+ , and are immediately close to Rpb1-D 485 and Rpb1-D 483 , respectively. The positively charged side chains of Rpb2-K 979 and Rpb2-K 987 face the last two nucleotides of RNA in the elongating bacterial and yeast RNAPs ( 7 ). In the E site, the γ-phosphate of the incoming NTPs is co-ordinated by Rpb2-K 987 and MgB. It then rotates around MgB to reach the A site, which brings its ribose ring close to side chain of Rpb1-R 446 ( Figure 5 A and B). The precise inter-atomic distance between Rpb2-K 979 and Rpb2-K 987 appears to be highly critical, given the lethality of the yeast rpb2-K979R or rpb2-K987R mutations ( 47 ). Moreover, the rpoβ-K1065R mutation of Escherichia coli (equivalent to rpb2-K979R ) produces dominant negative effect in vivo , with a mutant RNAP that synthesises dinucleotides but cannot enter elongation ( 51 ).
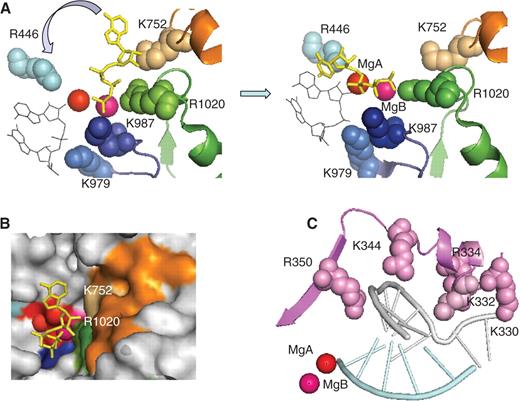
NTP loading and organization of the switch 2 domain in yeast RNAP II. ( A ) NTP loading. The left and right panel correspond to the NTP entry and addition configurations, respectively, and are based on the PDB 1R9T and 1R9S coordinates ( 8 ). The invariant Rpb1-R 446 (light blue), the Rpb2-K 979 and Rpb2-K 987 of the basic loop (dark blue), and the Rpb1-K 752 and Rpb2-R 1020 of the funnel domain (orange and green) are shown as space-filled spheres. The red and magenta spheres denote the catalytic MgA and MgB, respectively. The incoming NTP is shown in yellow. Thin black lines indicate the last two nucleotides of the RNA transcript. A blue arrow symbolises the rotation of the NTP toward the 3′-OH end of the transcript. ( B ) View of the entry site showing the surface of the RNAP II molecule at the end of the secondary channel (NTP entry pore). Same colour code as above, with additional indication of the DFDGD MgA loop. ( C ) Switch 2 domain: the switch 2 motif (Rpb1 327–351 ) is shown in magenta. Space-filled spheres denote positively charged residues. Crystallographic coordinates were taken from PDB 1R9T ( 8 ).
Beyond these eight amino acids, all DNA-dependent two-barrel RNAPs (including the putative RNAPs of Pseudomonas phages) share Rpb1-K 332, on the switch 2 domain, and Rpb1-G 750 , Rpb1-K 752 and Rpb2-R 1020 (forming a salt bridge with Rpb2-D837) on the funnel domain ( Figure 5 B and C). The switch 2 domain bears several conserved Arg and Lys (including Rpb1-K 332 ) which hold the template DNA strand upstream of the catalytic site. Accordingly, the corresponding yeast rpb1-K332A and rpb1-R344A mutations are highly prone to abortive transcription ( 49 ), suggesting a direct role in the formation of the DNA transcription bubble. The funnel domain is located beneath the catalytic site. It forms the most conserved part of the NTP pore mentioned above, and may thus be specifically required to load NTP. Thus, Rpb1-K 752 and Rpb2-R 1020 would delineate the NTP entry site, together with Rpb2-K 987 . This is supported by the fact that RNAP III mutations of the funnel domain ( rpc160-L792S and rpc128-V955M ) compensate a temperature-sensitive mutation altering the MgA loop, perhaps by favouring the delivery of NTPs to the catalytic site ( 52 ). We note that the funnel structure may be specifically imposed by the double-stranded nature of the RNAP template. This would be consistent with the fact that, in vitro , yeast RNAP II is also able to transcribe double-stranded RNA templates ( 53 ).
