





Abstract
Estrogen receptor-α contains two transactivation functions, a weak constitutive activation function (AF-1) and a hormone-dependent activation function (AF-2). AF-2 works by recruiting a large coactivator complex, composed of one or more p160s, CREB-binding protein (CBP)/p300, and P/CAF (p300 and CBP-associated factor), via direct contacts with the p160s. We report here that independent AF-1 activity also requires p160 contacts. Unlike AF-2, which binds signature NR boxes in the center of the p160 molecule, AF-1 binds to sequences near the p160 C terminus. We propose that the ability of AF-1 and AF-2 to interact with separate surfaces of the same coactivator is important for the ability of these transactivation functions to synergize.
INTRODUCTION
Estrogen signal transduction is mediated by two receptors, α (hereafter ER) and β (ERβ) (1–3). Both estrogen receptors (ERs) are members of the nuclear receptor family of conditional transcription factors (4, 5). In the absence of estrogen, the ERs are held in the nucleus in a complex with molecular chaperonins, such as HSP90. Upon estrogen binding, the receptors dissociate from the heat shock protein (HSP) complex and bind, as homodimers or heterodimers (6, 7), to a specific estrogen response element (ERE). From this location on DNA, the receptor enhances transcription from the nearby promoter.
Transactivation is best understood within the context of theα -receptor. It is mediated by two activation functions, a weak constitutive activation function, AF-1, that lies within the ER N-terminal (AB) domain and a stronger estrogen-dependent activation function, AF-2, that lies within the ER ligand-binding domain (LBD) (8–14). Together, AF-1 and AF-2 synergize strongly to give the final overall level of estrogen activation. The ER transactivation functions are believed to work by binding coactivators and bringing them to the promoter (15–18). The AF-2 surface consists of a cluster of residues from helices 3, 5, and 12 (13, 19), that form a hydrophobic patch on the surface of the liganded LBD (19A ). This hydrophobic patch binds a family of related proteins called the p160s, which include GRIP1/TIF2 (20–22), SRC-1 (23), and RAC3/p/CIP/ACTR/AIB1 (24–27). In each case, AF-2 recognizes a specific signature motif (LXXLL), termed the NR box, that is found repeated throughout the p160s (22, 25, 28–30), and also within proteins that act as AF-2 repressors, such as RIP140 (28, 31) and TIF1 (32). GRIP1, for example, and its human homolog TIF2, contain three separate NR boxes (I, II, and III, respectively), of which NR boxes II and III are the most important for ER binding (22, 29). The p160s, in turn, interact with other coactivator proteins, including CREB-binding protein (CBP)/p300 (22, 25, 33, 34) and p300 and CBP-associated factor (P/CAF) (26, 35, 36). Together, this large coactivator complex, whose subunits possess both histone acetyl-transferase activity (26, 35, 37–39) and the capacity to bind the basal transcription machinery (40, 41), is responsible for the ability of AF-2 to stimulate gene expression.
ER-AF-1, by contrast, is poorly understood. AF-1 usually displays little independent activity and serves only to synergize with AF-2 (10, 12, 14). For unknown reasons, however, AF-1 can also show strong independent activity in some cell types and on some promoters (12). Residues that are important for AF-1 activity are dispersed throughout a large region of the AB domain between amino acid 41 and amino acids 120–150, depending upon the cell type (13, 42, 43). Within this region, motifs that contribute to AF-1-independent activity (amino acids 41–64, hereafter Box 1) and synergism with the LBD (amino acids 87–108, hereafter Box 2) have been identified (44). AF-1 is also strong under conditions of mitogen-activated protein (MAP) kinase pathway stimulation (45–48). Several serine residues (S104, S106, S118) that are phosphorylated by MAP kinases, or cyclin-dependent kinases (49), have been identified, with the latter accounting for more than 90% of total ER phosphorylation (45, 46, 50). Each phosphorylated serine contributes to overall AF-1 activity (45, 46). Despite all this information, the AF-1 protein target or targets remain unknown. Furthermore, the reasons why AF-1 synergizes with AF-2 in some contexts, yet works independently in others, are also unknown.
While the exact nature of the prospective AF-1 coactivator complex is poorly defined, several lines of evidence suggest that it shares features with the AF-2 coactivator complex. First, AF-1 can mask weak mutations in AF-2, suggesting that AF-1 can compensate for reduced p160 recruitment (13). Second, AF-1 and AF-2 squelch each others activity, suggesting that they compete for common limiting target molecules (51). Third, the transcriptional activity of isolated AF-1 can be enhanced by introducing the free ER-LBD into the same cells (52). This activity requires that the LBD be liganded to estrogen and possesses an intact AF-2 surface. Furthermore, enhancement of AF-1 activity by the free LBD is potentiated by exogenous SRC-1 (53). Thus, the free LBD utilizes the AF-2 surface and p160 contacts to participate in the AF-1 coactivator complex.
In this paper, we ask whether AF-1 and AF-2 might indeed bind a similar complement of coactivator proteins. We find that overexpression of p160 proteins, especially GRIP1, increases the transcriptional activity of AF-1. Furthermore, AF-1 interacts with sequences near the GRIP1 C terminus, and this region is needed for the ability of GRIP1 to potentiate AF-1. We propose that p160s are a direct target for both AF-1 and AF-2.
RESULTS
GRIP1 Stimulates AF-1
To test whether AF-1 and AF-2 might utilize similar coactivators, we first examined whether GRIP1 would stimulate AF-1 activity. We transfected an ERE-dependent reporter [ERE-coll-CAT(choramphenicol acetyltransferase)] into HeLa cells along with expression vectors for ER and GRIP1. We then determined the transcriptional activity of the promoter in cells treated with either ethanolic vehicle, estrogen, or the antiestrogen tamoxifen, which is a drug that inhibits AF-2 (54) but allows full AF-1 activity (12).
In the absence of exogenous GRIP1, wild-type ER elicited a strong estrogen response, but no tamoxifen response, from the ERE-coll-CAT promoter (Fig. 1A). This is consistent with previous observations that show that AF-1 has little independent activity at classical EREs in Hela cells (10, 12, 55). In the presence of exogenous GRIP1, ER gave enhanced estrogen response but also gave significant activity both in the presence of tamoxifen and absence of ligand. The ability of GRIP1 to enhance the activity of tamoxifen-liganded ER was more clearly seen when we used an ER variant (ER-V400). This ER variant binds more tightly to the HSP complex than its wild-type counterpart and therefore lacks most of the spontaneous ligand-independent activity that ER usually displays in tissue culture (56, 57). Here, GRIP1 potentiated the estrogen response of ER-V400 and also elicited a large increase in tamoxifen response without the corresponding increase in basal activity. The tamoxifen response of ER-V400 was dependent upon the amounts of transfected GRIP1 and approached 40% of overall estrogen response at high amounts of GRIP1 (Fig. 1B). Several alternate ERE-responsive promoters also showed enhanced tamoxifen response in the presence of GRIP1 (Fig. 1C). We conclude that GRIP1 enhances the transcriptional activity of tamoxifen and estrogen-liganded ER.
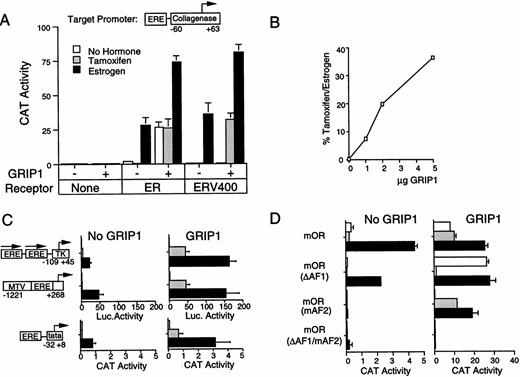
GRIP1 Potentiates Tamoxifen Response A, The effect of estrogen and tamoxifen upon expression of a transiently transfected ERE-coll-CAT reporter gene was examined in HeLa cells in the presence or absence of GRIP1. Where indicated, we also included empty expression vector SG5 (none) or ER expression vectors SG5-ER or SG5-ERV400. The transfected cells were treated with either ethanolic vehicle (), 5 μm tamoxifen (gray bars) or 10 nm estradiol (black bars). CAT activities represent averages from triplicate wells from a representative transfection. B, Relative activity of tamoxifen vs. estrogen increases as a function of GRIP1 levels. Transfections utilizing SG5-ERV400 expression vector were performed as in Fig. 1A in the presence of increasing amounts of GRIP1 expression vector. The data are expressed as the percentage of tamoxifen response relative to estrogen response (set at 100%). C, GRIP1 enhances the activity of tamoxifen- and estrogen-liganded ER at a variety of promoters. The experiment was performed as described in Fig. 1A using a variety of ER-responsive reporters (from top to bottom): EREII-LUC (GL45), two vitellogenin EREs upstream of the herpes simplex virus TK proximal promoter (−109/+45); MTV-ERE-LUC, the MTV long terminal repeat region (−1228/+268) with a single palindromic ERE replacing the complex multiple hormone response element; ERE-TATA-CAT, a single ERE 19-mer upstream of the collagenase TATA box region (−32/+8). D, Analysis of tamoxifen action on ER mutants. Expression vectors for mouse ERs (mORs) were transfected into HeLa cells and their effect on ERE-coll-CAT expression was determined in the absence or presence of GRIP1. mORs were full length, contained a deletion of the first 120 amino acids of the AB domain (mORΔAF-1), an AF-2-disruptive double alanine for leucine substitution at positions 543 and 544 in LBD activation helix 12 (mORmAF-2), or both (mORΔAF-1/mAF-2).
We then confirmed that the effect of GRIP1 on tamoxifen-liganded ER was mediated by AF-1 (Fig. 1D). We asked whether GRIP1 would enhance the activity of mouse ERs (mORs) with either a truncation of the AB domain that eliminates AF-1 (mORΔAF-1), a double-point mutation that eliminates AF-2 (mORmAF-2), or both (mORΔAF-1/mAF-2) (13). In the absence of exogenous GRIP1, wild-type mOR elicited a strong estrogen response from the ERE-coll-CAT promoter. This response was reduced 50% by the AF-1 deletion and eliminated by the AF-2 mutation, consistent with the idea that estrogen response in the absence of GRIP1 predominantly reflects AF-2 activity. In the presence of GRIP1, wild-type mOR gave enhanced estrogen response and significant activity both in the presence of tamoxifen and absence of ligand, just as did the human ER. The AF-1 deletion (mORΔAF-1) retained strong activity in the presence of estrogen and absence of ligand, but showed no activity in the presence of tamoxifen. In contrast, the AF-2 mutant (mORmAF-2) gave strong tamoxifen and estrogen responses, but gave no activity in the absence of ligand. The double mutant (mORΔAF-1/mAF-2) showed no activity under any conditions. Thus, the spontaneous ligand-independent activity that is obtained in the presence of exogenous GRIP1 requires AF-2, and not AF-1. We speculate that this AF-2 dependent, ligand-independent, activity reflects stabilization of an ER-LBD conformation that resembles that of the estrogen-liganded receptor by exogenous GRIP1. We also conclude that GRIP1 enhances the activity of the tamoxifen-liganded ER by enhancing the activity of AF-1 and not by enhancing the activity of AF-2.
p160s and p160-Associated Proteins Enhance AF-1 Activity
We next asked whether other coactivators would also enhance AF-1 activity. The p160 protein RAC3 enhanced both estrogen and tamoxifen response at an ERE, and SRC-1a showed similar but weaker activity (Fig. 2A). This weak activity of SRC-1a is consistent with previous observations that show that this coactivator possesses little activity in HeLa cells (58), perhaps because SRC-1a contains an inhibitory function within its C-terminal domain (30). The AF-2 repressor RIP140 failed to enhance tamoxifen response. Of the p160-associated proteins, CBP and p300 enhanced estrogen and tamoxifen induction, especially in combination with GRIP1 (Fig. 2B). P/CAF only weakly enhanced tamoxifen activation (data not shown).
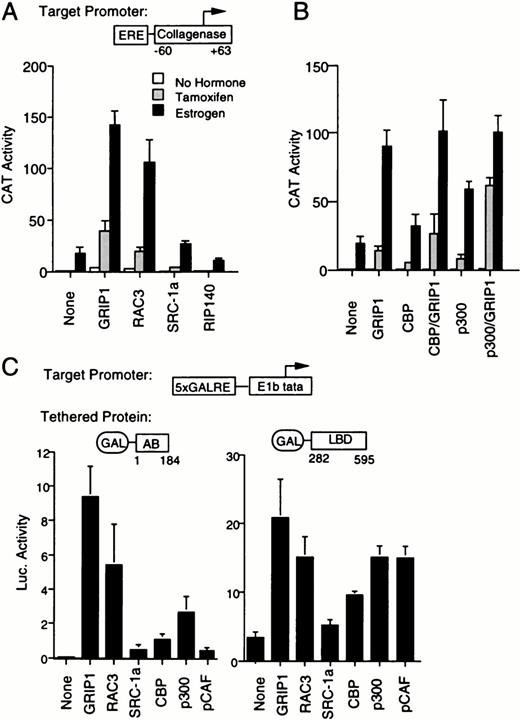
A Survey of the Effect of Coactivators on Tamoxifen Response A, Results of a transfection in which activity of ERE-coll-CAT reporter gene was monitored as above (Fig. 1A) in the presence of ER expression vector SG5-ERV400 and 5 μg of each coactivator expression vectors. B, As above, using expression vectors for GRIP1, CBP, or p300. C, The effect of transient transfection of a range of coactivators upon transcription of a GAL4 responsive reporter gene (GK1) was determined in the presence of GAL4-AB or GAL4-LBD expression vectors. Individual bars represent luciferase activities determined from triplicate wells. In the case of the GAL-LBD, the cells were treated with 100 nm estradiol.
We then examined whether the same coactivators would enhance the activity of isolated AF-1. We transfected an expression vector for a GAL4 DNA-binding domain (DBD)/ER-AB domain fusion protein (GAL4-AB) into HeLa cells, along with expression vectors for various coactivators (Fig. 2C). In accordance with their effects on tamoxifen-liganded ER, GRIP1, RAC3, and SRC-1a each enhanced AF-1-dependent transcription from a GAL4-responsive reporter gene. CBP, p300, and, to a lesser extent, P/CAF enhanced the transcriptional activity of AF-1. As expected, each of the coactivators also enhanced the estrogen-dependent activity of a GAL4-LBD fusion protein. Thus, the p160s and p300/CBP, and perhaps P/CAF, are involved in transactivation by both AF-1 and AF-2.
An Extended Region of AF-1 Is Required for GRIP1 Action
We next examined which region of AF-1 was needed for GRIP1 action. Three subregions of the AB domain have previously been implicated in AF-1 activity (see Fig. 3). Box 1 (amino acids 41–64) is needed for tamoxifen activation in MDA-231 breast cells (44). Box 2 (amino acids 87–108) is required for synergism with AF-2 in the same cell type. Sequences surrounding the main phosphorylated serine residue at position 118 [kinase target region (KTR), amino acids 108–129], also contribute to AF-1 activity (43). To test whether these subregions were needed for GRIP1 action, we transfected mutant human ERs into HeLa cells and assessed their ability to activate ERE-dependent transcription in the presence or absence of GRIP1 (Fig. 3A). In each case, the ERs contained mutations within the AB domain (amino acids 1–184), but retained an intact DBD and LBD. For simplicity, we compared the activity of mutant and wild-type ERs in the absence or presence of GRIP1 (see Fig. 1A) and set activity of wild-type ER at 100%.
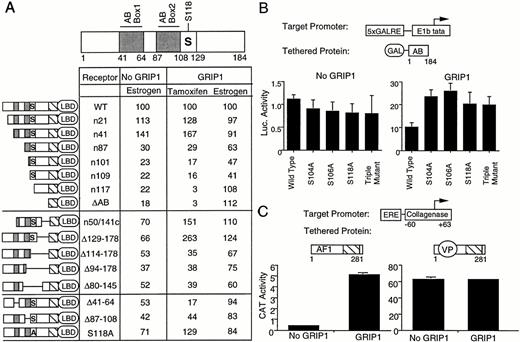
GRIP1 Action on AF-1 Requires an Extended Region of the AB Domain A, Effect of AB domain mutations upon ERE-dependent transcription in HeLa cells. A schematic of the ER AB domain showing each of the three AF-1 subregions, box 1 (41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64 ), box 2 87–108, and the KTR (108–129) is shown above. The locations of box 1 and box 2 are shown in gray. The KTR is boxed, and the major phosphorylation site Serine 118 is marked with S. A schematic of each mutation is shown on the left. In all cases, ERs contained mutations in the AB domain (amino acids 1–184) but retained an intact DBD and LBD. We tested the effect of mutations upon ER action on ERE-coll-CAT, as described in Fig. 1A, in the absence or presence of GRIP1. We set activity of wild-type ER (full length) at 100% in each column of data and compared the activity of mutant ER relative to this number under this condition. B, Effect of phosphorylation site mutations on GRIP1 action. Hela cells were transfected, as in Fig. 2C, with a GRIP1 expression vector, where indicated, and GAL4-AB expression vectors bearing alanine substitutions at each, or all three, phosphorylation sites. C, Effect of a VP16 substitution on GRIP1 action. Hela cells were transfected with expression vectors for either the ER AB-DBD region or a similar vector containing a VP16 transactivation domain inserted at amino acid 65, and either an empty expression vector or an expression vector for GRIP1.
We were able to confirm previous results (43) that showed that AF-1 activity in the absence of GRIP1 required an extended region of the AB domain (Fig. 3A, first column). Tamoxifen response in the presence of GRIP1 also required an extended region of the AB domain, but especially sequences in and around box 1 (Fig. 3A, second column). ERs lacking sequences upstream of amino acid 41 (n21, n41) showed normal, or enhanced, tamoxifen response, but an ER with an N-terminal deletion that eliminated box 1 (n87) showed markedly reduced tamoxifen response. ERs lacking box 1 and box 2 (n101, n109), or all three subregions (n117), gave further stepwise reductions in tamoxifen response. ERs lacking sequences downstream of amino acid 129 (n50/141c; Δ129–178) showed modestly enhanced tamoxifen response, but ERs with further C-terminal deletions that eliminate the KTR (Δ114–178), or both the KTR and box 2 (Δ93–178, Δ80–145), again showed reduced tamoxifen activation. Finally, an ER specifically lacking box 1 (Δ41–64) showed markedly reduced tamoxifen activation, and an ER specifically lacking box 2 (Δ87–108) showed a more modest reduction in tamoxifen activation. Thus, GRIP1 action requires each of the three AF-1 subregions and, especially, sequences within box 1.
We then specifically addressed the role of the AF-1 phosphorylation sites. An ER with an alanine substitution at serine 118 (S118A) actually showed enhanced tamoxifen activation in the presence of GRIP1 (Fig. 3A). Similarly, introduction of alanine residues at each MAP kinase target serine (S104, S106, S118) also enhanced GRIP1 activation of a GAL4-AB fusion protein (Fig. 3B). Thus, GRIP1 action at AF-1 is independent of the phosphorylation sites.
Next, we compared the effect of GRIP1 on the ER AB-DBD region (AB-DBD), which only contains AF-1, with a similar ER truncation that contained an in- frame insertion of the VP16 activation function, just downstream of box 1 (V281C, Fig. 3C) (55, 59). As expected, GRIP1 enhanced the weak AF-1 activity that was obtained with the AB-DBD truncation. In contrast, GRIP1 had no effect on the stronger activity that was obtained with the VP16-ER fusion protein. Thus, insertion of a heterologous transactivation function into the AB domain abolishes the effect of GRIP1, supporting the notion that integrity of the proximal part of AF-1 (box 1) is essential for GRIP1 action. Together, our results show that GRIP1 enhancement of AF-1 activity requires an extended region of the AB domain and, especially, sequences in and around box 1.
ER AF-1 Binds p160s
We then asked whether the AB domain binds p160s (Fig. 4A). In vitro translated GRIP1 and SRC-1a bound to both the AB domain and the ER-LBD in a glutathione-S-transferase (GST) pull-down assay. Over the course of this study, the bacterially expressed AB domain retained about 5–20% of the amounts of GRIP1 and SRC-1a that were retained by equimolar amounts of bacterially expressed LBD. We were unable to demonstrate binding of the AB domain to CBP (data not shown). We conclude that the AB domain, like the LBD, binds p160s.
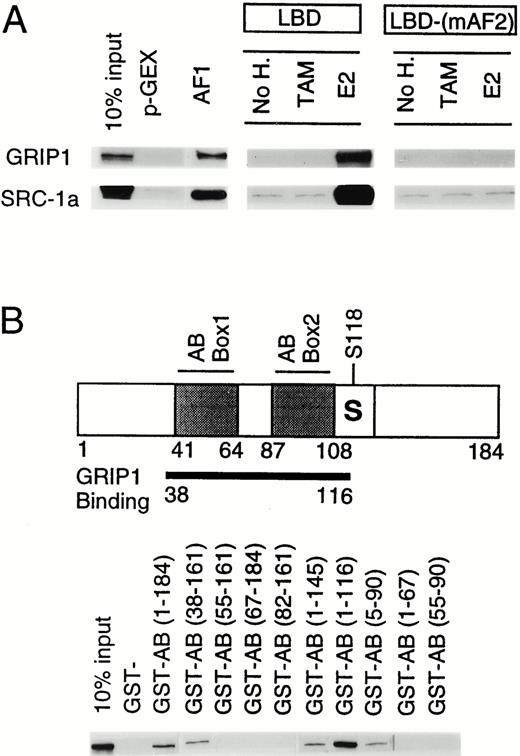
The Estrogen Receptor AB Domain Binds p160s A, The AB domain binds p160s. Autoradiograms of labeled proteins retained on beads in GST pull-down assays. The binding of either labeled GRIP1 or SRC-1a to beads coated with GST, GST-AB, or GST-LBD fusions. B, Binding of GRIP1 to the AB domain. A diagram of the AB domain is shown above with positions of previously described AB transactivation motifs, box 1, box 2, and the KTR-containing serine 118, marked. The extent of AB sequences implicated in GRIP1 binding is marked below with a thick black line. Below, autoradiography of labeled GRIP1 retained on GST beads is presented. Beads are coated with GST fused to portions of the AB domain between the amino acids that are described.
To examine which part of the AB domain was needed for GRIP1 binding, we produced a series of GST fusion proteins containing portions of the AB domain and asked whether they would bind in vitro translated GRIP1 (Fig. 4B). An AB domain fusion protein with an N-terminal deletion to amino acid 37 (38–161) bound GRIP1, but AB domains with larger N-terminal deletions (55–161; 67–184; 82–161) failed to bind GRIP1. Two AB fusion proteins with C-terminal deletions (1–145; 1–116) each bound GRIP1 efficiently. Indeed, the latter showed enhanced binding relative to the native AB domain. An AB fusion protein with a further deletion (5–90) retained some, albeit reduced, GRIP1 binding, but two portions of the AB domain that overlap sequences 5–90 (1–67 or 55–90) failed to bind GRIP1. Similar results were obtained with SRC-1a (data not shown). Thus, an extended region of the AB domain (amino acids 38–116), and especially sequences between amino acids 38–55 and upstream of amino acid 90, were needed to bind p160s. We conclude that both GRIP1 enhancement of AF-1 activity (Fig. 3A, second column), and GRIP1 binding (Fig. 4), require a similar region of the AB domain, and, especially, sequences in and around box 1. This suggests that AF-1, like AF-2, works by binding p160 proteins.
AF-1 Binds the GRIP C Terminus
We next asked how AF-1 recognized GRIP1 (Fig. 5A). Competition experiments revealed that a specific peptide that was homologous to GRIP1 NR box III blocked GRIP1 binding to the LBD but failed to block GRIP1 binding to the AB domain, or to the p160-binding region of CBP (35) (Fig. 5B). Similar results were also obtained with a peptide homologous to NR box II (data not shown), or when SRC-1a was substituted for GRIP1 and tested with either peptide (data not shown). Thus, the AB domain recognizes a region of GRIP1 and SRC-1a that is distinct from the NR boxes.
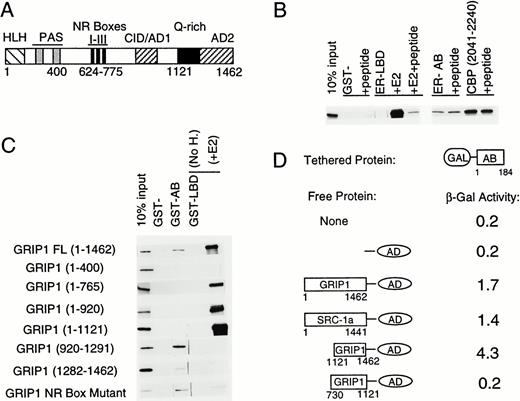
The ER-AB Domain Binds to the GRIP1 C Terminus A, Schematic of full-length GRIP1. Approximate positions of putative and defined functional domains are marked, including βHLH/PAS region, NR boxes I-III, CBP interaction domain (CID/AD1), Q-rich domain, and activation domain 2 (AD2). B, Autoradiography of GRIP1 protein retained on beads coated with either GST-LBD, GST-AB, or GST-CBP fusion proteins. The CBP fragment consists of amino acids 2041–2240 and contains the previously defined p160-binding motif. Where indicated (+peptide), binding to GRIP1 was performed in the presence of an excess of a synthetic peptide homologous to NR box III. We also performed controls with an NR box peptide that contained double alanine substitutions at key leucine residues (data not shown). C, Binding of GST-AB or GST-LBD to GRIP1 protein fragments. Autoradiograms show retention of labeled full-length GRIP1 or GRIP1 fragments on beads coated with GST-AB or GST-LBD fusion proteins. Amino acid numbers of all fragments were indicated. The GRIP1 NR box mutant has alanine substitutions in the key leucine doublets within NR boxes II and III (N-LXXLL-C to N-LXXAA-C). D, Interaction of ER-AF-1 and p160s in a yeast two-hybrid system. β-Galactosidase production from a GAL4-responsive reporter gene was determined in yeast strains containing GAL4DBD-AB and free GAL4AD-GRIP1 or GAL4AD-SRC-1a fusion proteins.
To localize the AB binding site within GRIP1, we analyzed binding of the AB domain and LBD to a series of GRIP1 truncations (Fig. 5C). The AB domain failed to bind GRIP1 truncations lacking amino acids beyond positions 400, 765, 920, or 1121, respectively, but did bind a GRIP1 fragment containing amino acids 920-1291 and, more weakly, to a fragment containing amino acids 1282–1462. In contrast, the LBD bound all C-terminal truncations that contained intact NR boxes (1–765; 1–920; 1–1121), but failed to bind either C-terminal GRIP1 fragment. As predicted, the AB domain bound to a GRIP1 NR box mutant (boxes II and III mutated) efficiently, and the LBD bound this molecule only weakly. Experiments using GST-GRIP1 fragments also indicated that GRIP1 sequences downstream of amino acid 1121, and especially sequences 1121–1282, were needed for ER binding (data not shown). These results confirm that AF-1 recognizes GRIP1 at a site that, most likely, lies between amino acids 1121 and 1462 and is distinct from the NR box cluster. Amino acids 1121–1282, which contribute most to AF-1 binding, overlap a glutamine rich (Q-rich) domain that is conserved throughout the p160 family (26).
We then confirmed the location of the putative GRIP1 AB domain-binding site in a yeast two-hybrid system (Fig. 5D). A GAL4-AB fusion protein weakly enhanced transcription of a GAL4-responsiveβ -galactosidase gene in yeast, relative to the levels of transcription obtained with the GAL4-DBD alone (data not shown). This is consistent with previous studies indicating AF-1 is weakly active in yeast cells (12). ER-AF-1 activity was unaffected by coexpression of the GAL4 acid activation domain (GAL-AD), but was enhanced by coexpression of GAL4-AD/full-length GRIP1 or SRC-1a fusion proteins. A GAL-AD/GRIP1 fusion protein, containing C-terminal amino acids 1121–1462, also enhanced AF-1 activity. In contrast, a GAL-AD/GRIP1 fusion protein containing amino acids 730-1121 did not enhance AF-1 activity. Similarly, GAL-AD fusions to GRIP1 amino acids 563-1121 and 1–766 also failed to enhance AF-1 activity (data not shown). We stress that the GAL-AD/GRIP1 fusions containing GRIP1 fragments 730-1121 (20), 563-1121, and 1–766 (data not shown) all strongly enhanced the activity of a GAL-ER-LBD fusion protein in yeast, in agreement with the fact that each of these fragments contains NR boxes, and confirming that these fragments are efficiently expressed in yeast cells. Thus, GRIP1 (and SRC-1a) target the GAL4-AD to the AB domain in yeast, and this activity is specifically recapitulated by the GRIP1 C terminus. Furthermore, both the GST pull-down and yeast two-hybrid assays indicate that the C-terminal region of GRIP1 is the only region that interacts with the ER-AB domain in these systems.
AF-1 Function Requires the GRIP1 C Terminus
We next tested whether the GRIP1 C terminus was needed for AF-1 activity in mammalian cells. We coexpressed GAL4-AB or GAL4-LBD fusion proteins with either wild-type GRIP1, a GRIP1 truncation that cannot bind AF-1 (GRIP-Δ1121C), or a GRIP1 molecule with NR box mutations (NR box mutant) (Fig. 6A). The GRIP1 truncation, GRIP-Δ1121C, was completely deficient for AF-1 enhancement, but showed increased AF-2 enhancement relative to wild-type GRIP1. In contrast, the GRIP1 NR box mutant strongly enhanced AF-1 activity, but failed to enhance AF-2 activity. Thus, GRIP1 needs an intact C terminus to enhance AF-1, but this region is dispensable for GRIP1 enhancement of AF-2. Furthermore, NR boxes II and III are not needed for GRIP1 enhancement of AF-1 activity.
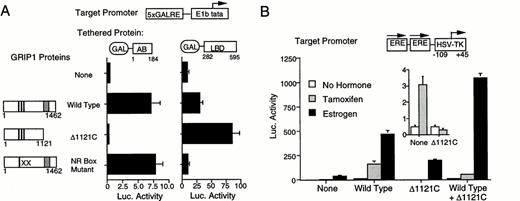
GRIP1 Action on AF-1 Requires the GRIP1 C Terminus A, Differential effects of GRIP1 mutations on AF-1 and AF-2. A schematic of the GRIP1 proteins is shown on the left of the figure. Intact NR boxes are marked with black lines, mutant NR boxes with an X, and the GRIP1 Q-rich domain with a dark gray box. The Δ1121C truncation lacks sequences downstream of position 1121 and will not bind the AB domain. The GRIP1 NR box mutant contains double leucine to alanine substitutions within NR boxes II and III. The effect of the GRIP1 mutants upon GAL-AB and GAL-LBD fusion proteins was determined as in Fig. 3A. B, Requirement of the GRIP1 C terminus for ER action. The effect of tamoxifen and estrogen upon expression of an EREII-LUC reporter gene was examined in HeLa cells in the presence of ERV400 and GRIP1 or the GRIP-Δ1121C truncation. The inset shows effect of wild-type and truncated GRIP1 upon EREII-LUC expression in the presence of tamoxifen, on an expanded scale.
We then asked how the GRIP1 truncation (Δ1121C) would affect the activity of full-length ER (Fig. 6B). For these experiments we used a promoter that shows some tamoxifen activation in the absence of exogenous p160s (ERE-II-LUC; Fig. 6B, inset). As expected, GRIP-Δ1121C enhanced the action of the estrogen-liganded ER at the ERE-II-LUC promoter, although this response was somewhat lower than that obtained with wild-type GRIP1. In contrast, GRIP-Δ1121C eliminated the tamoxifen response that was seen in the absence of exogenous p160s (see inset). A combination of wild-type GRIP1 and GRIP-Δ1121C gave increased estrogen response relative to the responses obtained with wild-type GRIP1 cotransfected with empty expression vector. [Note that this enhanced estrogen response occurs because wild-type GRIP1 and the GRIP-Δ1121C truncation both activate isolated AF-2; therefore, this transfection contains more coactivator protein that is capable of activating AF-2 than the transfection containing wild-type GRIP1 alone]. Nonetheless, the tamoxifen response that was obtained with this combination of coactivators was significantly reduced relative to wild-type GRIP1 alone, showing that the GRIP-Δ1121C truncation also inhibited the enhanced tamoxifen response that was obtained in the presence of exogenous GRIP1. We conclude that the GRIP1 C terminus is needed for enhancement of AF-1 activity and that GRIP-Δ1121C acts as a dominant negative for tamoxifen response. Because GRIP-Δ1121C is weakened in its ability to enhance estrogen response in the presence of full-length ER, yet strengthened in its ability to enhance isolated AF-2, we also suggest that the GRIP1 C terminus is needed for AF-1/AF-2 synergism.
GRIP1 Is a Weak Activator of ERβ AF-1
Previous observations have suggested that ERβ, unlike ER(α), shows little activity in the presence of tamoxifen (60, 61). We therefore examined the role of p160s in ERβ function. Wild-type GRIP1 strongly enhanced the activity of unliganded and estrogen-liganded ERβ (Fig. 7A), consistent with similar results that were obtained with SRC-1 (60), but only weakly increased the activity of tamoxifen-liganded ERβ. In parallel, GRIP1 markedly increased the activity of tamoxifen-liganded ER(α) and failed to affect the activity of an ER truncation lacking of the AB domain (ERΔAB). Thus, ERβ lacks an efficient AF-1 activity that responds to GRIP1.
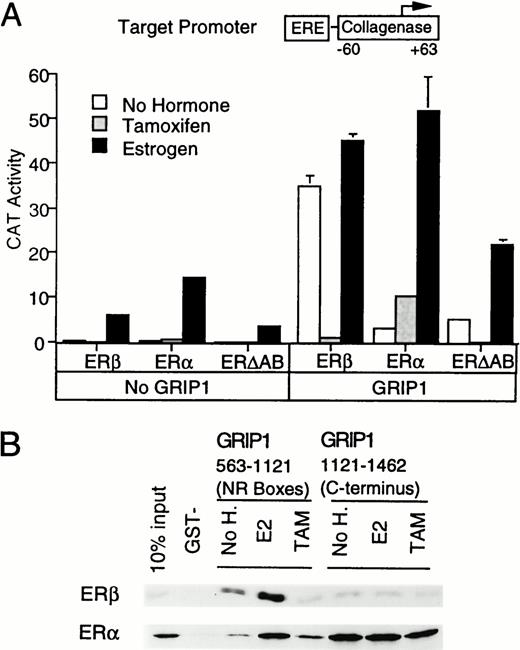
GRIP1 Is a Weak Activator of ERβ AF-1 A, GRIP1 action on ERβ. We tested the effect of GRIP1 upon ERE-coll-CAT expression in the presence of CMV-driven expression vectors for human ERβ, ER (α), or an ER lacking an AB domain (ERΔAB). B, The GRIP1 C terminus binds ER constitutively. Autoradiograms of in vitro translated full-length ERβ or ER (α) retained on glutathione beads coated with GST-GRIP1 fragments (563–1121 and 1121–1462) in the presence of ethanolic vehicle, tamoxifen, or estradiol.
We then compared binding of in vitro translated ERs to GST-GRIP1 fusion proteins (Fig. 7B). A GRIP1 fragment overlapping the NR boxes (563–1121) bound ERβ, but only in the presence of estrogen and not tamoxifen. In contrast, a GRIP1 C-terminal fragment (1121–1462) bound weakly to ERβ but strongly to the ER(α) control. As expected, ERΔAB also showed markedly reduced binding to this C-terminal fragment (data not shown). Thus, the strength of the interaction of the C terminus of GRIP1 with the two isoforms of ER parallels the ability of GRIP1 to potentiate the activity of either tamoxifen-liganded ERβ or ER(α). This result further underlines the importance of the interactions between the ER(α) AB domain and the GRIP1 C terminus.
DISCUSSION
AF-1 Works by Binding p160s
This paper presents evidence that describes a direct target for the ERα AF-1 function. We find that AF-1 activity, in the context of tamoxifen-liganded ER or the isolated AB domain, is strongly enhanced by overexpression of p160s and CBP/p300 and, somewhat weakly, by overexpression of P/CAF. We find that the AB domain binds GRIP1 and SRC-1a. Furthermore, a similar extended region of the AB domain is required for GRIP1 potentiation of AF-1 activity in vivo and for GRIP1 binding in vitro. We were also able to identify a GRIP1 truncation that does not bind the AB domain and acts as a specific dominant negative for tamoxifen-liganded ER, either in the absence of exogenous p160s or the presence of overexpressed GRIP1. We therefore propose that AF-1 action requires a p160/p300 complex and that direct contacts with p160s are essential for recruitment of this complex. Our study complements a recently published study which shows that microinjection of anti-p160 or anti-CBP antibodies block tamoxifen response in cells (HepG2) where AF-1 activity is high (62).
GRIP1 strongly enhances independent AF-1 activity. In the absence of GRIP1, however, AF-1 largely serves to synergize with AF-2. We suggest that AF-1/p160 interactions may also play a role in AF-1/AF-2 synergism. ER N-terminal deletions have similar effects on estrogen response in the absence of GRIP1 and tamoxifen activation in the presence of GRIP1. Likewise, box 1 (amino acids 41–64), which is responsible for tamoxifen activation in breast cells (44), is needed both for AF-1/AF-2 synergism in the absence of GRIP1 and tamoxifen activation in the presence of GRIP1. Finally, the GRIP1 truncation (Δ1121C), which does not bind the AB domain, does not potentiate the activity of estrogen-liganded ER as strongly as wild-type GRIP1, yet potentiates the activity of isolated AF-2 better than wild-type GRIP1 (Fig. 6). Thus, GRIP-Δ1121C is defective for potentiation of independent AF-1 activity and AF-1/AF-2 synergism. Together, these results suggest that the ability of AF-1 and AF-2 to bind separate surfaces of the same coactivator is important for the ability of these transactivation functions to synergize.
While we, and others (62), have readily detected protein-protein interactions between ER AF-1 and p160s, two other groups failed to do so. One set of authors failed to see interaction between AF-1 and TIF-2 in a yeast two-hybrid system (22). In that study, rather than full-length TIF-2 fused to the GAL-AD, the authors used a TIF-2 fragment spanning amino acids 624-1288. Another set of authors failed to see interactions between mouse AF-1 and SRC-1 in GST pull-downs (30). While we have no explanation for this discrepancy, we note that the second set of authors did see that a C-terminal fragment of SRC-1, which overlaps the Q-rich domain, bound full-length mouse ER in an AF-2-independent manner. We speculate that this interaction may be similar to the one that we have described in this paper.
The idea that both AF-1 and AF-2 directly bind p160s may account for several unexplained observations. First, AF-1 can compensate for weak AF-2 mutations (13). This could be explained if AF-1/p160 contacts assist weakened AF-2 interactions with p160s. Indeed, the ability of AF-1 to mask AF-2 mutations is lost in AB domain truncations that, we now know, lack the ability to bind GRIP1. Second, AF-1 and AF-2 squelch each others activity (51). This could be explained if AF-1 and AF-2 were both able to bind and to sequester limiting target p160s molecules. Third, free ER-LBD can enhance the activity of isolated AF-1 in trans (53). This could be explained if the LBD binds the NR boxes of p160s that, themselves, are bound to AF-1. Fourth, it has been previously shown that SRC-1 enhances the ability of tamoxifen-liganded ER to activate the C3 complement promoter in CV-1 cells (58). This could be explained if SRC-1 were simply enhancing AF-1 activity. Finally, our results may explain why tamoxifen behaves as a complete antagonist of ERβ action at EREs (60, 61). GRIP1 potentiates the activity of tamoxifen-liganded ERβ weakly and the GRIP1 C-terminus binds ERβ weakly. The lack of ERβ-AF-1 activity may reflect poor interaction between ERβ and the GRIP1 C terminus.
GRIP1 Action and GRIP1 Binding Are Mediated By A Similar Region of AF-1
In this study we asked which regions of the AB domain were needed for GRIP1 stimulation of AF-1 activity and for direct GRIP1 binding by the AB domain. We found marked similarities between AF-1 and the p160-binding unit. First, a similar extended region of the AB domain is required for GRIP1 activation (amino acids 41–129) and GRIP1 binding (amino acids 38–116). Second, box 1 (amino acids 41–64), which is also required for tamoxifen response in breast cells (44), is important for both GRIP1 activation and GRIP1 binding. Third, the kinase target residue serine 118 is dispensable for both GRIP1 action and GRIP1 binding. Lastly, sequences toward the C terminus of the AB domain inhibit both GRIP1 activation and GRIP1 binding. These results strongly suggest that GRIP1 activation of AF-1 is mediated by GRIP1 binding. We speculate that this extended region of the AB domain forms a folded AF-1 structure that, like AF-2 (19A ), contains p160-binding residues along the length of its primary sequence. Our data particularly implicate box 1 in GRIP1 activation and GRIP1 binding. While we favor the possibility that box 1 contains residues that contact GRIP1, it remains possible that mutations in box 1 are particularly deleterious to overall AF-1 structure. It will be important to identify and mutate specific residues on the surface of the folded AF-1 structure to fully define the p160-binding site.
The fact that a large region of the AB domain is required for GRIP1 action raises the question of whether AF-1 might bind other target proteins. Our in vitro binding studies reveal that sequences within box 1 are absolutely required for GRIP1 binding (Fig. 4), yet ERs with deletions of box 1 (Δ41–64; n87, Fig. 3) retain 17–29% of the GRIP1-induced tamoxifen response. One explanation for this discrepancy is that, because transcriptional activity was determined in the presence of AF-2, then residual interaction between GRIP1 and the LBD masks the phenotypes of the AF-1 deletions. Alternatively, the AF-1 mutants might fold differently in vivo and in vitro. More interestingly, if regions of AF-1 downstream of box 1 make contacts with other constituents of the coactivator complex, such as p300 or P/CAF, then transfected GRIP1 might stimulate AF-1 activity through these intermediate proteins, even in the absence of its own binding site.
Two other lines of evidence point to the existence of alternate AF-1 targets. While the AF-1 phosphorylation sites themselves are dispensable for GRIP1 action, they are needed for AF-1 activity under conditions of MAP kinase stimulation (Refs. 45, 46 and W. J. Feng, P. Webb, J. Li, M. Karin, J. D. Baxter, and P. J. Kushner, manuscript submitted). This suggests that the AF-1 phosphorylation sites make contacts with other proteins. The existence of a subregion of the AB domain that inhibits GRIP1 action is also suggestive. Previous reports have indicated that tamoxifen- liganded ER binds the corepressors N-COR and SMRT (63) and that the overall level of activity of tamoxifen-liganded ER is regulated by the levels of corepressors (58, 62, 64, 65). Indeed, one group has shown that ER/corepressor interactions involve serine 118 (64). We are exploring the possibility that the inhibitory region of the AB domain binds corepressors.
The GRIP1 C Terminus Is Needed for AF-1 Activity
AF-1/GRIP1 interaction requires sequences at the GRIP1 C terminus and especially sequences that overlap the Q-rich domain. This region is conserved within the p160 family, suggesting that it may also be involved in the interaction of other p160s with the AB domain. The exact nature of the AF-1- binding site in the GRIP1 C terminus is likely to be complex. While the Q-rich domain is essential for strong AF-1 binding, a GRIP1 fragment (1282–1462), which is downstream of the Q-rich region, also binds weakly to AF-1. Thus, the AF-1-binding site may be spread across a large region of GRIP1. We also find that the AB domain readily binds free GRIP1 in GST pull-down assays, but the reciprocal interaction between tethered GRIP1 and the free AB domain is weak (data not shown). We do see strong hormone-independent binding of full-length ER to the GRIP C terminus in GST pull-down assays (Fig. 8B), and this binding requires the AB domain (data not shown). Thus, contributions from the rest of the ER molecule assist AF-1/GRIP1 interactions. One explanation for this phenomenon is that interaction between the GRIP1 C terminus and ER-AF1 requires receptor dimerization functions, as does interaction between the p160 NR box region and ER-AF2 (30). Another explanation is that the ER DBD-LBD region contributes weak GRIP1 contacts that help stabilize the AF-1/GRIP1 interaction. We are actively investigating these possibilities.
Our work does not address whether AF-1 binding is the only role of the GRIP1 C terminus in AF-1 activity. Recent studies have suggested that transcription factors may bind the same coactivator, but require different functions within that coactivator (66, 67). It is therefore possible that AF-1 and AF-2 bind p160s but, at the same time, require different p160 functions. TIF2 (the human homolog of GRIP1) has two independent transactivation functions, AD1 and AD2 (22). AD1, which binds CBP/p300, lies downstream of the AF-2-binding site (NR boxes) in the middle of the p160 molecule. Intriguingly, AD2, whose target is unknown, lies downstream of the AF-1-binding site within the TIF2 C terminus. Likewise, p160 HAT activity (26, 39), which is dispensable for AF-2 (66, 67), maps to the same p160 Q-rich region that is involved in AF-1 binding. Finally, SRC-1a contains an inhibitory function within its C terminus (30). A GRIP1 (Δ1121C) truncation activated isolated AF-2 better than wild-type GRIP1, suggesting that the GRIP1 C terminus may also contain inhibitory sequences. AF-1 action could specifically require any of these C-terminal GRIP1 functions.
The Choice Between AF-1/AF-2 Synergism and Independent AF1 Activity May Be Regulated by p160s
GRIP1 enhances the ability of AF-1 to work independently of AF-2. This suggests a simple hypothesis to explain why AF-1 synergizes with AF-2 in some cells, but works independently in others. We propose that the relatively weak AF-1/p160 contacts ordinarily support the stronger interaction of the ER-LBD with p160s, and this explains why AF-1 synergizes with AF-2. At increased p160 levels, however, AF-1/p160 contacts become sufficient to recruit p160s independently. Thus, the balance between AF-1/AF-2 synergism and AF-1 independent activity would be regulated by p160 levels. In a sense, the behavior of AF-1 in this model is analogous to the previously described behavior of weak AF-2 mutations that reduce GRIP1 binding, and whose phenotype is suppressed at high levels of GRIP1 (19A, 68).
It was previously suggested that the agonist/antagonist behavior of tamoxifen in breast cells is influenced by cell-specific differences in factors that interact with subregions of the AB domain, such as box 1 (44). The results presented here suggest that those factors are p160 proteins. It is known that p160 levels are increased in certain breast and ovarian cancers because one of the p160 genes, AIB1, lies in an area of the genome that is commonly amplified in tumors (27). We have previously examined the properties of ERE-coll-CAT in MCF-7 and MDA-MB-453 breast cells that, we now know, contain amplified AIB1 (55). In accordance with our latest hypothesis, we found strong AF-1 activity. The similarity between the behavior of AF-1 in MDA-MB-231 cells (53) and HeLa cells in the presence of GRIP1 (present paper) may suggest that MDA-MB-231 cells also contain amplified AIB1. It will be interesting to test how p160 overexpression might affect the tamoxifen or estrogen responsiveness of genes with different promoter architectures, such as spaced half-sites (69) or AP-1 sites (55), whose transcription may correlate better with estrogen and antiestrogen effects on growth.
MATERIALS AND METHODS
Mammalian Reporter Genes and Expression Vectors
ERE-coll60-CAT (55), ERE-II-LUC (61), MTV(ERE)-LUC (70), and ERE-tata-CAT (14) have been previously described. The GAL4-responsive reporter gene GK1 contains five GAL4 response elements upstream of a minimal adenovirus E1b promoter that has been previously described (68) but, for the purposes of these studies, was moved into a pUC vector backbone devoid of the pUC AP1 site (71).
ER expression vectors have been previously described. For ease of comparison, we have given ER expression vectors a consistent nomenclature. The previous names and sources of each construct are as follows: SG5-ER = HEG0; SG5-ERV400 = HE0 (57); n21; n41= E41; n87 = A87; n109 = M109; ERΔAB; Δ41–64; Δ87–108 (44), n101 = HE302; n117 = HE303; n50/141c = HE344/368;Δ 129–178 = HE316 (43). MORΔAF-1 = mOR121–599; MORmAF-2 = mORL543A,L544A; MORΔAF-1/mAF-2 = MOR121–599/543A,L544A (13).
Coactivator expression vectors (GRIP1, RAC3, SRC-1a, RIP-140, p300, CBP, and P/CAF) have each been previously described (24, 29, 33–35, 39, 72).
Cell Culture and Transfection
HeLa cells were maintained and transfected as previously described (55). Unless otherwise indicated, the transfections contained 5 μg CAT or luciferase reporter, 1 μg pJ3 β-galactosidase control, 1 μg ER expression vector, and 5 μg coactivator expression vector. Cell lysates were prepared 20–40 h after transfection and CAT, luciferase, and β-galactosidase assays were performed using standard methods (55). In each case CAT and luciferase activities were corrected for variations in transfection efficiency using theβ -galactosidase activity.
GST-Fusion Proteins
GST-AB and GST-LBD fusions have been previously described (55). GST AB truncations were prepared by amplification of AB sequences with standard PCR methods and the resulting fragments were cloned in frame into pGEX-5x-1 using appropriate restriction sites. GST-AB fragment 1–67 was prepared by digesting GST-AB with NotI and self-ligating. Fragment 65–184 was prepared by cloning the excised NotI fragment from GST-AB into pGEX-5X-2.
The GST-GRIP1 fragments were amplified by standard PCR methods and cloned into pGEX-2TK (563–1121) as a BamHI/EcoRI fragment or pGEX-4T1 (1121–1462) as a EcoRI/XhoI fragment.
In Vitro Translation Vectors
GRIP1, GRIP1 NR mutant, and SRC-1a transcription/translation vectors have been previously described (29, 39). GRIP1 C-terminal truncations (400, 765, 920) were prepared by restriction digestion and run-off transcription/translation. The GRIP1 C-terminal truncationΔ 1121C was prepared by digesting full-length GRIP1 with XhoI and XbaI and inserting a comparable fragment from an original partial GRIP1 cDNA clone 730-1121 (20). GRIP1 fragments 920-1291 and 1282–1462 were prepared by standard PCR methods. In both cases, the 5′-oligonucleotide used for amplification contained an in-frame Kozak consensus sequence. These fragments were inserted into pSG5 between the EcoRI and BamHI sites.
Protein-Binding Assays
GST-pull downs were performed as previously described (55). Production and use of competitor peptides and yeast two-hybrid assays are described elsewhere (29). For these studies a cDNA fragment coding for the ER-AB domain (amino acids 1–184) was PCR amplified and cloned into the EcoRI/BamHI sites of pGBT9 (CLONTECH, Palo Alto, CA). GRIP1 and SRC-1a/GAL4-AD fusions were also previously described (29).
We thank Drs. P. Chambon, D. Metzger, C. Glass, M. Parker, Y. Nakatani, J. D. Chen, and B. W. O’Malley for generous gifts of plasmids.
This work was supported by grants from the California Breast Cancer Research Program (to P.J.K., P.W., and M.S.) and from the NIH (to P.J.K., M.S., and B.K.)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}