CONCLUSIONS
The diversity of the two-barrel RNAP family is now well documented, as first suggested by Iyer et al. ( 13 ), and extends far beyond the two main classes of DNA- or RNA-dependent RNAPs. Most of the newly discovered members of this protein super-family are proven ( 15 ) or putative non-canonical RNAPs encoded by eukaryotic DNA viruses and bacterial phages (or inserted prophages DNA), which are much under-represented in current databases. Likewise, their apparent lack in the archaeal kingdom could merely reflect our very limited knowledge of archaeal viruses ( 54 ). Thus, their real diversity may be greater than currently meet the eyes. Except for the well-characterized DNA-dependent RNAPs of Baculoviruses ( 15 ), little is known of their biological role(s), notably in the case of the enigmatic Yono-like proteins or NcGl702 gene product of C. glutamicum ( 13 ). We have assumed here that they act as nucleotide polymerases, but this evidently calls for direct experimental studies.
All known members of the two-barrel protein family share eight closely spaced amino acids, of which seven have negatively (Asp) or positively charged side chains (Arg, Lys). In DNA-dependent RNAPs, four invariant Asp coordinate two catalytic Mg 2+ , whereas the incoming NTP and the growing end of the RNA transcript interact with the Mg 2+ ions and with the positively charged side chains of Rpb2-K 979 , Rpb2-K 987 and Rpb1-R 446 . Other positively charged residues, belonging to the switch 2 and funnel domains, are highly conserved or even strictly invariant (Rpb1-K 332, Rpb1-K 752 and Rpb2-K 1020 ) in all cellular or viral DNA-dependent two-barrel RNAPs and thus probably determine the transcription of double-stranded DNA templates. Such combinations of catalytic Mg 2+ , Asp, Arg and Lys are not specific of the two-barrel RNAPs, but are a common pattern of other, structurally unrelated families of nucleotide polymerases. All these enzymes probably emerged during the early steps of pre-cellular evolution, but have distinct ways of loading their NTPs and show substantial differences in the architecture of their catalytic domain ( 6 , 8 , 17 , 55–58 ).
Unlike the sporadic distribution of the eukaryotic RNA-dependent RNAPs and bacterial Yono-like proteins, multi-subunit DNA-dependent RNAPs operate in all bacterial, archaeal and eukaryotic lineages, with no known exception. Remarkably, these enzymes have an almost invariant core structure of ∼500 strongly conserved amino acids, also found in most virus-encoded RNAPs ( 12 , 21 ), in two recently discovered plant RNAPs ( 59 , 60 ) and, to some extent, in the simplified Orf6 protein of yeast ‘killer’ plasmids ( 24 , 25 ). Hence, the LUCA of all cellular organisms must itself have had an elaborated DNA-dependent RNAP with the same complex active site of ∼500 amino acids, possibly distributed on several polypeptide subunits. The ulterior evolution of this RNAP thus primarily concerned its DNA recruitment factors (corresponding to bacterial σ subunits or to pre-initiation complexes with a TATA box binding protein in the archaea and eukaryotes), along with the acquisition of backtracking properties mediated by the bacterial GreA/B or archaeo-eukaryotic TFIIS-like factors.
We have seen above that the putative RNAPs encoded by Pseudomonas phages have some homology to bacterial-specific β and β′ domains and thus presumably derive from bacterial ancestors. Likewise, the insect-specificity of the Baculo- and Nudiviruses argues for a eukaryotic ancestry of their LEF-8 and LEF-9 RNAP subunits. Hence, these proteins are unlikely to be archaic forms of DNA-dependent RNAPs, but probably result from the highly divergent evolution of bacterial and eukaryotic DNA-dependent RNAPs ancestors, which obliterated most of their ∼500 strongly conserved positions but retained what was strictly for DNA transcription, including the switch 2 and funnel domains. In sharp contrast to the conserved active site of all cellular and nucleo-cytoplasmic viral RNAP, this implies a largely unconstrained evolution of these atypical phage and viral RNAPs that, moreover, may only be competent for the transcription of a small number of genes ( 15 ). It will therefore be interesting to know how far the catalytic properties of these enzymes have diverged from their cellular counterparts in terms of elongation rate, processivity or promoter recognition mechanisms.
FUNDING
French Agence Nationale de la Recherche (grant BLAN08-3-309259). Funding for open access charge: Commissariat à l'energie atomique and French Agence Nationale de la Recherche (grant BLAN08-3-309259).
Conflict of interest statement . None declared.
ACKNOWLEDGEMENTS
Part of this article was written whilst one of us (P.T.) was a guest of Dr Austen Ganley and his colleagues at the Institute of Natural Science, Massey University (New Zealand). We also thank Dr Michel Werner for useful comments and discussion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments